Identification of PARP-1, Histone H1 and SIRT-1 as New Regulators of Breast Cancer-Related Aromatase Promoter I.3/II


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Aromatase Activity Testing
2.3. NAD+/NADH Quantification
2.4. Preparation of Soluble Nuclear Extracts
2.5. Isolation and Identification of Putative DNA-Binding Proteins
2.6. Electrophoretic Mobility Shift Assays
2.7. Immunoprecipitation-Based DNA-Binding Assay
2.8. Western Blotting
2.9. Transfection and Luciferase Reporter Gene Assays
2.10. Chromatin Immunoprecipitation (ChIP)
2.11. Quantification of Aromatase mRNA-Expression in BAFs
2.12. Quantification of Aromatase Promoter-Utilization in Transfected PARP-1 Wild-Type and Knock-Out MEFs
2.13. Statistical Analyses
3. Results
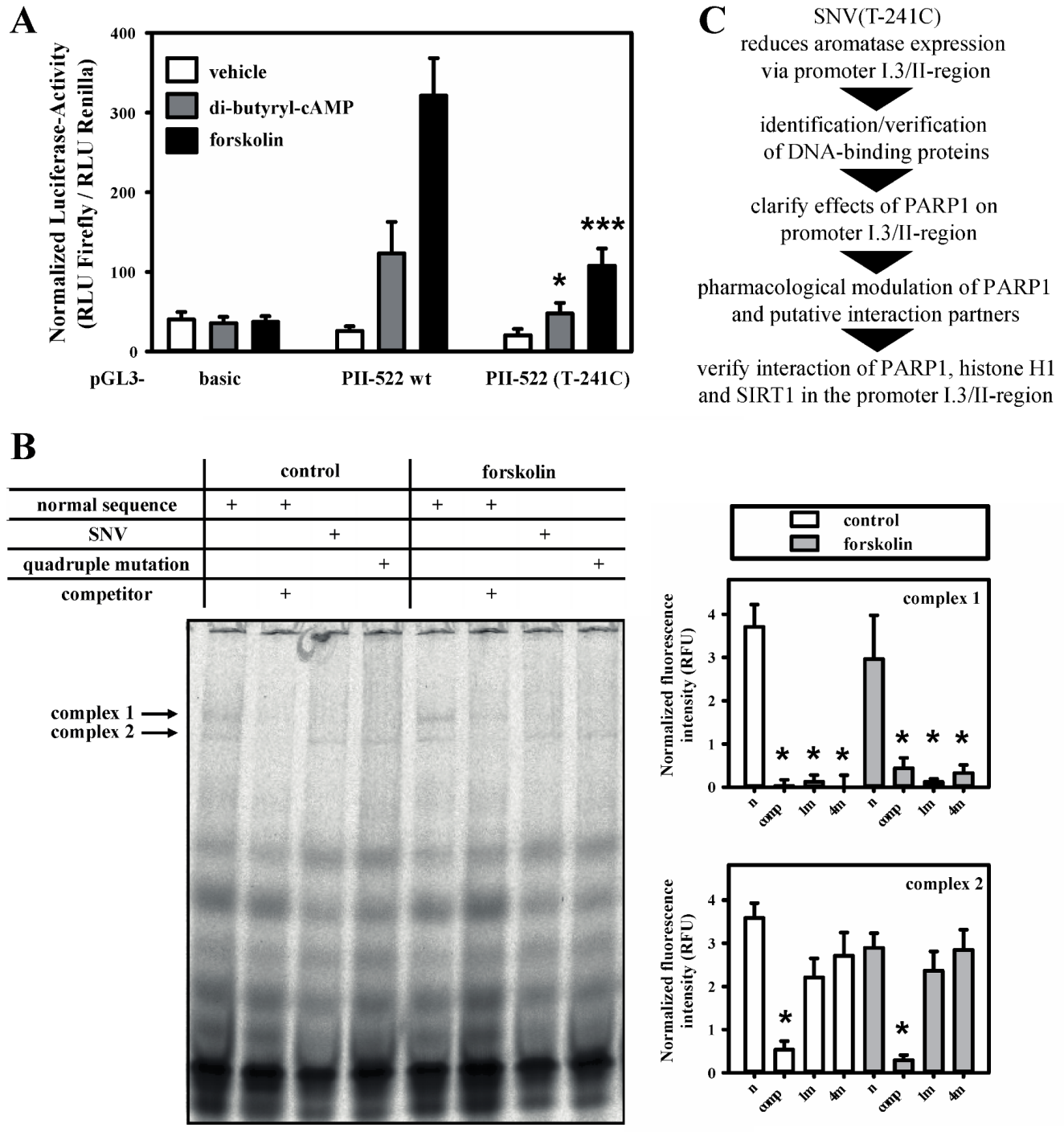
3.1. SNV-Dependent Protein Complex Formation in the Aromatase Promoter I.3/II Region
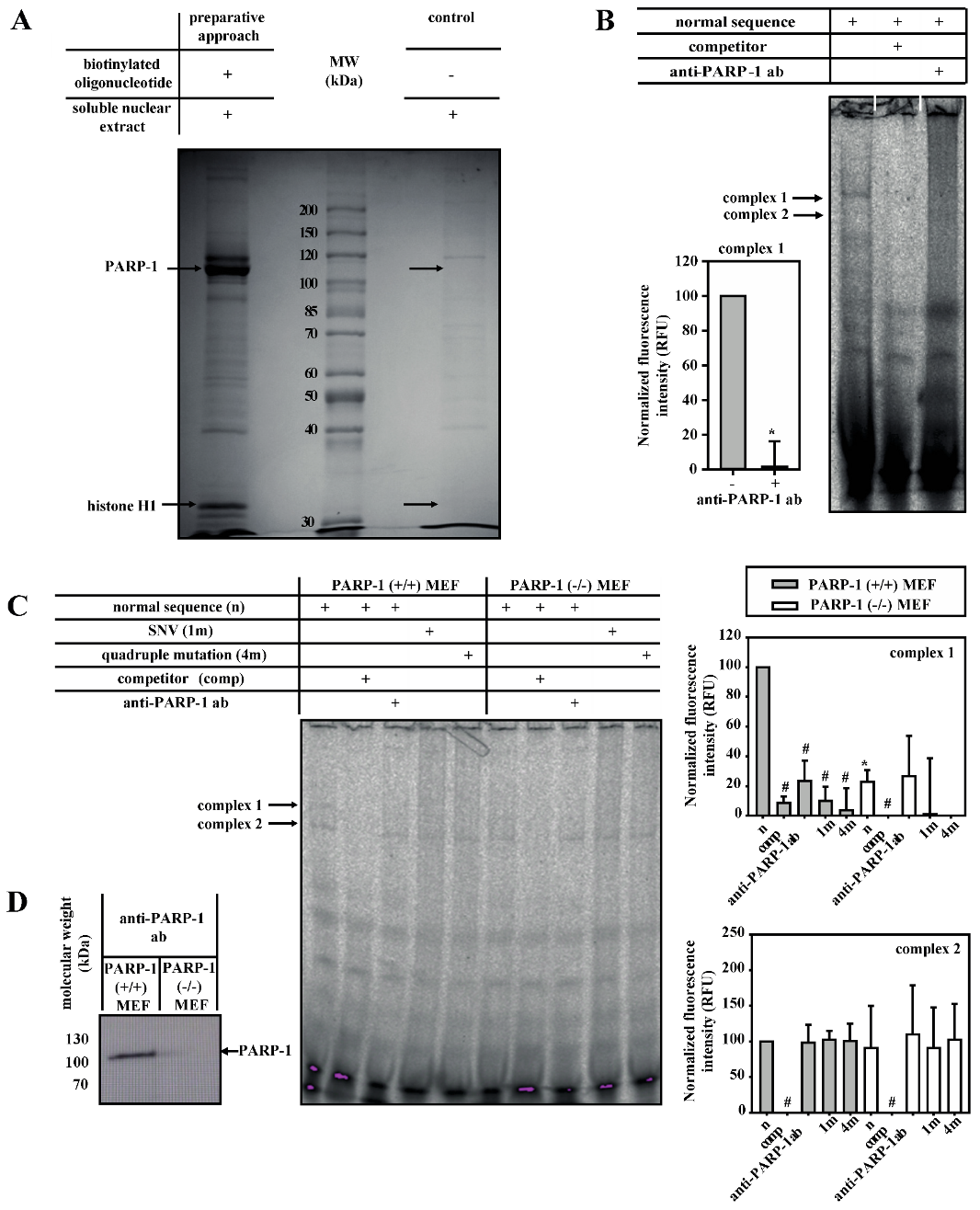
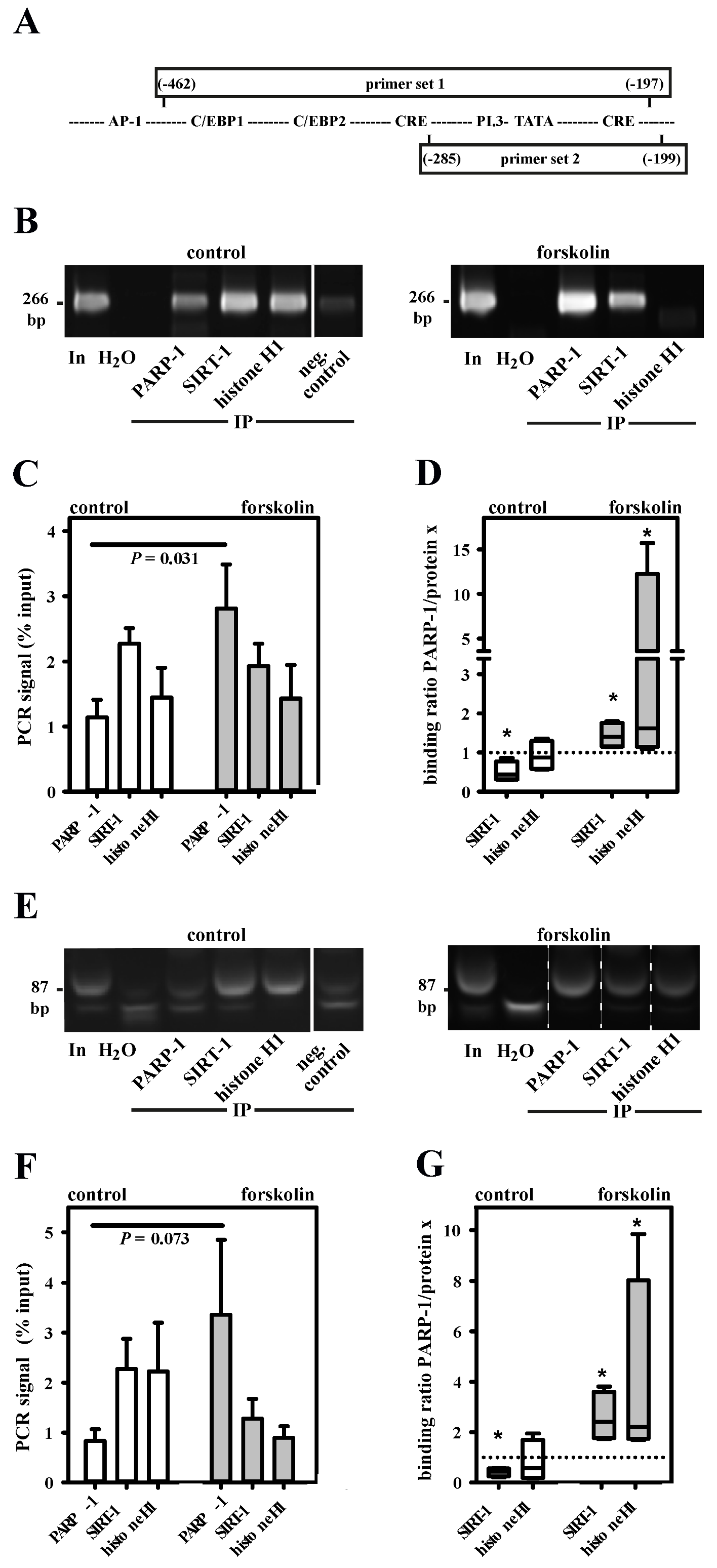
3.2. PARP-1 and Histone H1-Isoforms Bind to the SNV-Region
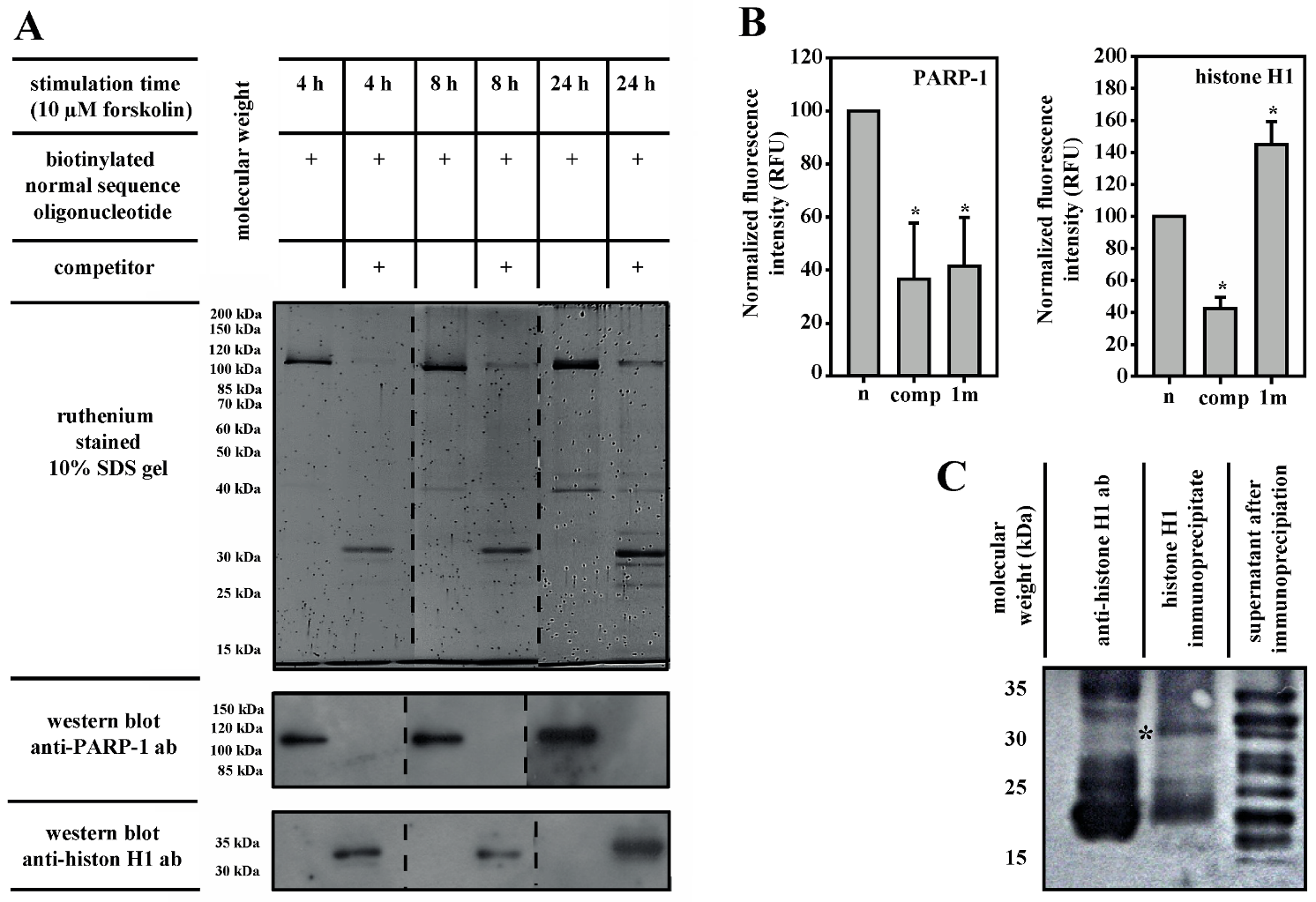
3.3. PARP-1 and Histone H1 Compete for Binding to the Aromatase Promoter I.3/II-Region
3.4. Histone H1 Is Parylated
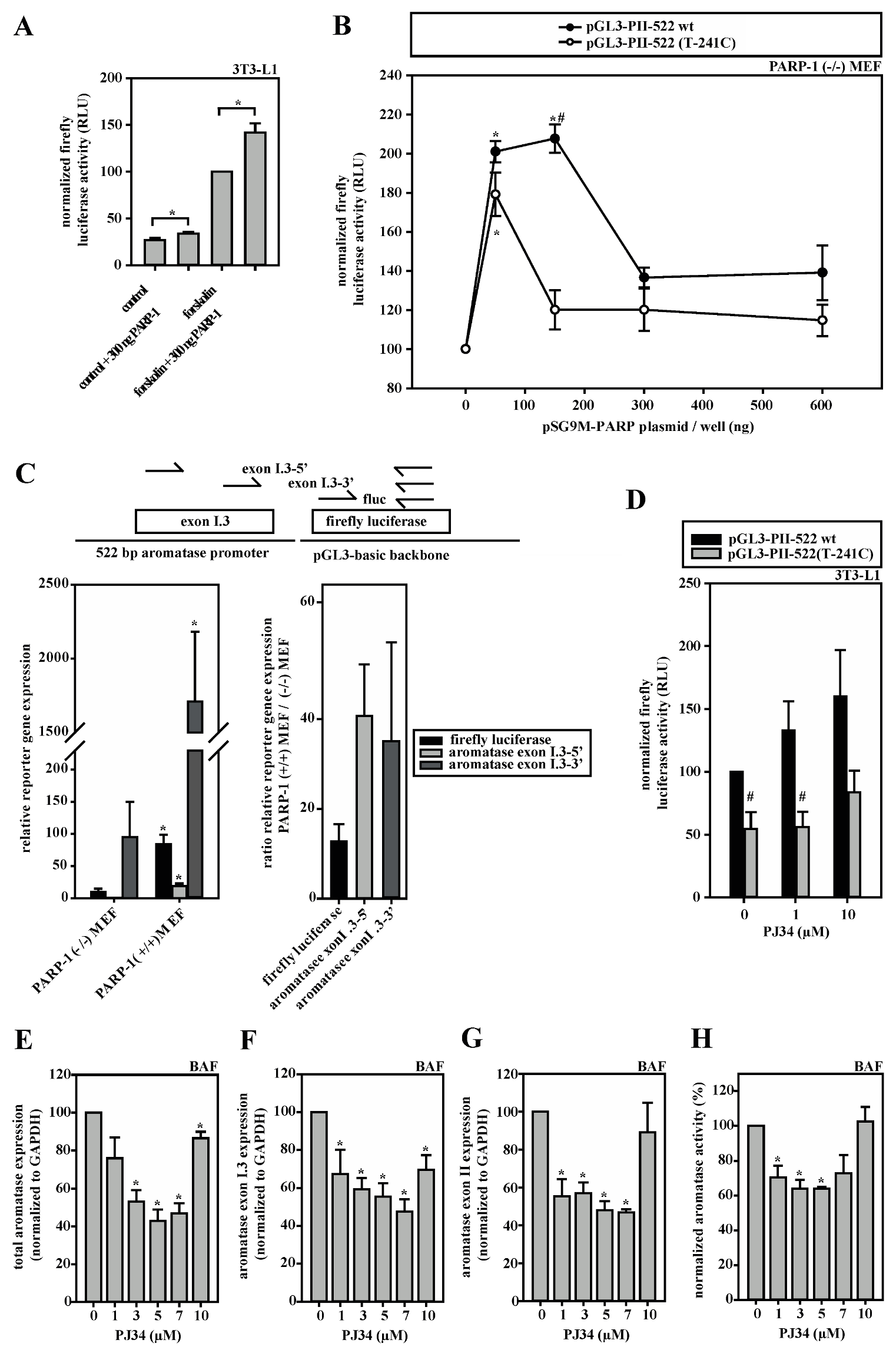
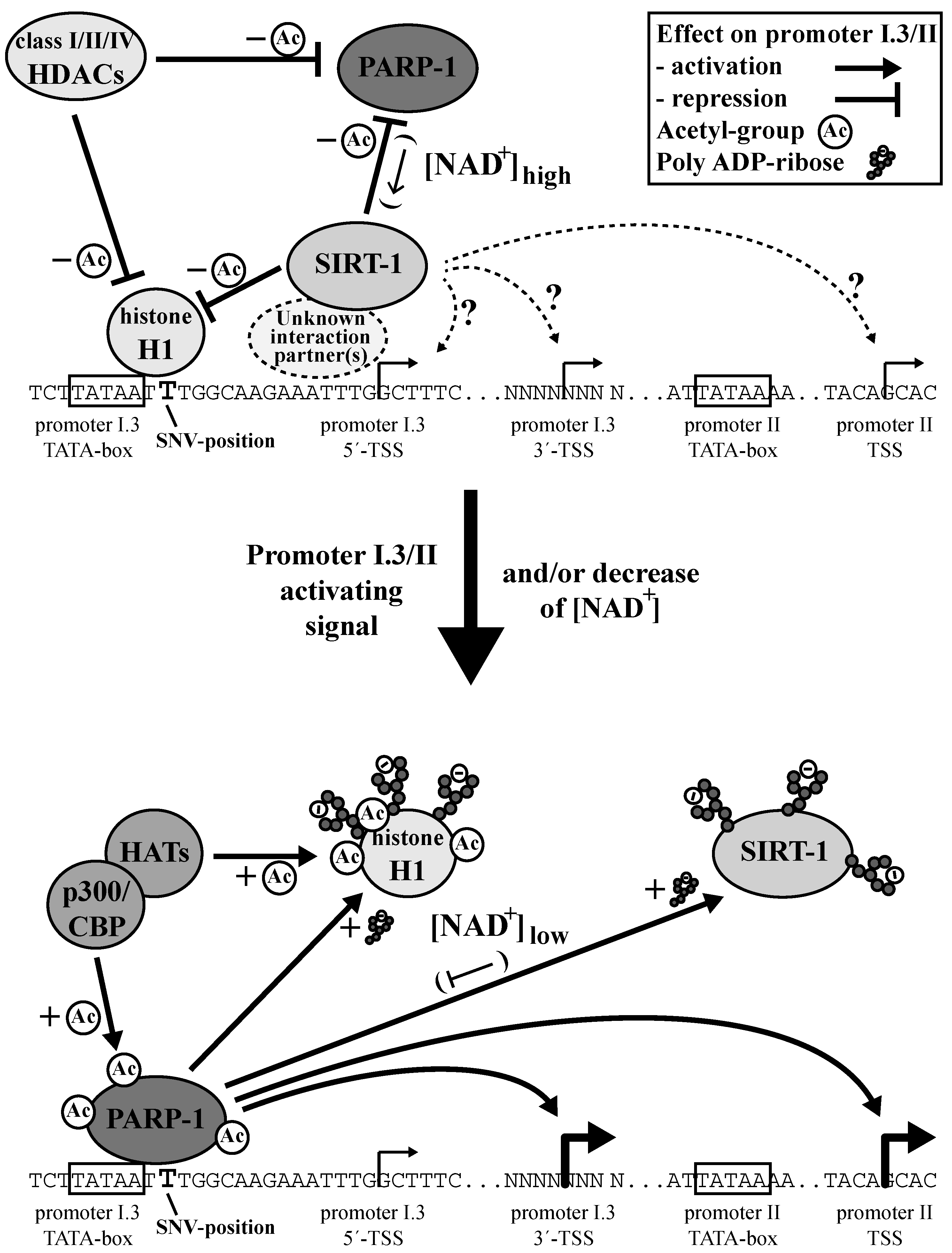
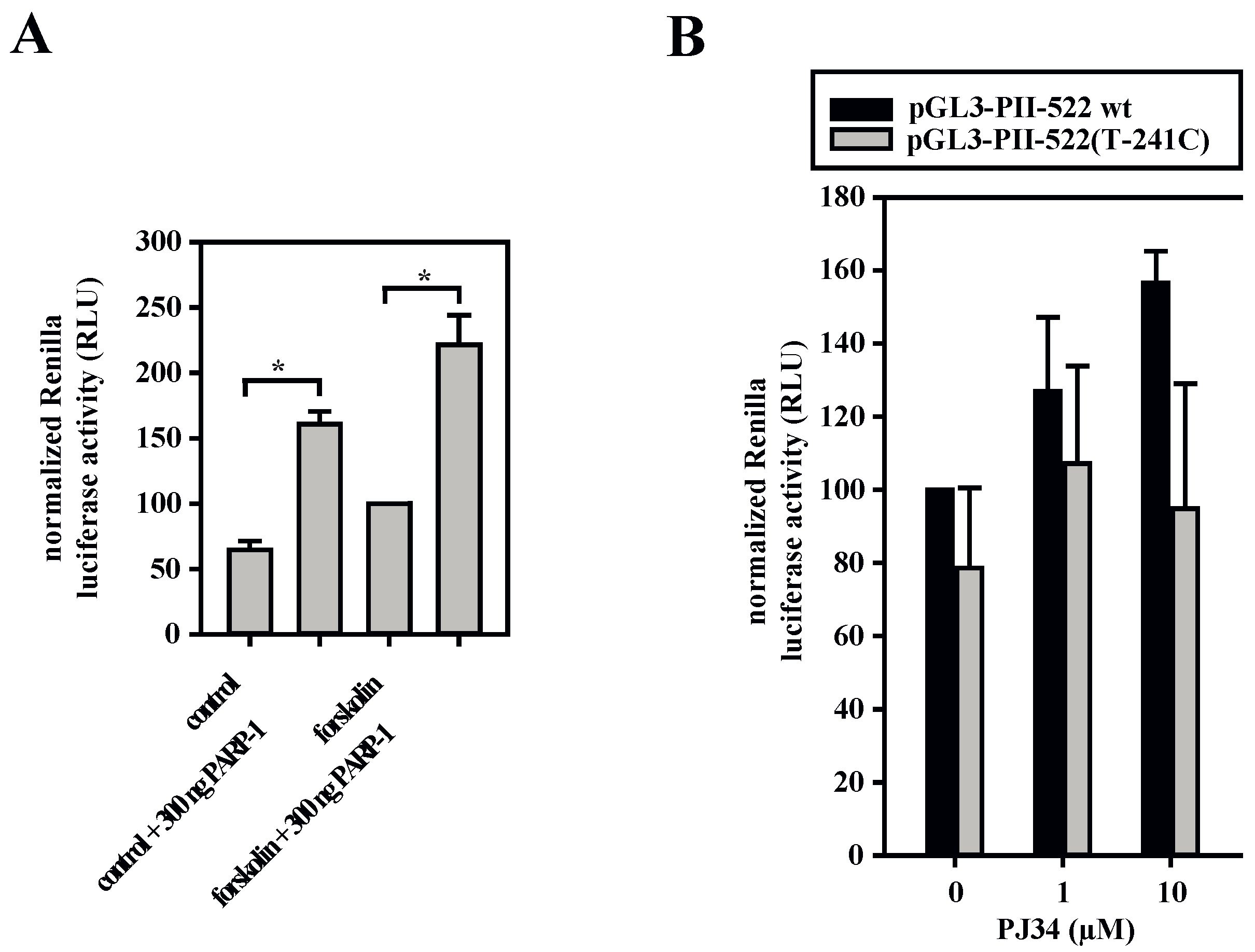
3.5. A Dual Role for PARP-1 on the Aromatase Promoter I.3/II
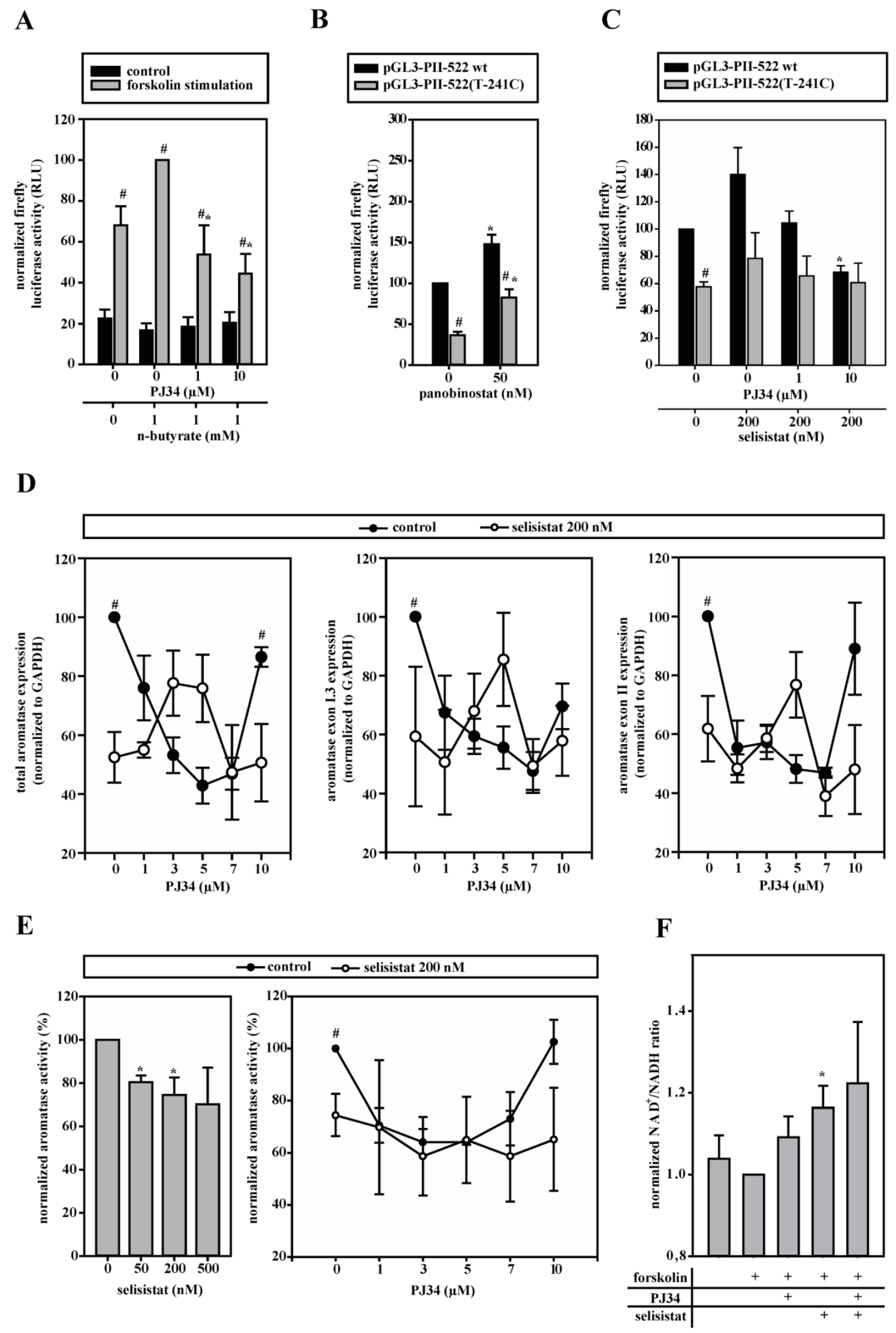
3.6. HDACs Modify Aromatase Promoter I.3/II Activity
3.7. Aromatase Expression Is Regulated by an Antagonizing System Including PARP-1, Histone H1 and SIRT-1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BAFs | breast adipose fibroblasts |
| CAFs | cancer-associated fibroblasts |
| ChIP | chromatin immunoprecipitation |
| DMEM | Dulbecco´s modified Eagle´s medium |
| EMSA | electrophoretic mobility shift assay |
| ER+ | estrogen receptor-positive |
| FBS | fetal bovine serum |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| H1 | histone H1 |
| HDAC | histone deacetylase |
| KM | Michaelis constant |
| MALDI-TOF MS | matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry |
| MEF | murine embryonic fibroblast |
| Nano-ESI-Quadrupol-TOF MS | nano-electrospray ionization-quadrupol-time-of-flight mass spectrometry |
| PARP-1 | poly (ADP-ribose) polymerase 1 |
| p300/CBP | p300/cAMP response element-binding protein acetylase |
| qRT-PCR | quantitative real-time PCR |
| SIRT-1 | NAD-dependent protein deacetylase sirtuin 1 |
| SNV | single nucleotide variant |
| TNBC | triple-negative breast cancer |
| TSS | transcriptional start-site |
| Wt | wild-type |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide/Label | Forward | Reverse |
|---|---|---|
| normal sequence /label | 5′-ctt ata att tgg caa gaa att-3′ Cy5 | 5′-att ttc ttg cca aat tat aag-3′ - |
| competitor with normal sequence | 5′-ctt ata att tgg caa gaa att-3′ | 5′-att ttc ttg cca aat tat aag-3′ |
| sequence with SNV /label | 5′-ctt ata atc tgg caa gaa att-3′ Cy5 | 5′-aat ttc ttg cca gat tat aag-3′ - |
| sequence with quadruple mutation /label | 5′-ctt ata atc cgg cgg gaa att-3′ Cy5 | 5′-aat ttc ccg ccg gat tat aag-3′ - |
| normal sequence /label | 5′-ctt ata att tgg caa gaa att-3′ biotin | 5′-att ttc ttg cca aat tat aag-3′ - |
| Target Protein | Antibodies |
|---|---|
| histone H1 | anti-histone H1 (AE-4): sc-8030, monoclonal mouse-antibody IgG2a (Santa Cruz Biotechnology; Dallas, TX, USA) |
| PARP-1 | mouse anti-PARP (C2-10): 556362, monoclonal mouse-antibody IgG1 (BD PharmingenTM; Franklin Lakes, NJ, USA) |
| SIRT-1 | anti-SIRT-1 (H-300): sc-15404, polyclonal rabbit-antibody (Santa Cruz Biotechnology; Dallas, TX, USA) |
| parylated proteins | anti-poly (ADP-ribose): 4335-MC-100, monoclonal mouse-antibody IgG3 (Trevigen; Gaithersburg, MD, USA) |
| Plasmid Name | Function | Amount Per Well |
|---|---|---|
| pGL3-basic | empty vector of reporter gene plasmid with firefly luciferase activity | 800 ng |
| pGL3-PII-522 wt | wild-type-aromatase-promoter I.3/II-reporter gene plasmid with firefly-luciferase-activity | 800 ng |
| pGL3-PII-522 (T-241C) | SNV containing aromatase-promoter I.3/II-reporter gene plasmid with firefly-luciferase-activity | 800 ng |
| pRL-SV40 | constitutively active Renilla-luciferase vector | 50 ng |
| pSG9M | empty vector | 50 ng |
| pSG9M-PARP-1 | constitutively active PARP-1 expression plasmid | 50–600 ng |
Appendix B

References
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Krebs in Deutschland 2007/2008. 8. Ausgabe. Robert Koch-Institut (Ed.) und die Gesellschaft der epidemiologischen Krebsregister in Deutschland e.V. (Ed). Berlin, 2012. Available online: http://dx.doi.org/10.25646/3161 (accessed on 12 September 2017).
- Huang, B.; Warner, M.; Gustafsson, J.A. Estrogen receptors in breast carcinogenesis and endocrine therapy. Mol. Cell Endocrinol. 2015, 418, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.T.; Phuong, T.N.T.; Tien, N.L.B.; Tran, D.K.; Nguyen, T.T.; Thanh, V.V.; Quang, T.L.; Minh, L.B.; Pham, V.H.; Ngoc, V.T.N.; et al. The Effects of Adipocytes on the Regulation of Breast Cancer in the Tumor Microenvironment: An Update. Cells 2019, 8, 857. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Lin, Z.; Imir, G.; Amin, S.; Demura, M.; Yilmaz, B.; Martin, R.; Utsunomiya, H.; Thung, S.; Gurates, B.; et al. Regulation of aromatase expression in estrogen-responsive breast and uterine disease: From bench to treatment. Pharmacol. Rev. 2005, 57, 359–383. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.R.; Mahendroo, M.S.; Means, G.D.; Kilgore, M.W.; Hinshelwood, M.M.; Graham-Lorence, S.; Amarneh, B.; Ito, Y.; Fisher, C.R.; Michael, M.D.; et al. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr. Rev. 1994, 15, 342–355. [Google Scholar] [CrossRef]
- Chen, D.; Reierstad, S.; Lu, M.; Lin, Z.; Ishikawa, H.; Bulun, S.E. Regulation of breast cancer-associated aromatase promoters. Cancer Lett. 2009, 273, 15–27. [Google Scholar] [CrossRef]
- Chen, D.; Reierstad, S.; Lin, Z.; Lu, M.; Brooks, C.; Li, N.; Innes, J.; Bulun, S.E. Prostaglandin E(2) induces breast cancer related aromatase promoters via activation of p38 and c-Jun NH(2)-terminal kinase in adipose fibroblasts. Cancer Res. 2007, 67, 8914–8922. [Google Scholar] [CrossRef]
- Wang, X.; Docanto, M.M.; Sasano, H.; Lo, C.; Simpson, E.R.; Brown, K.A. Prostaglandin E2 inhibits p53 in human breast adipose stromal cells: A novel mechanism for the regulation of aromatase in obesity and breast cancer. Cancer Res. 2015, 75, 645–655. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef]
- Santen, R.J.; Brodie, H.; Simpson, E.R.; Siiteri, P.K.; Brodie, A. History of aromatase: Saga of an important biological mediator and therapeutic target. Endocr. Rev. 2009, 30, 343–375. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Kolthur-Seetharam, U.; Dantzer, F.; McBurney, M.W.; de Murcia, G.; Sassone-Corsi, P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle 2006, 5, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, L.; Garber, J.E. PARP inhibitors in the management of breast cancer: Current data and future prospects. BMC Med. 2015, 13, 188. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Auer, B.; Stingl, L.; Berghammer, H.; Haidacher, D.; Schweiger, M.; Wagner, E.F. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995, 9, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Loffler, G. Induction of aromatase activity in human adipose tissue stromal cells by extracellular nucleotides--evidence for P2-purinoceptors in adipose tissue. Eur. J. Biochem. 1998, 252, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, G.E.; Smith, M.E.; Mendelson, C.R.; MacDonald, P.C.; Simpson, E.R. Aromatization of androstenedione by human adipose tissue stromal cells in monolayer culture. J. Clin. Endocrinol. Metab. 1981, 53, 412–417. [Google Scholar] [CrossRef]
- Pogrebniak, A.; Schemainda, I.; Azzam, K.; Pelka-Fleischer, R.; Nussler, V.; Hasmann, M. Chemopotentiating effects of a novel NAD biosynthesis inhibitor, FK866, in combination with antineoplastic agents. Eur. J. Med. Res. 2006, 11, 313–321. [Google Scholar]
- Wilde, J.; Erdmann, M.; Mertens, M.; Eiselt, G.; Schmidt, M. Aromatase activity induction in human adipose fibroblasts by retinoic acids via retinoic acid receptor alpha. J. Mol. Endocrinol. 2013, 51, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Schagger, H. Tricine-SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef]
- Lamanda, A.; Cheaib, Z.; Turgut, M.D.; Lussi, A. Protein buffering in model systems and in whole human saliva. PLoS ONE 2007, 2, e263. [Google Scholar] [CrossRef] [PubMed]
- Baldin, C.; Valiante, V.; Kruger, T.; Schafferer, L.; Haas, H.; Kniemeyer, O.; Brakhage, A.A. Comparative proteomics of a tor inducible Aspergillus fumigatus mutant reveals involvement of the Tor kinase in iron regulation. Proteomics 2015, 15, 2230–2243. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.D.; Ackroyd, A.J.; Halford, S.E. The gel shift assay for the analysis of DNA-protein interactions. Methods Mol. Biol. 1994, 30, 263–279. [Google Scholar] [CrossRef]
- Kyhse-Andersen, J. Electroblotting of multiple gels: A simple apparatus without buffer tank for rapid transfer of proteins from polyacrylamide to nitrocellulose. J. Biochem. Bioph. Meth. 1984, 10, 203–209. [Google Scholar] [CrossRef]
- Herceg, Z.; Wang, Z.Q. Failure of poly(ADP-ribose) polymerase cleavage by caspases leads to induction of necrosis and enhanced apoptosis. Mol. Cell Biol. 1999, 19, 5124–5133. [Google Scholar] [CrossRef]
- Hampf, M.; Gossen, M. A protocol for combined Photinus and Renilla luciferase quantification compatible with protein assays. Anal. Biochem. 2006, 356, 94–99. [Google Scholar] [CrossRef]
- Weiske, J.; Huber, O. The histidine triad protein Hint1 triggers apoptosis independent of its enzymatic activity. J. Biol. Chem. 2006, 281, 27356–27366. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Hassa, P.O.; Haenni, S.S.; Buerki, C.; Meier, N.I.; Lane, W.S.; Owen, H.; Gersbach, M.; Imhof, R.; Hottiger, M.O. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J. Biol. Chem. 2005, 280, 40450–40464. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, A.; Palyga, J. Modulation of chromatin function through linker histone H1 variants. Biol. Cell 2016, 108, 339–356. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, R.; Gamble, M.J.; Frizzell, K.M.; Berrocal, J.G.; Kininis, M.; Kraus, W.L. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 2008, 319, 819–821. [Google Scholar] [CrossRef] [PubMed]
- Pazzaglia, S.; Pioli, C. Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases. Cells 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Kurebayashi, J.; Kanomata, N.; Yamashita, T.; Kozuka, Y.; Moriya, T.; Sonoo, H. Antitumor and anticancer stem cell activity of a poly ADP-ribose polymerase inhibitor olaparib in breast cancer cells. Breast Cancer 2014, 21, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, R.; Hu, Y. The alternative noncoding exons 1 of aromatase (Cyp19) gene modulate gene expression in a posttranscriptional manner. Endocrinology 2009, 150, 3301–3307. [Google Scholar] [CrossRef]
- Thomas, C.; Tulin, A.V. Poly-ADP-ribose polymerase: Machinery for nuclear processes. Mol. Aspects Med. 2013, 34, 1124–1137. [Google Scholar] [CrossRef]
- Aubin, R.J.; Frechette, A.; de Murcia, G.; Mandel, P.; Lord, A.; Grondin, G.; Poirier, G.G. Correlation between endogenous nucleosomal hyper(ADP-ribosyl)ation of histone H1 and the induction of chromatin relaxation. Embo. J. 1983, 2, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Huletsky, A.; de Murcia, G.; Muller, S.; Hengartner, M.; Menard, L.; Lamarre, D.; Poirier, G.G. The effect of poly(ADP-ribosyl)ation on native and H1-depleted chromatin. A role of poly(ADP-ribosyl)ation on core nucleosome structure. J. Biol. Chem. 1989, 264, 8878–8886. [Google Scholar] [PubMed]
- Parbin, S.; Kar, S.; Shilpi, A.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Histone deacetylases: A saga of perturbed acetylation homeostasis in cancer. J. Histochem. Cytochem. 2014, 62, 11–33. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Rajamohan, S.B.; Pillai, V.B.; Gupta, M.; Sundaresan, N.R.; Birukov, K.G.; Samant, S.; Hottiger, M.O.; Gupta, M.P. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol. Cell Biol. 2009, 29, 4116–4129. [Google Scholar] [CrossRef]
- Canto, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspects Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Auwerx, J. NAD(+) metabolism: A therapeutic target for age-related metabolic disease. Crit Rev. Biochem. Mol. Biol. 2013, 48, 397–408. [Google Scholar] [CrossRef]
- Mendoza-Alvarez, H.; Alvarez-Gonzalez, R. Poly(ADP-ribose) polymerase is a catalytic dimer and the automodification reaction is intermolecular. J. Biol. Chem. 1993, 268, 22575–22580. [Google Scholar]
- Smith, B.C.; Hallows, W.C.; Denu, J.M. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal. Biochem. 2009, 394, 101–109. [Google Scholar] [CrossRef]
- Pillai, J.B.; Isbatan, A.; Imai, S.; Gupta, M.P. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J. Biol. Chem. 2005, 280, 43121–43130. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Flomenberg, N.; Tsirigos, A.; Howell, A.; et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010, 9, 2412–2422. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Tu, G.; Liu, Z.; Liu, M. Cancer-associated fibroblasts: A multifaceted driver of breast cancer progression. Cancer Lett. 2015, 361, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Winkler, U.; Hirrlinger, J. Crosstalk of Signaling and Metabolism Mediated by the NAD(+)/NADH Redox State in Brain Cells. Neurochem. Res. 2015, 40, 2394–2401. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaiser, A.; Krüger, T.; Eiselt, G.; Bechler, J.; Kniemeyer, O.; Huber, O.; Schmidt, M. Identification of PARP-1, Histone H1 and SIRT-1 as New Regulators of Breast Cancer-Related Aromatase Promoter I.3/II. Cells 2020, 9, 427. https://doi.org/10.3390/cells9020427
Kaiser A, Krüger T, Eiselt G, Bechler J, Kniemeyer O, Huber O, Schmidt M. Identification of PARP-1, Histone H1 and SIRT-1 as New Regulators of Breast Cancer-Related Aromatase Promoter I.3/II. Cells. 2020; 9(2):427. https://doi.org/10.3390/cells9020427
Chicago/Turabian StyleKaiser, Alexander, Thomas Krüger, Gabriele Eiselt, Joachim Bechler, Olaf Kniemeyer, Otmar Huber, and Martin Schmidt. 2020. "Identification of PARP-1, Histone H1 and SIRT-1 as New Regulators of Breast Cancer-Related Aromatase Promoter I.3/II" Cells 9, no. 2: 427. https://doi.org/10.3390/cells9020427
APA StyleKaiser, A., Krüger, T., Eiselt, G., Bechler, J., Kniemeyer, O., Huber, O., & Schmidt, M. (2020). Identification of PARP-1, Histone H1 and SIRT-1 as New Regulators of Breast Cancer-Related Aromatase Promoter I.3/II. Cells, 9(2), 427. https://doi.org/10.3390/cells9020427