Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance

 ,
,  ,
, 
Abstract
1. Introduction
2. EMT Mechanisms, Its Interactions with Tumor Microenvironment and Its Targeting
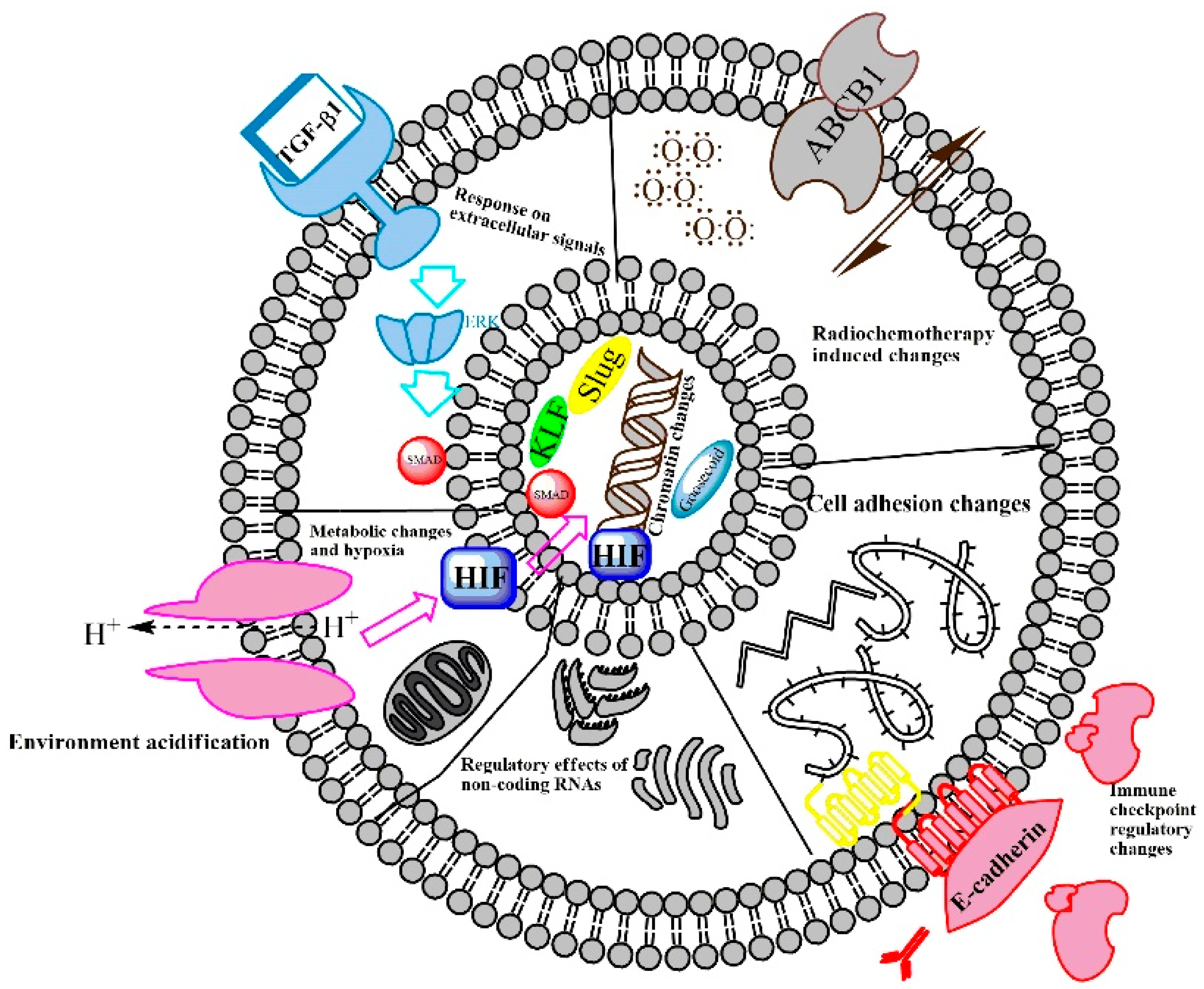
2.1. EMT-Regulating Mechanisms
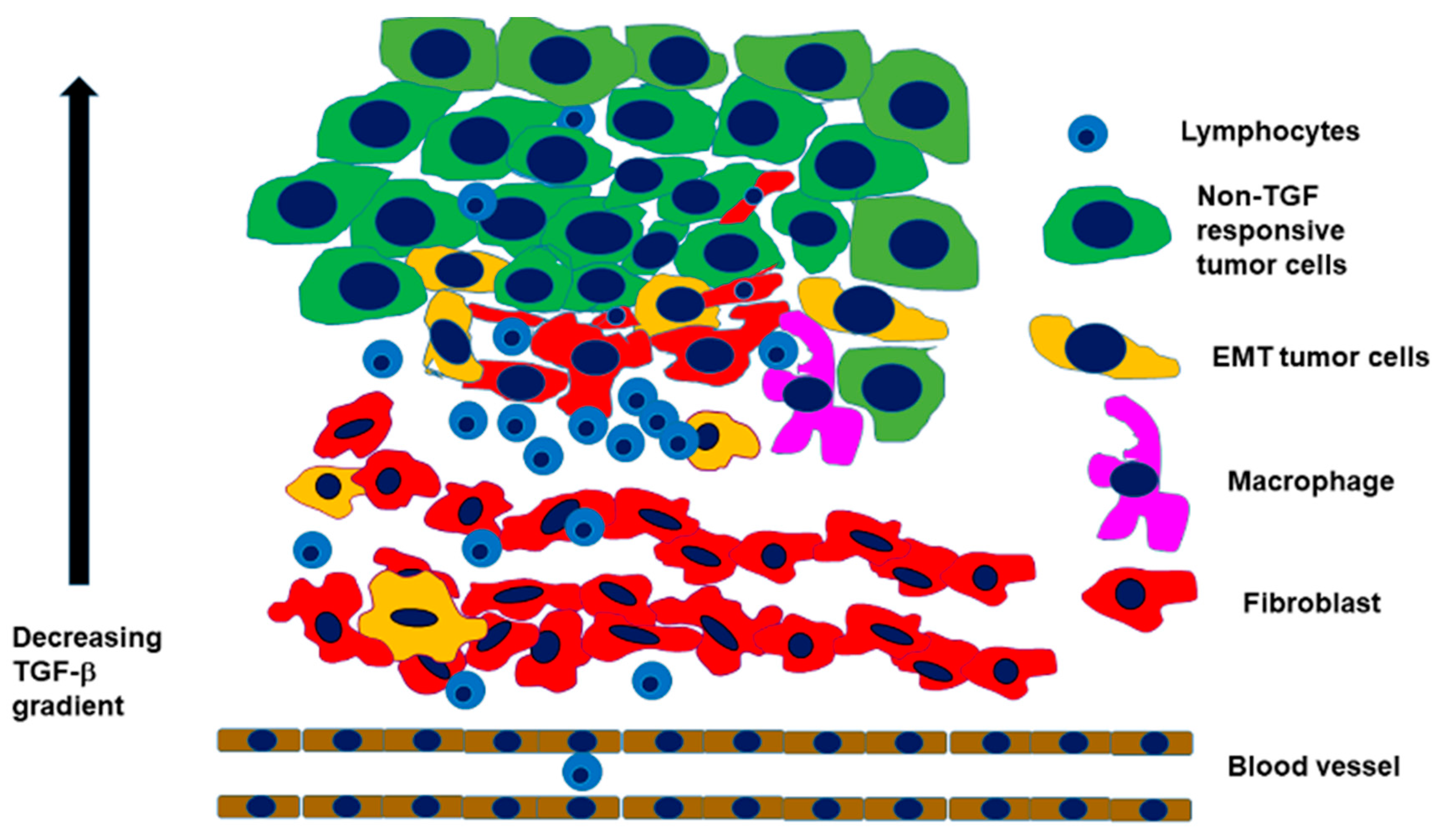
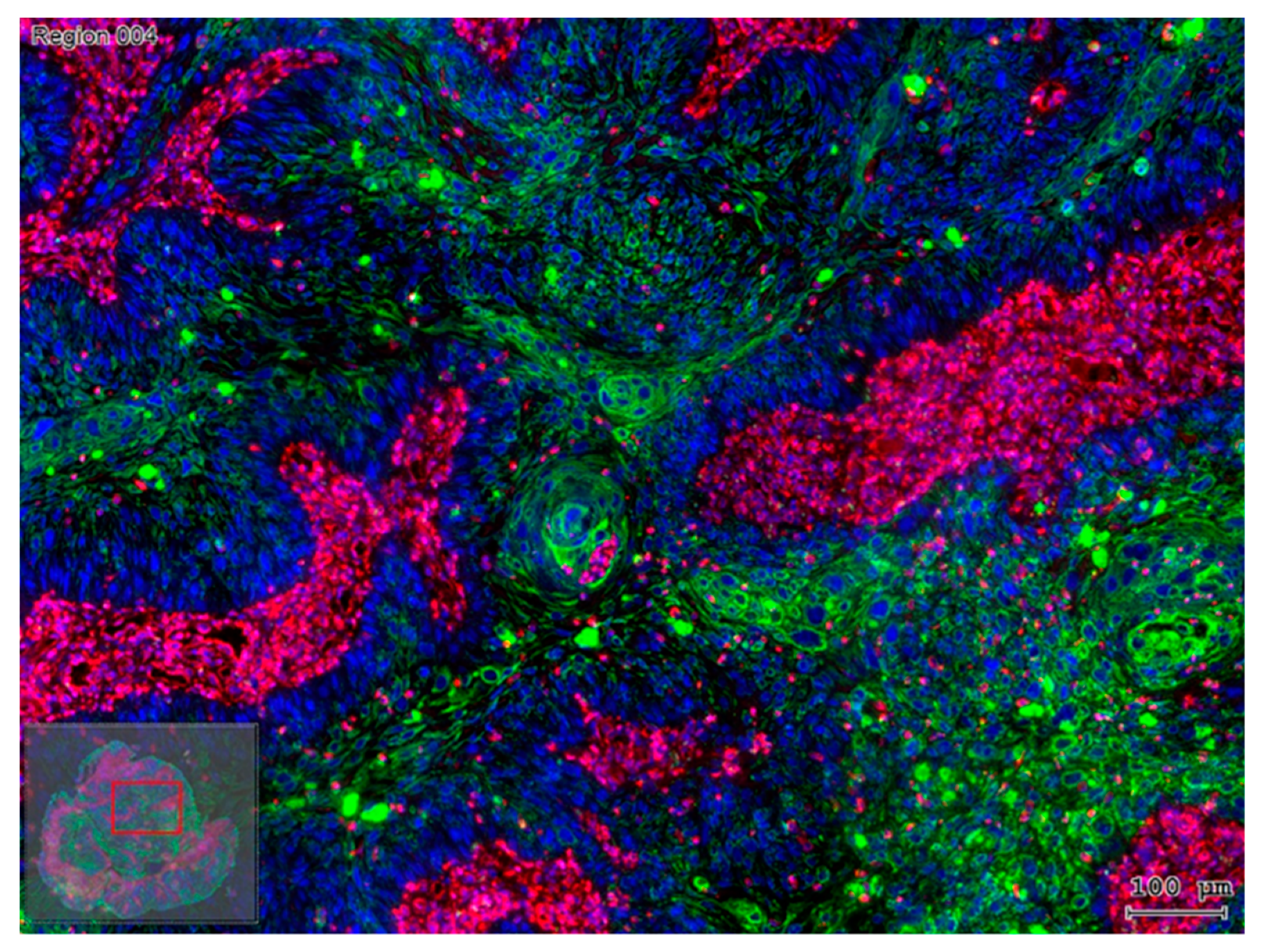
2.2. Interaction with an Inflammatory Microenvironment
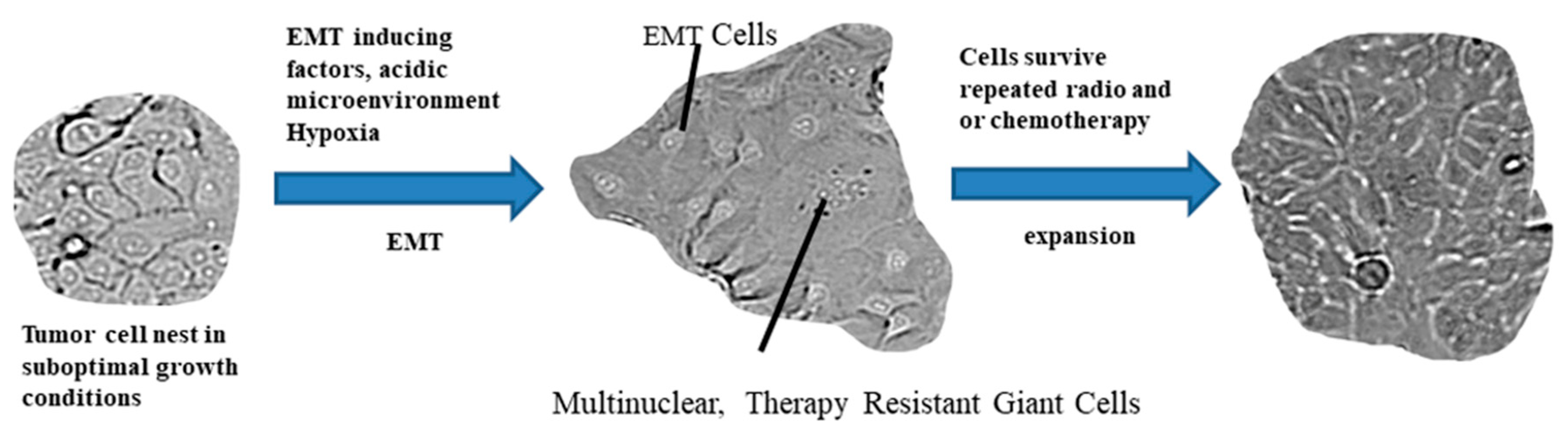
2.3. The Path from EMT to Radio/Chemoresistance
2.4. Therapeutic Targeting of the EMT Fueling Radio/Chemoresistance
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | alanine aminotransferase |
| Arg1 | arginase |
| B7-H3 | CD276, an immune checkpoint molecule |
| BDNF | brain-derived neurotrophic factor |
| Bmi-1 | Polycomb Group RING finger protein 4 (PCGF4) |
| CCL18 | CC-chemokine ligand 18 |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| EMT | epithelial to mesenchymal transition |
| FAK | focal adhesion kinase |
| FoxP3 | forkhead box P3 |
| HCC | hepatocellular carcinoma |
| HIF | hypoxia-inducible factor |
| IFN | interferon |
| IL | interleukin |
| KLF | Krüppel-like factors |
| MHC | major histocompatibility complex |
| miRNA | microRNA |
| MITA1 | metabolism-induced tumor activator 1 |
| MRX34 | miR-34a microRNA mimics |
| MTD | maximum tolerated dose |
| Nanog | stem cell homeobox transcription factor |
| NF-κB | nuclear transcription factor-kappa B |
| NK | natural killer |
| NKG2D | NK group 2, member D |
| NRF2 | nuclear factor erythroid-2-related factor 2 |
| OSCC | oral squamous cell carcinoma |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed cell death ligand 1 |
| PGCCs | polyploid giant cancer cells |
| PSA | prostate specific antigen |
| Ras | G-protein Ras (rat sarcoma) |
| Slug | snail family zinc finger 2 |
| Smad | The name is a combination of Drosophila melanogaster, MAD (mothers against decapentaplegic) and “small body size” |
| STAT | signal transducer and activator of transcription |
| STC1 | stanniocalcin-1 |
| TEC | tubular epithelial cells |
| TFs | transcription factors |
| TG2 | tissue transglutaminase 2 |
| TGF-β1 | transforming growth factor-beta 1 |
| TIM-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TNF | tumor necrosis factor |
| VEGF | vascular endothelial growth factor |
| ZEB | zinc finger E-box-binding homeobox 1 |
References
- Hay, E.D.; Zuk, A. Transformations between epithelium and mesenchyme: Normal, pathological, and experimentally induced. Am. J. Kidney Dis. 1995, 26, 678–690. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T. EMT and MET in metastasis: Where are the cancer stem cells? Cancer Cell 2012, 22, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Gruszka, A.M.; Valli, D.; Restelli, C.; Alcalay, M. Adhesion Deregulation in Acute Myeloid Leukaemia. Cells 2019, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Gao, A.; Zhang, G.; Han, H.; Zhou, Q. Decoding the knots of initiation of oncogenic epithelial-mesenchymal transition in tumor progression. Curr. Cancer Drug Targets 2013, 13, 996–1011. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. Organization and fine structure of epithelium and mesenchyme in the developing chick embryo. In Epithelial-Mesenchymal Interactions; Fleischmajer, R., Billingham, R.E., Eds.; Williams & Wilkins Co: Baltimore, MD, USA, 1968; pp. 31–55. [Google Scholar]
- Hay, E.D. Role of cell-matrix contacts in cell migration and epithelial-mesenchymal transformation. Cell Differ. Dev. 1990, 32, 367–375. [Google Scholar] [CrossRef]
- LaGamba, D.; Nawshad, A.; Hay, E.D. Microarray analysis of gene expression during epithelial-mesenchymal transformation. Dev. Dyn. 2005, 234, 132–142. [Google Scholar] [CrossRef]
- Nawshad, A.; Lagamba, D.; Polad, A.; Hay, E.D. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: Implications for embryogenesis and tumor metastasis. Cells Tissues Organs 2005, 179, 11–23. [Google Scholar] [CrossRef]
- Tang, H.M.; Kuay, K.T.; Koh, P.F.; Asad, M.; Tan, T.Z.; Chung, V.Y.; Lee, S.C.; Thiery, J.P.; Huang, R.J. An epithelial marker promoter induction screen identifies histone deacetylase inhibitors to restore epithelial differentiation and abolishes anchorage independence growth in cancers. Cell Death Discov. 2016, 2, 16041. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Li, Q.; Shi, G.; Li, N. Involvement of epithelial-mesenchymal transition in liver fibrosis. Saudi J. Gastroenterol. 2018, 24, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Ballhause, T.M.; Soldati, R.; Mertens, P.R. Sources of myofibroblasts in kidney fibrosis: All answers are correct, however to different extent! Int. Urol. Nephrol. 2014, 46, 659–664. [Google Scholar] [CrossRef]
- Ivanova, L.; Butt, M.J.; Matsell, D.G. Mesenchymal transition in kidney collecting duct epithelial cells. Am. J. Physiol. Renal. Physiol. 2008, 294, F1238–F1248. [Google Scholar] [CrossRef]
- Rastaldi, M.P.; Ferrario, F.; Giardino, L.; Dell’Antonio, G.; Grillo, C.; Grillo, P.; Strutz, F.; Muller, G.A.; Colasanti, G.; D’Amico, G. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002, 62, 137–146. [Google Scholar] [CrossRef]
- Ikegami, T.; Zhang, Y.; Matsuzaki, Y. Liver fibrosis: Possible involvement of EMT. Cells Tissues Organs 2007, 185, 213–221. [Google Scholar] [CrossRef]
- Gilles, C.; Polette, M.; Piette, J.; Birembaut, P.; Foidart, J.M. Epithelial-to-mesenchymal transition in HPV-33-transfected cervical keratinocytes is associated with increased invasiveness and expression of gelatinase A. Int. J. Cancer 1994, 59, 661–666. [Google Scholar] [CrossRef]
- Dudas, J.; Bitsche, M.; Schartinger, V.; Falkeis, C.; Sprinzl, G.M.; Riechelmann, H. Fibroblasts produce brain-derived neurotrophic factor and induce mesenchymal transition of oral tumor cells. Oral Oncol. 2011, 47, 98–103. [Google Scholar] [CrossRef]
- Dudas, J.; Dietl, W.; Romani, A.; Reinold, S.; Glueckert, R.; Schrott-Fischer, A.; Dejaco, D.; Johnson Chacko, L.; Tuertscher, R.; Schartinger, V.H.; et al. Nerve Growth Factor (NGF)-Receptor Survival Axis in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 1771. [Google Scholar] [CrossRef]
- Dudas, J.; Fullar, A.; Romani, A.; Pritz, C.; Kovalszky, I.; Hans Schartinger, V.; Mathias Sprinzl, G.; Riechelmann, H. Curcumin targets fibroblast-tumor cell interactions in oral squamous cell carcinoma. Exp. Cell Res. 2013, 319, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Dudas, J.; Riml, A.; Tuertscher, R.; Pritz, C.; Steinbichler, T.B.; Schartinger, V.H.; Sprung, S.; Glueckert, R.; Schrott-Fischer, A.; Johnson Chacko, L.; et al. Brain-Derived Neurotrophin and TrkB in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 272. [Google Scholar] [CrossRef] [PubMed]
- Metzler, V.M.; Pritz, C.; Riml, A.; Romani, A.; Tuertscher, R.; Steinbichler, T.; Dejaco, D.; Riechelmann, H.; Dudas, J. Separation of cell survival, growth, migration, and mesenchymal transdifferentiation effects of fibroblast secretome on tumor cells of head and neck squamous cell carcinoma. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Alshaimaa, A.; Maria, M.V.; Daniel, D.; Herbert, R.; Jozsef, D.; Ira-Ida, S. Epithelial-mesenchymal crosstalk induces radioresistance in HNSCC cells. Oncotarget 2018, 9, 3641–3652. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Metzler, V.; Pritz, C.; Riechelmann, H.; Dudas, J. Tumor-associated fibroblast-conditioned medium induces CDDP resistance in HNSCC cells. Oncotarget 2016, 7, 2508–2518. [Google Scholar] [CrossRef] [PubMed]
- Tarin, D.; Thompson, E.W.; Newgreen, D.F. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005, 65, 5996–6000. [Google Scholar] [CrossRef]
- Nieto, M.A. Context-specific roles of EMT programmes in cancer cell dissemination. Nat. Cell Biol. 2017, 19, 416–418. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Labernadie, A.; Kato, T.; Brugues, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; Gonzalez-Tarrago, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef]
- Lopez-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Savic, D.; Dejaco, D.; Romani, A.; Kofler, B.; Skvortsova, I.I.; Riechelmann, H.; Dudas, J. Pleiotropic Effects of Epithelial Mesenchymal Crosstalk on Head and Neck Cancer: EMT and beyond. Cancer Microenviron. 2019. [Google Scholar] [CrossRef]
- Prakash, V.; Carson, B.B.; Feenstra, J.M.; Dass, R.A.; Sekyrova, P.; Hoshino, A.; Petersen, J.; Guo, Y.; Parks, M.M.; Kurylo, C.M.; et al. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat. Commun. 2019, 10, 2110. [Google Scholar] [CrossRef]
- Fazilaty, H.; Rago, L.; Kass Youssef, K.; Ocana, O.H.; Garcia-Asencio, F.; Arcas, A.; Galceran, J.; Nieto, M.A. A gene regulatory network to control EMT programs in development and disease. Nat. Commun. 2019, 10, 5115. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, Y.; Liu, W.; Zhao, G.; Lee, S.; Balogh, A.; Zou, Y.; Guo, Y.; Zhang, Z.; Gu, W.; et al. Doxycycline inducible Kruppel-like factor 4 lentiviral vector mediates mesenchymal to epithelial transition in ovarian cancer cells. PLoS ONE 2014, 9, e105331. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.; Meyer-Schaller, N.; Arnold, P.; Antoniadis, H.; Pachkov, M.; van Nimwegen, E.; Christofori, G. Klf4 is a transcriptional regulator of genes critical for EMT, including Jnk1 (Mapk8). PLoS ONE 2013, 8, e57329. [Google Scholar] [CrossRef]
- Micke, P.; Ostman, A. Exploring the tumour environment: Cancer-associated fibroblasts as targets in cancer therapy. Expert Opin. Ther. Targets 2005, 9, 1217–1233. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.D.; Parvani, J.G.; Schiemann, W.P. The relevance of the TGF-beta Paradox to EMT-MET programs. Cancer Lett. 2013, 341, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef]
- Mayorca-Guiliani, A.; Erler, J.T. The potential for targeting extracellular LOX proteins in human malignancy. Onco Targets Ther. 2013, 6, 1729–1735. [Google Scholar] [CrossRef]
- Yang, X.G.; Zhu, L.C.; Wang, Y.J.; Li, Y.Y.; Wang, D. Current Advance of Therapeutic Agents in Clinical Trials Potentially Targeting Tumor Plasticity. Front. Oncol. 2019, 9, 887. [Google Scholar] [CrossRef]
- Jin, Y.; Xu, K.; Chen, Q.; Wang, B.; Pan, J.; Huang, S.; Wei, Y.; Ma, H. Simvastatin inhibits the development of radioresistant esophageal cancer cells by increasing the radiosensitivity and reversing EMT process via the PTEN-PI3K/AKT pathway. Exp. Cell Res. 2018, 362, 362–369. [Google Scholar] [CrossRef]
- Zang, C.; Liu, X.; Li, B.; He, Y.; Jing, S.; He, Y.; Wu, W.; Zhang, B.; Ma, S.; Dai, W.; et al. IL-6/STAT3/TWIST inhibition reverses ionizing radiation-induced EMT and radioresistance in esophageal squamous carcinoma. Oncotarget 2017, 8, 11228–11238. [Google Scholar] [CrossRef]
- Kidan, N.; Khamaisie, H.; Ruimi, N.; Roitman, S.; Eshel, E.; Dally, N.; Ruthardt, M.; Mahajna, J. Ectopic Expression of Snail and Twist in Ph+ Leukemia Cells Upregulates CD44 Expression and Alters Their Differentiation Potential. J. Cancer 2017, 8, 3952–3968. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cogle, C.R.; Bosse, R.C.; Brewer, T.; Migdady, Y.; Shirzad, R.; Kampen, K.R.; Saki, N. Acute myeloid leukemia in the vascular niche. Cancer Lett. 2016, 380, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kim, D.; Kim, D.; Park, J.; Koh, Y.; Yoon, S.S. Truncation of MYH8 tail in AML: A Novel prognostic marker with increase cell migration and epithelial-mesenchymal transition utilizing RAF/MAPK pathway. Carcinogenesis 2019. [Google Scholar] [CrossRef] [PubMed]
- De Coninck, S.; Berx, G.; Taghon, T.; Van Vlierberghe, P.; Goossens, S. ZEB2 in T-cells and T-ALL. Adv. Biol. Regul. 2019. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Stavropoulou, V.; Kaspar, S.; Brault, L.; Sanders, M.A.; Juge, S.; Morettini, S.; Tzankov, A.; Iacovino, M.; Lau, I.J.; Milne, T.A.; et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 2016, 30, 43–58. [Google Scholar] [CrossRef]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef]
- Sun, H.; Peng, Z.; Tang, H.; Xie, D.; Jia, Z.; Zhong, L.; Zhao, S.; Ma, Z.; Gao, Y.; Zeng, L.; et al. Loss of KLF4 and consequential downregulation of Smad7 exacerbate oncogenic TGF-beta signaling in and promote progression of hepatocellular carcinoma. Oncogene 2017, 36, 2957–2968. [Google Scholar] [CrossRef]
- Ding, X.; Wang, X.; Gong, Y.; Ruan, H.; Sun, Y.; Yu, Y. KLF7 overexpression in human oral squamous cell carcinoma promotes migration and epithelial-mesenchymal transition. Oncol. Lett. 2017, 13, 2281–2289. [Google Scholar] [CrossRef]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Beltran, M.; Puig, I.; Pena, C.; Garcia, J.M.; Alvarez, A.B.; Pena, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.; Garcia, J.M.; Silva, J.; Garcia, V.; Rodriguez, R.; Alonso, I.; Millan, I.; Salas, C.; de Herreros, A.G.; Munoz, A.; et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: Clinicopathological correlations. Hum. Mol. Genet. 2005, 14, 3361–3370. [Google Scholar] [CrossRef] [PubMed]
- Poser, I.; Dominguez, D.; de Herreros, A.G.; Varnai, A.; Buettner, R.; Bosserhoff, A.K. Loss of E-cadherin expression in melanoma cells involves up-regulation of the transcriptional repressor Snail. J. Biol. Chem. 2001, 276, 24661–24666. [Google Scholar] [CrossRef] [PubMed]
- Papiewska-Pajak, I.; Kowalska, M.A.; Boncela, J. Expression and activity of SNAIL transcription factor during Epithelial to Mesenchymal Transition (EMT) in cancer progression. Postepy. Hig. Med. Dosw. 2016, 70, 968–980. [Google Scholar] [CrossRef]
- Ma, M.; Xu, H.; Liu, G.; Wu, J.; Li, C.; Wang, X.; Zhang, S.; Xu, H.; Ju, S.; Cheng, W.; et al. Metabolism-induced tumor activator 1 (MITA1), an Energy Stress-Inducible Long Noncoding RNA, Promotes Hepatocellular Carcinoma Metastasis. Hepatology 2019, 70, 215–230. [Google Scholar] [CrossRef]
- Andreucci, E.; Peppicelli, S.; Carta, F.; Brisotto, G.; Biscontin, E.; Ruzzolini, J.; Bianchini, F.; Biagioni, A.; Supuran, C.T.; Calorini, L. Carbonic anhydrase IX inhibition affects viability of cancer cells adapted to extracellular acidosis. J. Mol. Med. (Berl) 2017, 95, 1341–1353. [Google Scholar] [CrossRef]
- Tian, X.P.; Wang, C.Y.; Jin, X.H.; Li, M.; Wang, F.W.; Huang, W.J.; Yun, J.P.; Xu, R.H.; Cai, Q.Q.; Xie, D. Acidic Microenvironment Up-Regulates Exosomal miR-21 and miR-10b in Early-Stage Hepatocellular Carcinoma to Promote Cancer Cell Proliferation and Metastasis. Theranostics 2019, 9, 1965–1979. [Google Scholar] [CrossRef]
- Joseph, J.P.; Harishankar, M.K.; Pillai, A.A.; Devi, A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef]
- Zhang, J.; Cheng, Q.; Zhou, Y.; Wang, Y.; Chen, X. Slug is a key mediator of hypoxia induced cadherin switch in HNSCC: Correlations with poor prognosis. Oral Oncol. 2013, 49, 1043–1050. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Peng, Y.; Wu, X.; Meng, S.; Yu, W.; Zhao, J.; Zhang, H.; Wang, J.; Li, W. CCL18 secreted from M2 macrophages promotes migration and invasion via the PI3K/Akt pathway in gallbladder cancer. Cell Oncol. (Dordr.) 2019, 42, 81–92. [Google Scholar] [CrossRef]
- Lv, N.; Gao, Y.; Guan, H.; Wu, D.; Ding, S.; Teng, W.; Shan, Z. Inflammatory mediators, tumor necrosis factor-alpha and interferon-gamma, induce EMT in human PTC cell lines. Oncol. Lett. 2015, 10, 2591–2597. [Google Scholar] [CrossRef]
- Long, X.; Ye, Y.; Zhang, L.; Liu, P.; Yu, W.; Wei, F.; Ren, X.; Yu, J. IL-8, a novel messenger to cross-link inflammation and tumor EMT via autocrine and paracrine pathways (Review). Int. J. Oncol. 2016, 48, 5–12. [Google Scholar] [CrossRef]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef]
- Lou, Y.; Diao, L.; Cuentas, E.R.; Denning, W.L.; Chen, L.; Fan, Y.H.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.A.; Behrens, C.; et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef]
- Chen, L.; Xiong, Y.; Li, J.; Zheng, X.; Zhou, Q.; Turner, A.; Wu, C.; Lu, B.; Jiang, J. PD-L1 Expression Promotes Epithelial to Mesenchymal Transition in Human Esophageal Cancer. Cell Physiol. Biochem. 2017, 42, 2267–2280. [Google Scholar] [CrossRef]
- Wangpaichitr, M.; Kandemir, H.; Li, Y.Y.; Wu, C.; Nguyen, D.; Feun, L.G.; Kuo, M.T.; Savaraj, N. Relationship of Metabolic Alterations and PD-L1 Expression in Cisplatin Resistant Lung Cancer. Cell Dev. Biol. 2017, 6. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Saci, A.; Szabo, P.M.; Chasalow, S.D.; Castillo-Martin, M.; Domingo-Domenech, J.; Siefker-Radtke, A.; Sharma, P.; Sfakianos, J.P.; Gong, Y.; et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat. Commun. 2018, 9, 3503. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.C.; Hwang, W.T.; Davis, C.; Deshpande, C.; Jeffries, S.; Rajpurohit, Y.; Krishna, V.; Smirnov, D.; Verona, R.; Lorenzi, M.V.; et al. Gene signatures of tumor inflammation and epithelial-to-mesenchymal transition (EMT) predict responses to immune checkpoint blockade in lung cancer with high accuracy. Lung Cancer 2019, 139, 1–8. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef]
- Noman, M.Z.; Van Moer, K.; Marani, V.; Gemmill, R.M.; Tranchevent, L.C.; Azuaje, F.; Muller, A.; Chouaib, S.; Thiery, J.P.; Berchem, G.; et al. CD47 is a direct target of SNAI1 and ZEB1 and its blockade activates the phagocytosis of breast cancer cells undergoing EMT. Oncoimmunology 2018, 7, e1345415. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Huergo-Zapico, L.; Galvan, J.A.; Rodrigo, L.; de Herreros, A.G.; Astudillo, A.; Gonzalez, S. Epithelial-mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. J. Immunol. 2013, 190, 4408–4419. [Google Scholar] [CrossRef]
- Chargari, C.; Clemenson, C.; Martins, I.; Perfettini, J.L.; Deutsch, E. Understanding the functions of tumor stroma in resistance to ionizing radiation: Emerging targets for pharmacological modulation. Drug Resist. Updat. 2013, 16, 10–21. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Donnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef]
- Guan, J.; Zhang, H.; Wen, Z.; Gu, Y.; Cheng, Y.; Sun, Y.; Zhang, T.; Jia, C.; Lu, Z.; Chen, J. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett. 2014, 345, 132–139. [Google Scholar] [CrossRef]
- Sui, H.; Zhu, L.; Deng, W.; Li, Q. Epithelial-mesenchymal transition and drug resistance: Role, molecular mechanisms, and therapeutic strategies. Oncol. Res. Treat. 2014, 37, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Menicacci, B.; Laurenzana, A.; Chilla, A.; Margheri, F.; Peppicelli, S.; Tanganelli, E.; Fibbi, G.; Giovannelli, L.; Del Rosso, M.; Mocali, A. Chronic Resveratrol Treatment Inhibits MRC5 Fibroblast SASP-Related Protumoral Effects on Melanoma Cells. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Nwani, N.G.; Deguiz, M.L.; Jimenez, B.; Vinokour, E.; Dubrovskyi, O.; Ugolkov, A.; Mazar, A.P.; Volpert, O.V. Melanoma Cells Block PEDF Production in Fibroblasts to Induce the Tumor-Promoting Phenotype of Cancer-Associated Fibroblasts. Cancer Res. 2016, 76, 2265–2276. [Google Scholar] [CrossRef] [PubMed]
- Sommers, C.L.; Heckford, S.E.; Skerker, J.M.; Worland, P.; Torri, J.A.; Thompson, E.W.; Byers, S.W.; Gelmann, E.P. Loss of epithelial markers and acquisition of vimentin expression in adriamycin- and vinblastine-resistant human breast cancer cell lines. Cancer Res. 1992, 52, 5190–5197. [Google Scholar]
- Chang, T.H.; Tsai, M.F.; Su, K.Y.; Wu, S.G.; Huang, C.P.; Yu, S.L.; Yu, Y.L.; Lan, C.C.; Yang, C.H.; Lin, S.B.; et al. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am. J. Respir. Crit. Care Med. 2011, 183, 1071–1079. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef]
- Munoz, M.; Henderson, M.; Haber, M.; Norris, M. Role of the MRP1/ABCC1 multidrug transporter protein in cancer. IUBMB Life 2007, 59, 752–757. [Google Scholar] [CrossRef]
- Saxena, M.; Stephens, M.A.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef]
- Ghosh, R.D.; Ghuwalewala, S.; Das, P.; Mandloi, S.; Alam, S.K.; Chakraborty, J.; Sarkar, S.; Chakrabarti, S.; Panda, C.K.; Roychoudhury, S. MicroRNA profiling of cisplatin-resistant oral squamous cell carcinoma cell lines enriched with cancer-stem-cell-like and epithelial-mesenchymal transition-type features. Sci. Rep. 2016, 6, 23932. [Google Scholar] [CrossRef]
- Skvortsova, I.; Skvortsov, S.; Raju, U.; Stasyk, T.; Riesterer, O.; Schottdorf, E.M.; Popper, B.A.; Schiestl, B.; Eichberger, P.; Debbage, P.; et al. Epithelial-to-mesenchymal transition and c-myc expression are the determinants of cetuximab-induced enhancement of squamous cell carcinoma radioresponse. Radiother. Oncol. 2010, 96, 108–115. [Google Scholar] [CrossRef]
- Lu, J.; Zhong, Y.; Chen, J.; Lin, X.; Lin, Z.; Wang, N.; Lin, S. Radiation Enhances the Epithelial- Mesenchymal Transition of A549 Cells via miR3591-5p/USP33/PPM1A. Cell Physiol. Biochem. 2018, 50, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Tsai, J.T.; Chao, T.Y.; Ma, H.I.; Liu, W.H. The STAT3/Slug Axis Enhances Radiation-Induced Tumor Invasion and Cancer Stem-like Properties in Radioresistant Glioblastoma. Cancers (Basel) 2018, 10, 512. [Google Scholar] [CrossRef] [PubMed]
- Kupferman, M.E.; Jiffar, T.; El-Naggar, A.; Yilmaz, T.; Zhou, G.; Xie, T.; Feng, L.; Wang, J.; Holsinger, F.C.; Yu, D.; et al. TrkB induces EMT and has a key role in invasion of head and neck squamous cell carcinoma. Oncogene 2010, 29, 2047–2059. [Google Scholar] [CrossRef]
- Agnihotri, N.; Kumar, S.; Mehta, K. Tissue transglutaminase as a central mediator in inflammation-induced progression of breast cancer. Breast Cancer Res. 2013, 15, 202. [Google Scholar] [CrossRef]
- Tato-Costa, J.; Casimiro, S.; Pacheco, T.; Pires, R.; Fernandes, A.; Alho, I.; Pereira, P.; Costa, P.; Castelo, H.B.; Ferreira, J.; et al. Therapy-Induced Cellular Senescence Induces Epithelial-to-Mesenchymal Transition and Increases Invasiveness in Rectal Cancer. Clin. Colorectal Cancer 2016, 15, 170–178. [Google Scholar] [CrossRef]
- Marie-Egyptienne, D.T.; Lohse, I.; Hill, R.P. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: Potential role of hypoxia. Cancer Lett 2013, 341, 63–72. [Google Scholar] [CrossRef]
- Jiao, M.; Nan, K.J. Activation of PI3 kinase/Akt/HIF-1alpha pathway contributes to hypoxia-induced epithelial-mesenchymal transition and chemoresistance in hepatocellular carcinoma. Int. J. Oncol. 2012, 40, 461–468. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Hanash, S.; Liu, J. iTRAQ-based proteomic analysis of polyploid giant cancer cells and budding progeny cells reveals several distinct pathways for ovarian cancer development. PLoS ONE 2013, 8, e80120. [Google Scholar] [CrossRef]
- Antoon, J.W.; Lai, R.; Struckhoff, A.P.; Nitschke, A.M.; Elliott, S.; Martin, E.C.; Rhodes, L.V.; Yoon, N.S.; Salvo, V.A.; Shan, B.; et al. Altered death receptor signaling promotes epithelial-to-mesenchymal transition and acquired chemoresistance. Sci. Rep. 2012, 2, 539. [Google Scholar] [CrossRef]
- Boone, J.D.; Arend, R.C.; Johnston, B.E.; Cooper, S.J.; Gilchrist, S.A.; Oelschlager, D.K.; Grizzle, W.E.; McGwin, G., Jr.; Gangrade, A.; Straughn, J.M., Jr.; et al. Targeting the Wnt/beta-catenin pathway in primary ovarian cancer with the porcupine inhibitor WNT974. Lab. Investig. 2016, 96, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Finkelstein, D.M.; Barrios, C.; Martin, M.; Iwata, H.; Hegg, R.; Glaspy, J.; Perianez, A.M.; Tonkin, K.; Deleu, I.; et al. Adjuvant denosumab in early breast cancer (D-CARE): An international, multicentre, randomised, controlled, phase 3 trial. Lancet Onco. 2020, 21, 60–72. [Google Scholar] [CrossRef]
- Nambiar, D.K.; Rajamani, P.; Singh, R.P. Silibinin attenuates ionizing radiation-induced pro-angiogenic response and EMT in prostate cancer cells. Biochem. Biophys. Res. Commun. 2015, 456, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Flaig, T.W.; Gustafson, D.L.; Su, L.J.; Zirrolli, J.A.; Crighton, F.; Harrison, G.S.; Pierson, A.S.; Agarwal, R.; Glode, L.M. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Investig. New Drugs 2007, 25, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Koochekpour, S.; Strup, S.E.; Kyprianou, N. Reversion of epithelial-mesenchymal transition by a novel agent DZ-50 via IGF binding protein-3 in prostate cancer cells. Oncotarget 2017, 8, 78507–78519. [Google Scholar] [CrossRef]
- Cuyas, E.; Perez-Sanchez, A.; Micol, V.; Menendez, J.A.; Bosch-Barrera, J. STAT3-targeted treatment with silibinin overcomes the acquired resistance to crizotinib in ALK-rearranged lung cancer. Cell Cycle 2016, 15, 3413–3418. [Google Scholar] [CrossRef]
- Nambiar, D.K.; Rajamani, P.; Deep, G.; Jain, A.K.; Agarwal, R.; Singh, R.P. Silibinin Preferentially Radiosensitizes Prostate Cancer by Inhibiting DNA Repair Signaling. Mol. Cancer Ther. 2015, 14, 2722–2734. [Google Scholar] [CrossRef]
- Flaig, T.W.; Glode, M.; Gustafson, D.; van Bokhoven, A.; Tao, Y.; Wilson, S.; Su, L.J.; Li, Y.; Harrison, G.; Agarwal, R.; et al. A study of high-dose oral silybin-phytosome followed by prostatectomy in patients with localized prostate cancer. Prostate 2010, 70, 848–855. [Google Scholar] [CrossRef]
- Antoon, J.W.; Nitzchke, A.M.; Martin, E.C.; Rhodes, L.V.; Nam, S.; Wadsworth, S.; Salvo, V.A.; Elliott, S.; Collins-Burow, B.; Nephew, K.P.; et al. Inhibition of p38 mitogen-activated protein kinase alters microRNA expression and reverses epithelial-to-mesenchymal transition. Int. J. Oncol. 2013, 42, 1139–1150. [Google Scholar] [CrossRef]
- Li, X.L.; Hara, T.; Choi, Y.; Subramanian, M.; Francis, P.; Bilke, S.; Walker, R.L.; Pineda, M.; Zhu, Y.; Yang, Y.; et al. A p21-ZEB1 complex inhibits epithelial-mesenchymal transition through the microRNA 183-96-182 cluster. Mol. Cell Biol. 2014, 34, 533–550. [Google Scholar] [CrossRef]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Kothari, A.N.; Mi, Z.; Zapf, M.; Kuo, P.C. Novel clinical therapeutics targeting the epithelial to mesenchymal transition. Clin. Transl. Med. 2014, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Carducci, M.; Sepulveda-Sanchez, J.M.; Azaro, A.; Calvo, E.; Seoane, J.; Brana, I.; Sicart, E.; Gueorguieva, I.; Cleverly, A.; et al. Pharmacokinetic, pharmacodynamic and biomarker evaluation of transforming growth factor-beta receptor I kinase inhibitor, galunisertib, in phase 1 study in patients with advanced cancer. Investig. New Drugs 2015, 33, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Cufi, S.; Del Barco, S.; Martin-Castillo, B.; Menendez, J.A. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 2010, 9, 3807–3814. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Cheng, X.; Wang, Y.; Han, R.; Li, L.; Xiang, T.; He, L.; Long, H.; Zhu, B.; He, Y. Metformin inhibits the IL-6-induced epithelial-mesenchymal transition and lung adenocarcinoma growth and metastasis. PLoS ONE 2014, 9, e95884. [Google Scholar] [CrossRef]
- Arrieta, O.; Barron, F.; Padilla, M.S.; Aviles-Salas, A.; Ramirez-Tirado, L.A.; Arguelles Jimenez, M.J.; Vergara, E.; Zatarain-Barron, Z.L.; Hernandez-Pedro, N.; Cardona, A.F.; et al. Effect of Metformin Plus Tyrosine Kinase Inhibitors Compared With Tyrosine Kinase Inhibitors Alone in Patients With Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, e192553. [Google Scholar] [CrossRef]
- Toden, S.; Okugawa, Y.; Jascur, T.; Wodarz, D.; Komarova, N.L.; Buhrmann, C.; Shakibaei, M.; Boland, C.R.; Goel, A. Curcumin mediates chemosensitization to 5-fluorouracil through miRNA-induced suppression of epithelial-to-mesenchymal transition in chemoresistant colorectal cancer. Carcinogenesis 2015, 36, 355–367. [Google Scholar] [CrossRef]
- Kanai, M.; Otsuka, Y.; Otsuka, K.; Sato, M.; Nishimura, T.; Mori, Y.; Kawaguchi, M.; Hatano, E.; Kodama, Y.; Matsumoto, S.; et al. A phase I study investigating the safety and pharmacokinetics of highly bioavailable curcumin (Theracurmin) in cancer patients. Cancer Chemother. Pharmacol. 2013, 71, 1521–1530. [Google Scholar] [CrossRef]
- Teknos, T.N.; Grecula, J.; Agrawal, A.; Old, M.O.; Ozer, E.; Carrau, R.; Kang, S.; Rocco, J.; Blakaj, D.; Diavolitsis, V.; et al. A phase 1 trial of Vorinostat in combination with concurrent chemoradiation therapy in the treatment of advanced staged head and neck squamous cell carcinoma. Investig. New Drugs 2019, 37, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Willis, A.; Kornhauser, N.; Ward, M.M.; Lee, S.B.; Nackos, E.; Seo, B.R.; Chuang, E.; Cigler, T.; Moore, A.; et al. Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases. Clin. Cancer Res. 2017, 23, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Di Cosimo, S.; La Verde, N.; Moretti, A.; Cazzaniga, M.E.; Generali, D.; Bianchi, G.V.; Mariani, L.; Torri, V.; Crippa, F.; Paolini, B.; et al. Neoadjuvant eribulin mesylate following anthracycline and taxane in triple negative breast cancer: Results from the HOPE study. PLoS ONE 2019, 14, e0220644. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Gordon, M.; Tsai, F.; Papadopoulos, K.P.; Rasco, D.; Beeram, M.; Fu, S.; Janku, F.; Hynes, S.M.; Gundala, S.R.; et al. A phase I study of LY3164530, a bispecific antibody targeting MET and EGFR, in patients with advanced or metastatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 407–418. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial ID | Compound | Compound Type | Phase | EMT Target | Tumor Type | Side Effects | Outcome | Comments | Reference |
|---|---|---|---|---|---|---|---|---|---|
| NCT00487721 | silibinin | flavonoid | I | signal transduction | Prostate cancer | elevation of ALT | no OR | low tissue penetration | Flaig et al. 2010 [119] |
| NCT01077154 | denosumab | monoclonal antibody | III | NF-κB-alpha | breast cancer | neutropenia, leukopenia | no improvement | Coleman et al. [113] | |
| NCT01829971 | MRX34 | miRNA | II | miRNA-related regulation | solid tumors refractory to standard treatment | fatigue, back pain, diarrhea, lymphopenia, neutropenia | PR, SD | dexamethasone pretreatment required | Zhang, Liao Tang 2019 [122] |
| NCT02536469 | BMS-986253 | monoclonal antibody | II | tumor microenvironment | locally advanced solid tumors | fatigue, hypophosphatemia hypersomnia | SD | safe, well tolerated | Chan et al. 2017 [133] |
| EudraCT 2012-004956-12 | eribulin mesylate | taxane-based cytostatics | I | mesenchymal expression | primary triple-negative breast cancer | neutropenia | pCR | biological activity in triple-negative breast cancer | Di Cosimo et al. 2019 [134] |
| LY3164530 | bispecific antibody | I | inhibition of EMT in tumor cells | patients with advanced or metastatic cancer | rash, acneiform dermatitis, hypomagnesemia, paronychia, fatigue, skin fissures, hypokalemia | ORR: 10.3%, DCR: 57.7% | significant toxicities associated with EGFR inhibition | Patnaik et al. 2018 [135] | |
| LY2157299 | TGF-β receptor kinase inhibitor | I | TGF-β1 | refractory/relapsed malignant glioma | DCR: 15% | favorable toxicity profile and no cardiac adverse events | Rodon et al. 2015 [125] | ||
| NCT00356460 | fresolimumab | monoclonal antibody | I | TGF-β1 | advanced malignant melanoma and renal carcinoma | keratoacanthomas, hyperkeratosis | PR: 3% | no dose limiting toxicity | Morris et al. [126] |
| NCT03071705 | metformin hydrochloride | biguanide | II | modifies EMT machinery | advanced lung adenocarcinoma | median PFS and OS significantly longer | Arrieta et al. 2019 [129] | ||
| ID: 000002950 | Theracurmin | polyphenol | I | SNAIL | pancreatic or biliary tract cancer patients | no unexpected adverse events | DCR: 25% | significantly available curcumin | Kanai et al. 2013 [131] |
| NCT01064921 | vorinostat | cytostatics | I | reversal of EMT | stage III, IVa, IVb HNSCC | anemia, leukopenia, lymphopenia | complete response 96.2% | with concurrent chemoradiation, safe and effective | Teknos et al. 2019 [132] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dudás, J.; Ladányi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells 2020, 9, 428. https://doi.org/10.3390/cells9020428
Dudás J, Ladányi A, Ingruber J, Steinbichler TB, Riechelmann H. Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells. 2020; 9(2):428. https://doi.org/10.3390/cells9020428
Chicago/Turabian StyleDudás, József, Andrea Ladányi, Julia Ingruber, Teresa Bernadette Steinbichler, and Herbert Riechelmann. 2020. "Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance" Cells 9, no. 2: 428. https://doi.org/10.3390/cells9020428
APA StyleDudás, J., Ladányi, A., Ingruber, J., Steinbichler, T. B., & Riechelmann, H. (2020). Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells, 9(2), 428. https://doi.org/10.3390/cells9020428