
Curcumin-Mediated Resistance to Lenvatinib via EGFR Signaling Pathway in Hepatocellular Carcinoma
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell Viability Assays
2.3. Development of Lenvatinib-Resistant Cell Lines
2.4. Invasion Assays
2.5. Colony Formation Assays
2.6. Apoptosis and Oxidase Stress Assay
2.7. Spheroid Formation Assays
2.8. Identification of Differentially Expressed Genes and Functional Pathway Enrichment in Lenvatinib-Resistant HCC Cells
2.9. RNA Extraction and Real-Time Quantitative Reverse Transcription PCR (RT-qPCR) Assays
2.10. Western Blotting
2.11. Statistical Analysis
3. Results
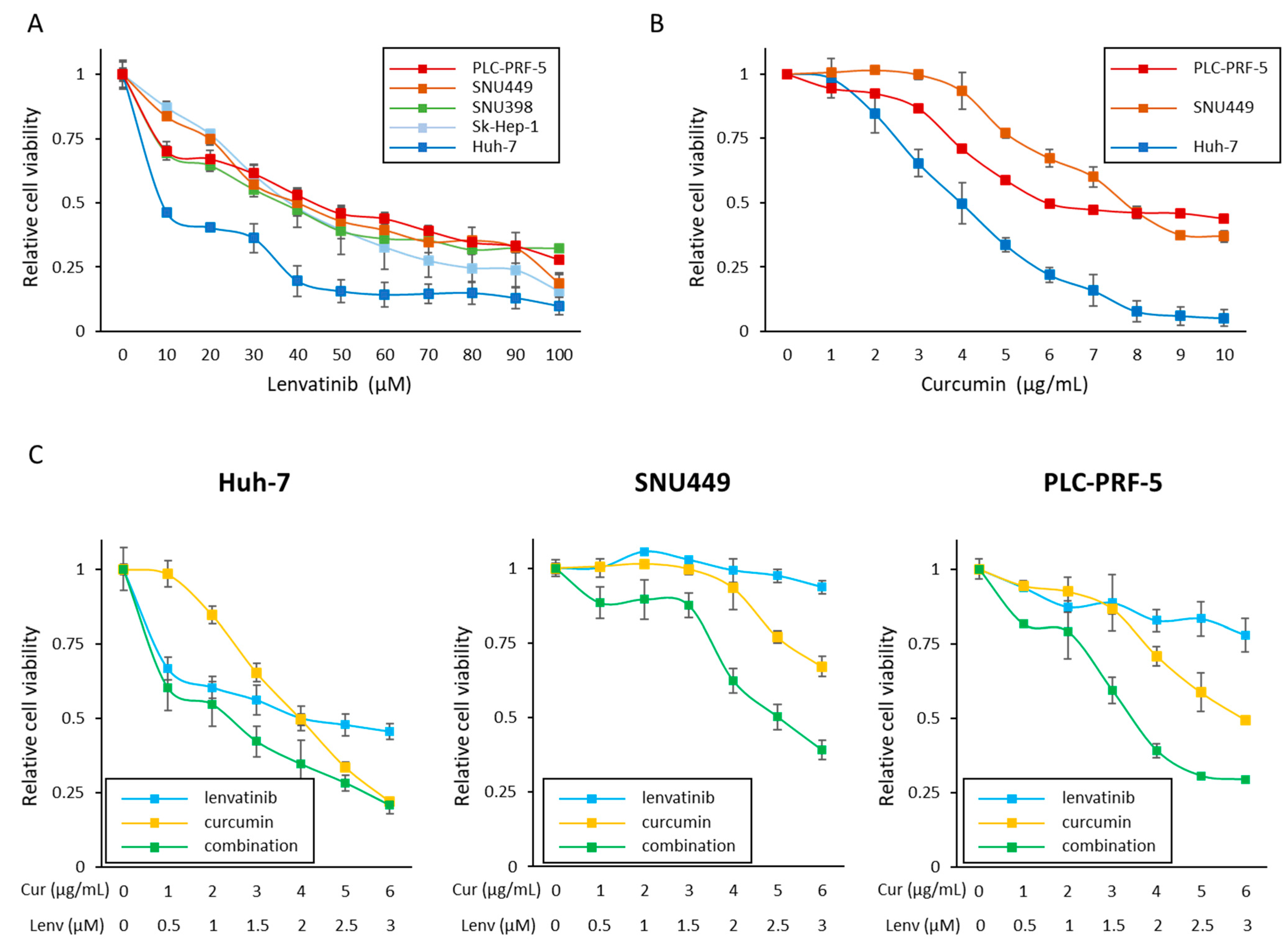
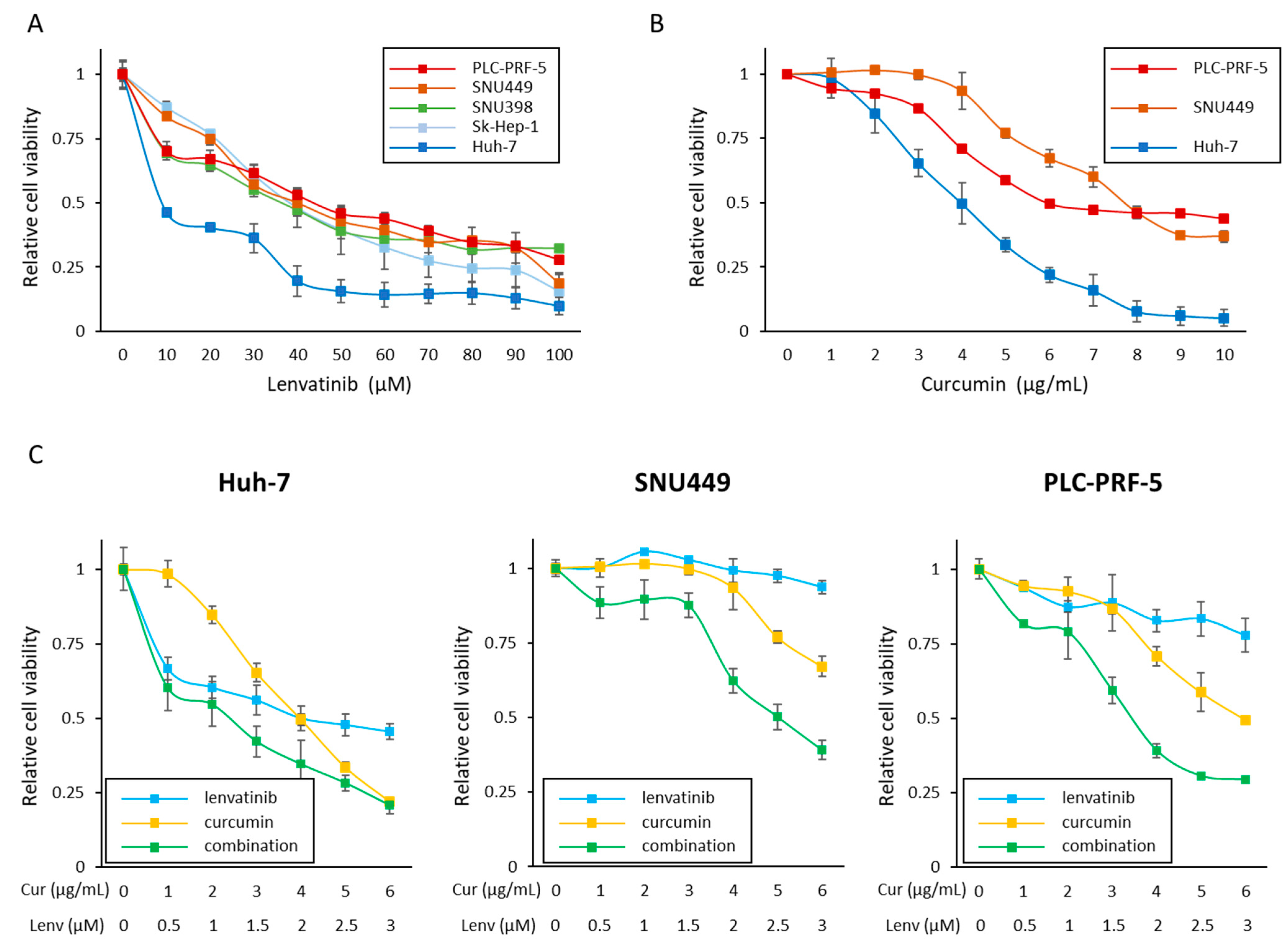
3.1. Curcumin Enhances the Anti-Proliferative Effect of Lenvatinib in HCC Cells
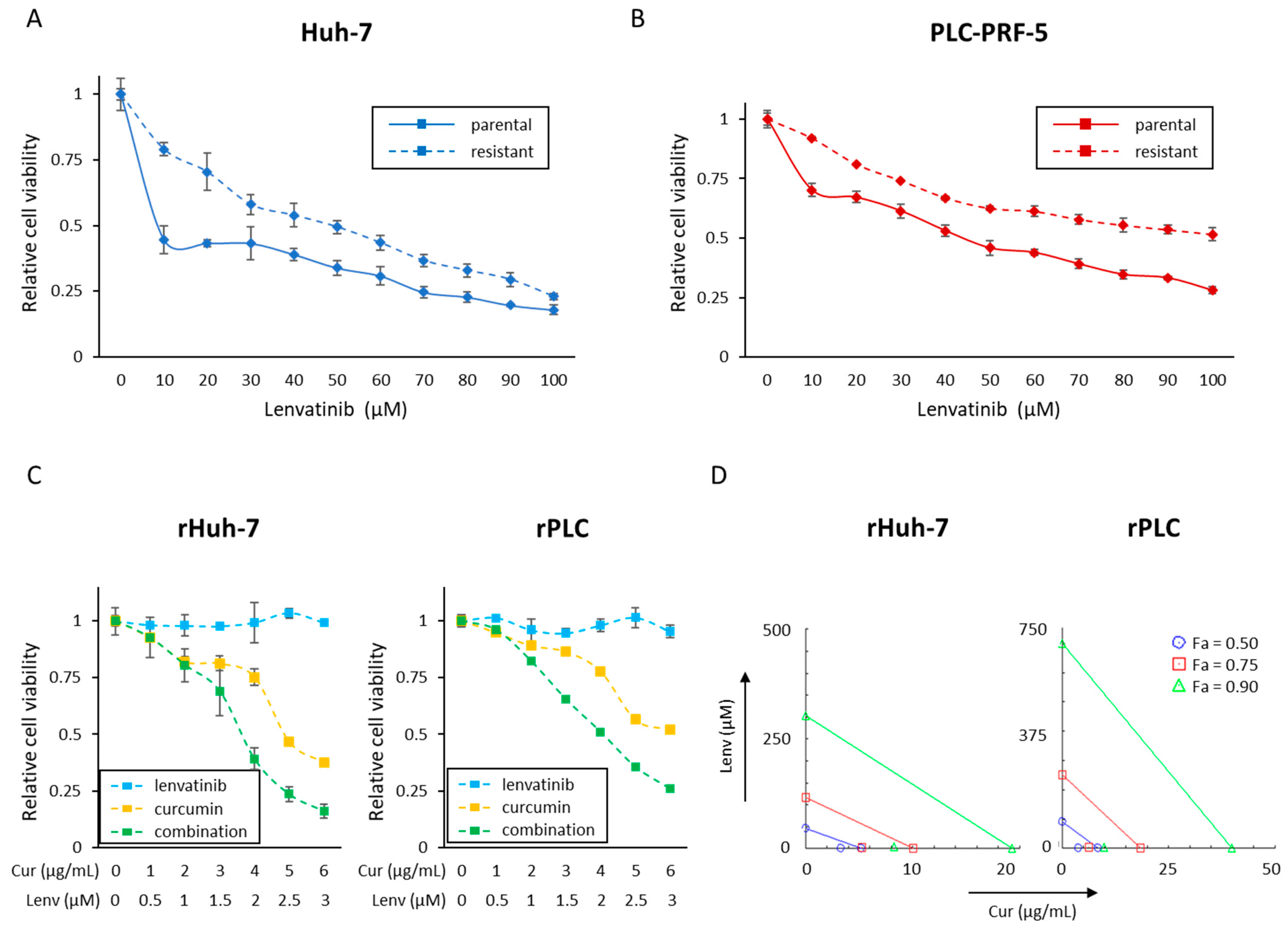
3.2. Successful Establishment of Lenvatinib-Resistant HCC Cell Lines
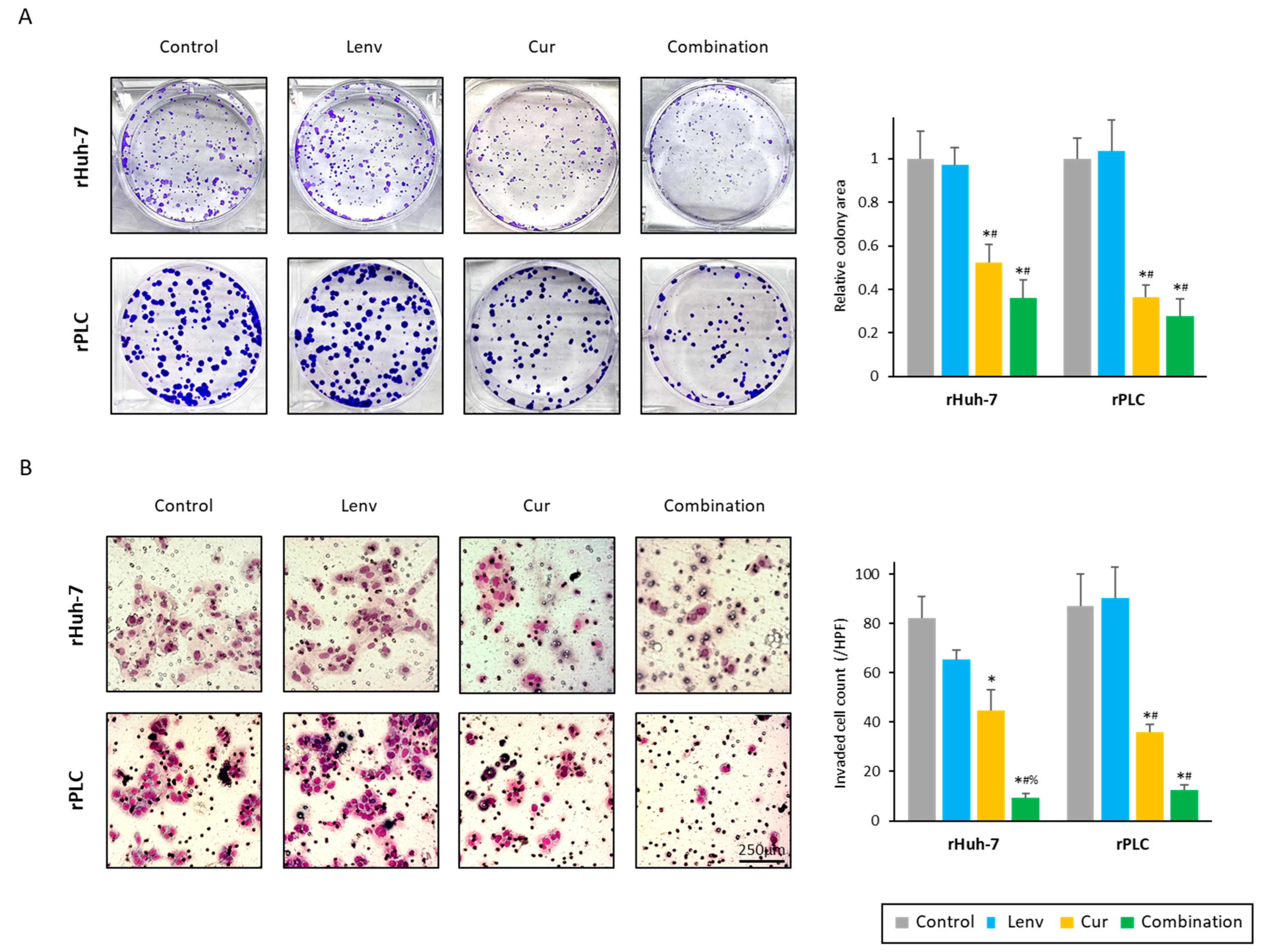
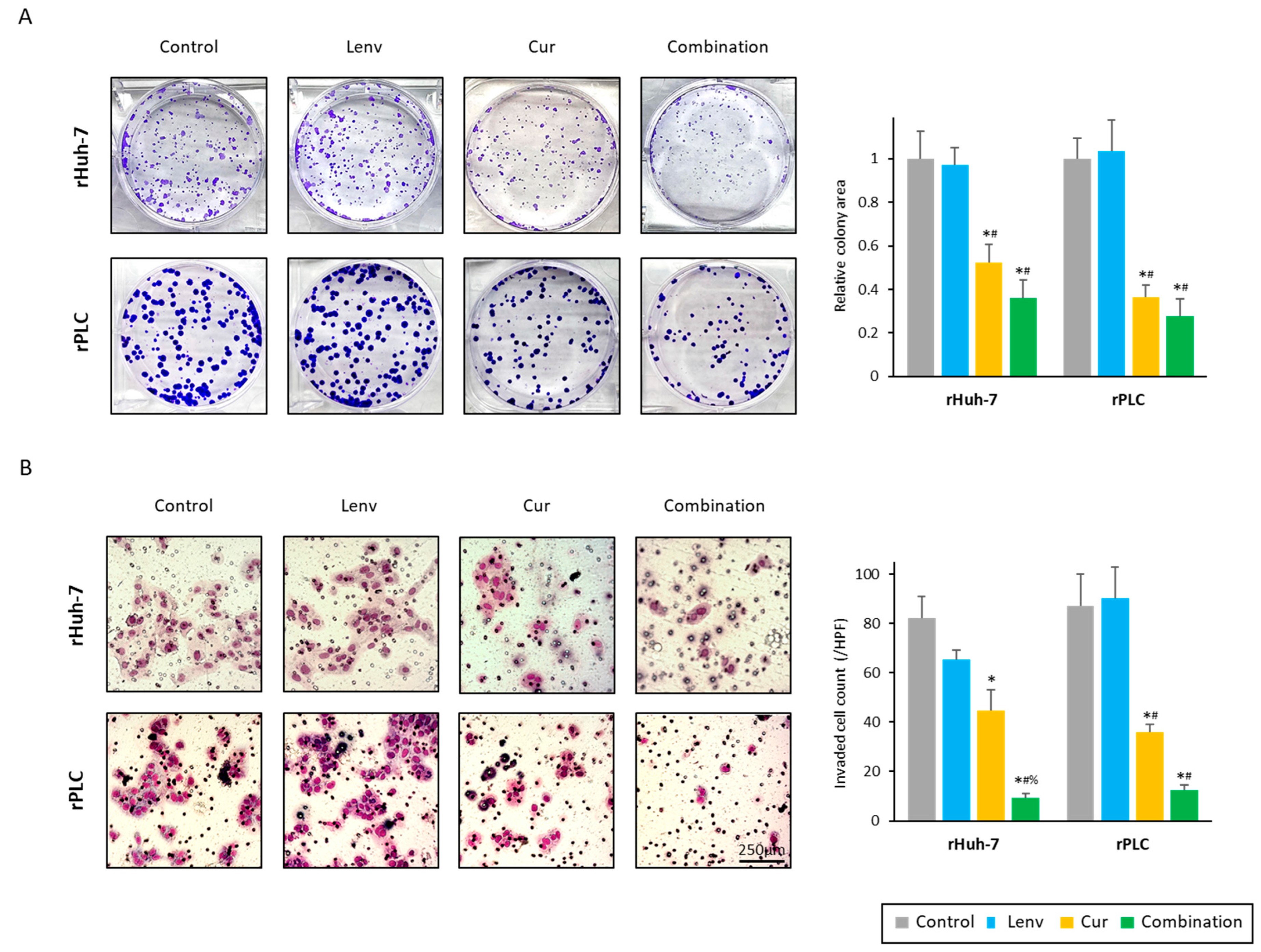
3.3. Curcumin Treatment Overcomes Acquired Lenvatinib Resistance via Inhibition of Proliferation, Invasion, and Colony Formation in HCC Cells
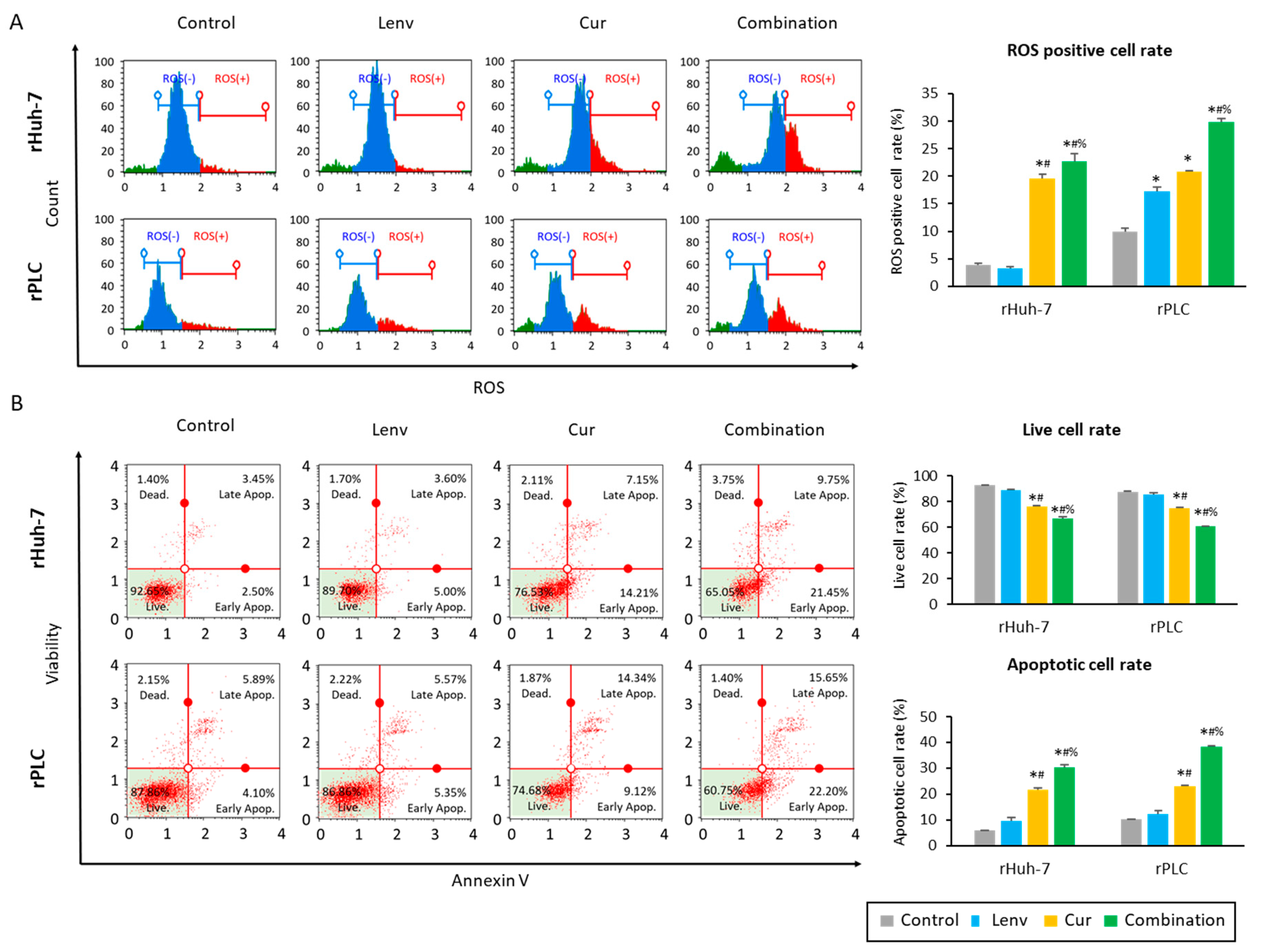
3.4. Curcumin Treatment in Lenvatinib-Resistant Cells Led to Increased Accumulation of Reactive Oxygen Species and Induction of Apoptosis in HCC Cells
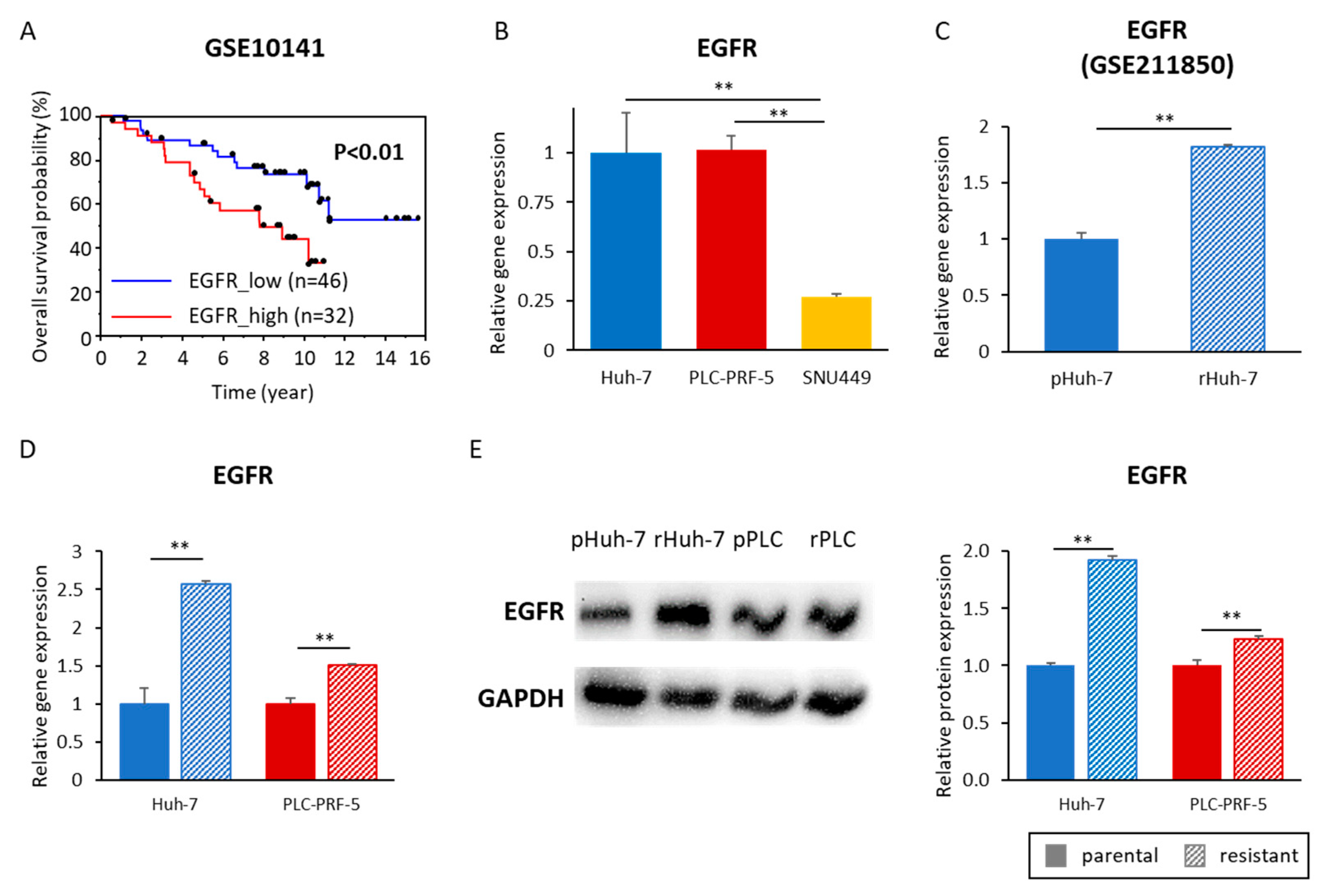
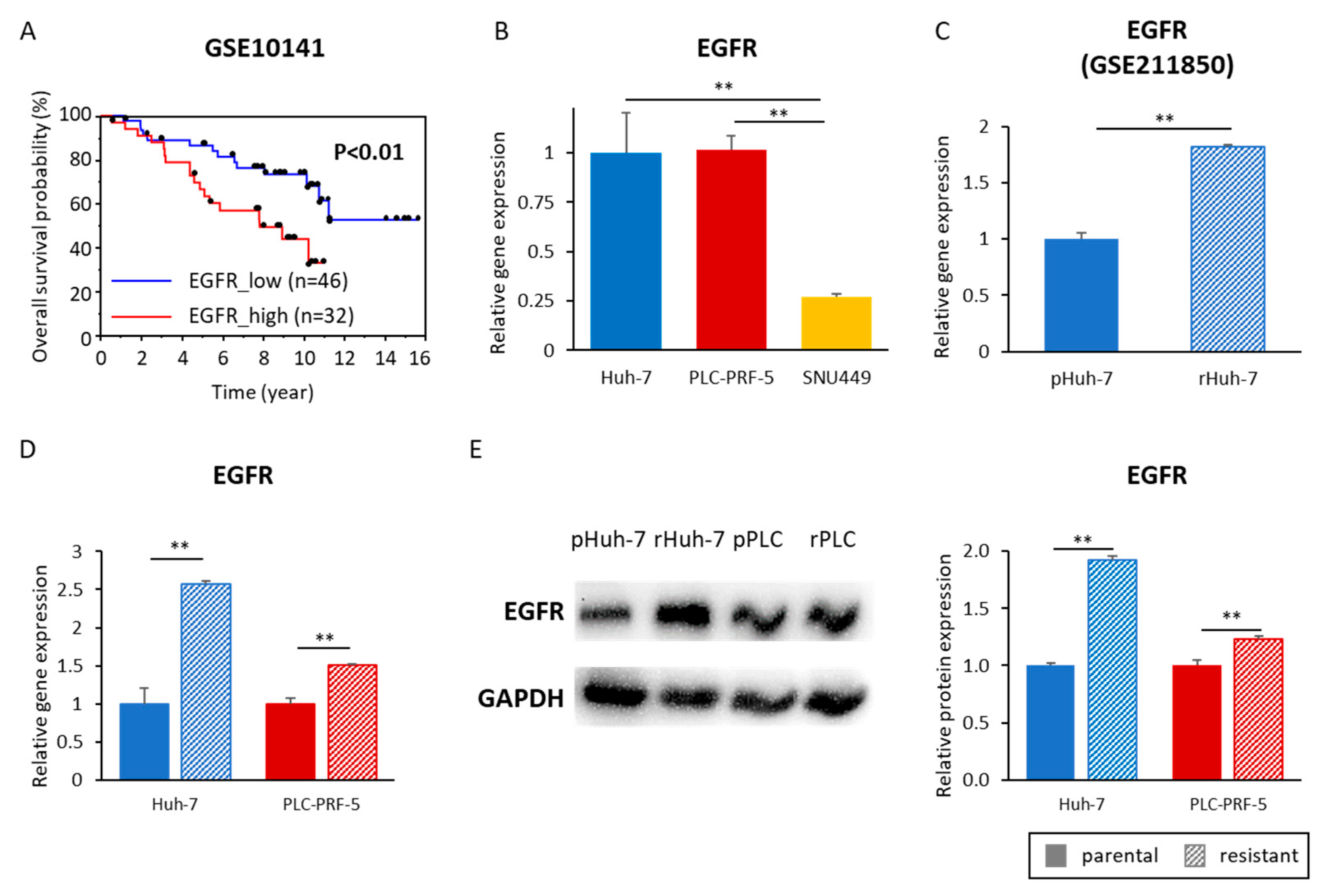
3.5. Acquired Lenvatinib Resistance Is Mediated by Activation of the EGFR Pathway in HCC Cells
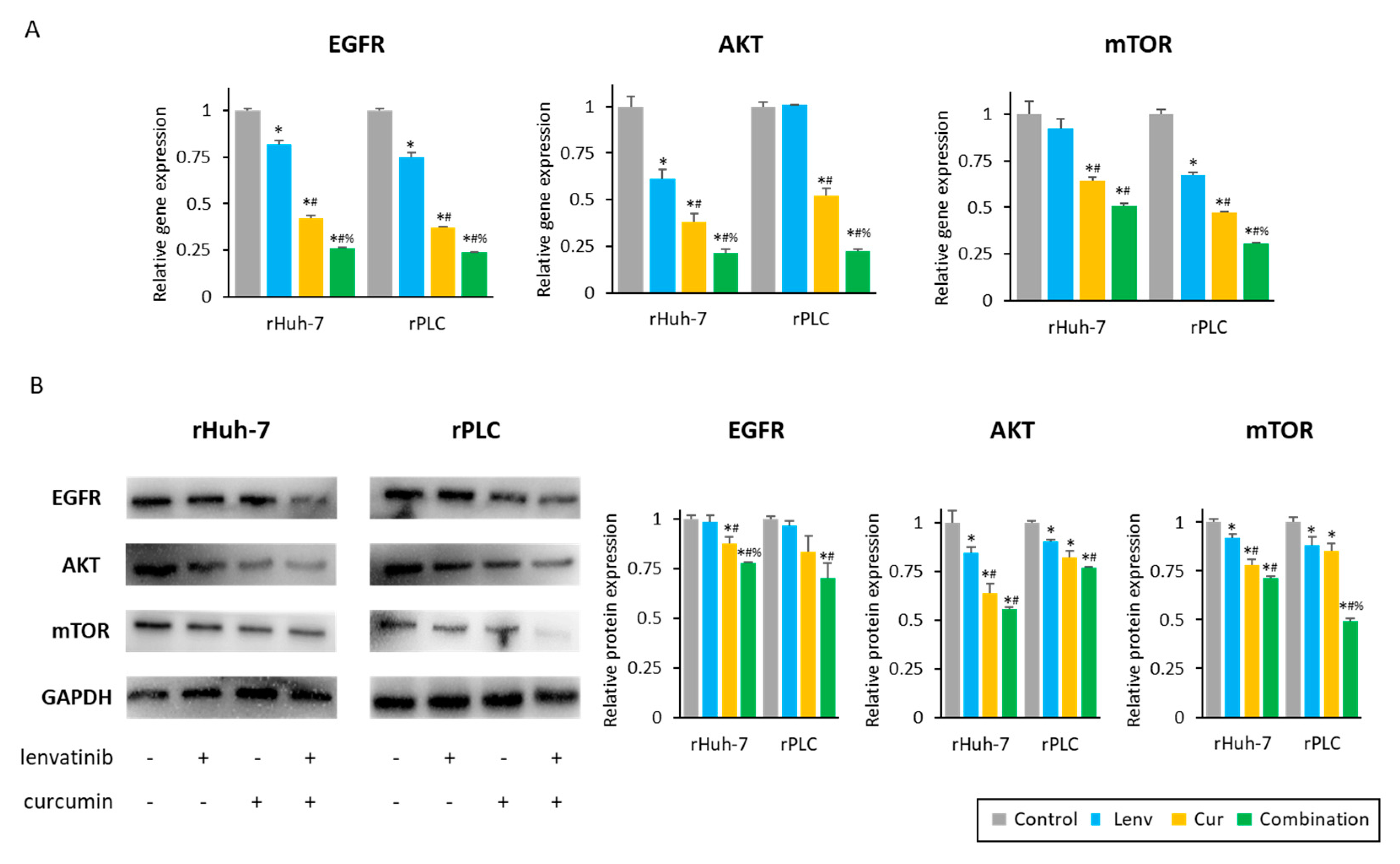
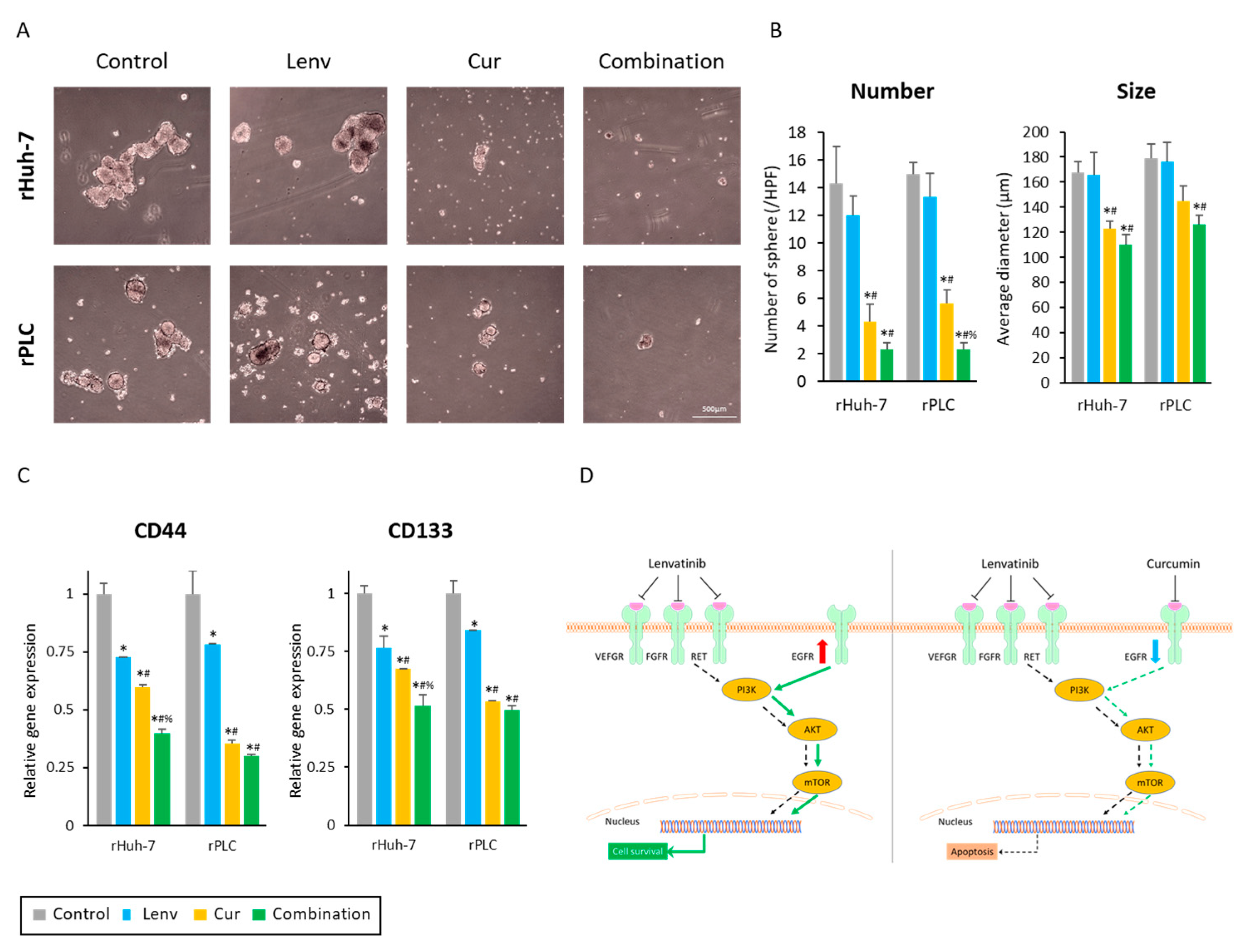
3.6. Curcumin-Mediated Sensitization to Lenvatinib Is Orchestrated via Suppression of EGFR and Its Downstream PI3K-AKT Pathway
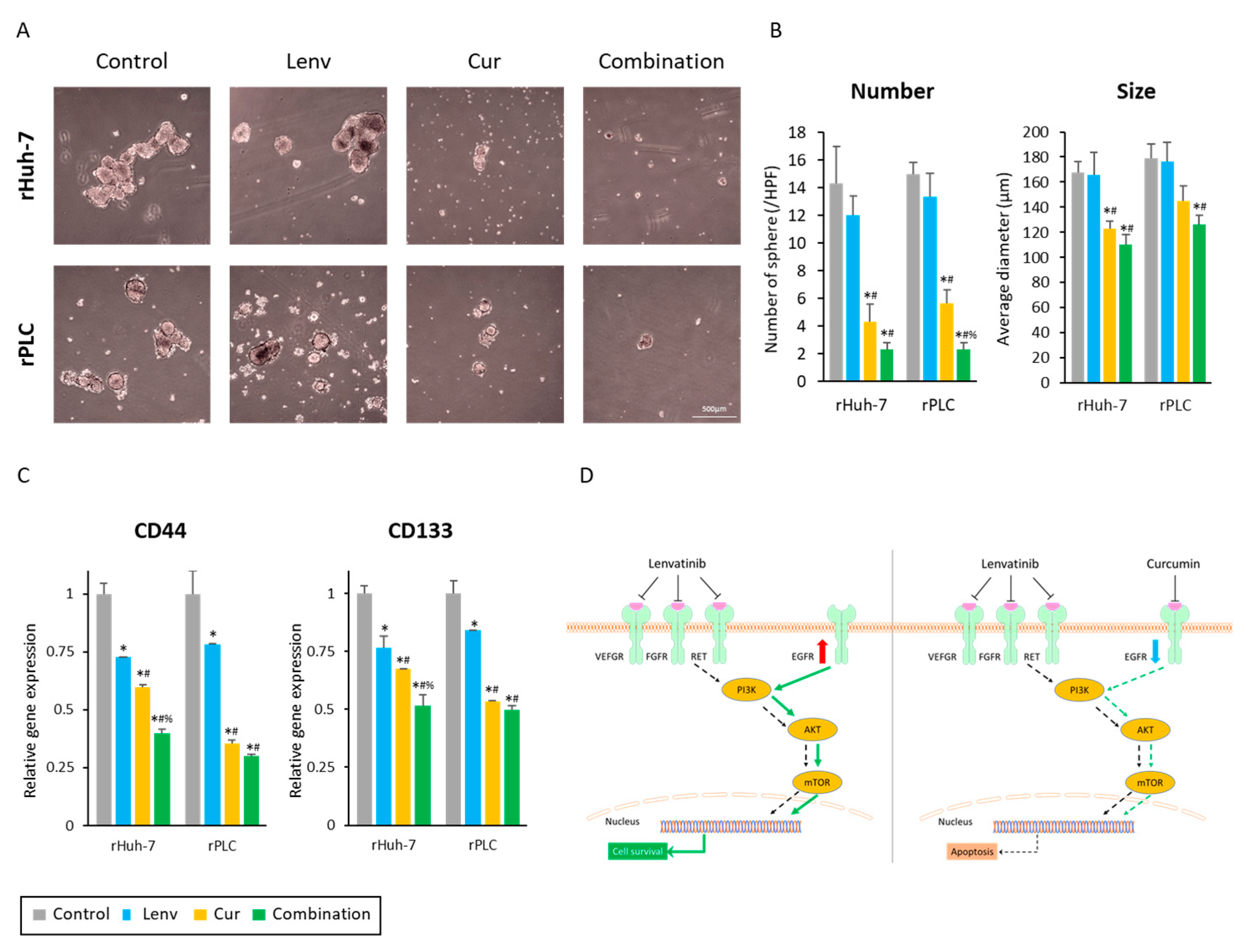
3.7. Curcumin Treatment Suppresses Cancer Stemness in Lenvatinib-Resistant HCC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Qin, S.; Kudo, M.; Su, Y.; Hudgens, S.; Yamashita, T.; Yoon, J.H.; Fartoux, L.; Simon, K.; López, C.; et al. Lenvatinib versus sorafenib for first-line treatment of unresectable hepatocellular carcinoma: Patient-reported outcomes from a randomised, open-label, non-inferiority, phase 3 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Su, G.L.; Altayar, O.; O’Shea, R.; Shah, R.; Estfan, B.; Wenzell, C.; Sultan, S.; Falck-Ytter, Y. AGA Clinical Practice Guideline on Systemic Therapy for Hepatocellular Carcinoma. Gastroenterology 2022, 162, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Maesaka, K.; Sakamori, R.; Yamada, R.; Doi, A.; Tahata, Y.; Miyazaki, M.; Ohkawa, K.; Mita, E.; Iio, S.; Nozaki, Y.; et al. Comparison of atezolizumab plus bevacizumab and lenvatinib in terms of efficacy and safety as primary systemic chemotherapy for hepatocellular carcinoma. Hepatol. Res. 2022, 52, 630–640. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Haber, P.K.; Puigvehí, M.; Castet, F.; Lourdusamy, V.; Montal, R.; Tabrizian, P.; Buckstein, M.; Kim, E.; Villanueva, A.; Schwartz, M.; et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-analysis of Randomized Controlled Trials (2002–2020). Gastroenterology 2021, 161, 879–898. [Google Scholar] [CrossRef]
- Rimini, M.; Rimassa, L.; Ueshima, K.; Burgio, V.; Shigeo, S.; Tada, T.; Suda, G.; Yoo, C.; Cheon, J.; Pinato, D.J.; et al. Atezolizumab plus bevacizumab versus lenvatinib or sorafenib in non-viral unresectable hepatocellular carcinoma: An international propensity score matching analysis. ESMO Open 2022, 7, 100591. [Google Scholar] [CrossRef]
- Casadei-Gardini, A.; Rimini, M.; Tada, T.; Suda, G.; Shimose, S.; Kudo, M.; Cheon, J.; Finkelmeier, F.; Lim, H.Y.; Rimassa, L.; et al. Atezolizumab plus bevacizumab versus lenvatinib for unresectable hepatocellular carcinoma: A large real-life worldwide population. Eur. J. Cancer 2022, 180, 9–20. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Shi, J.; Jia, X.; Dang, S.; Wang, W. Cost-effectiveness of Atezolizumab Plus Bevacizumab vs. Sorafenib for Patients With Unresectable or Metastatic Hepatocellular Carcinoma. JAMA Netw. Open 2021, 4, e214846. [Google Scholar] [CrossRef]
- Su, D.; Wu, B.; Shi, L. Cost-effectiveness of Atezolizumab Plus Bevacizumab vs. Sorafenib as First-Line Treatment of Unresectable Hepatocellular Carcinoma. JAMA Netw. Open 2021, 4, e210037. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, J.; Mantoan, B.; Bonetti, A. The cost-effectiveness of new first-line therapies approved in advanced hepatocellular carcinoma. J. Oncol. Pharm. Pract. 2022, 28, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; McFarlane, T.; Tully, S.; Wong, W.W.L. Lenvatinib Versus Sorafenib as First-Line Treatment of Unresectable Hepatocellular Carcinoma: A Cost-Utility Analysis. Oncologist 2020, 25, e512–e519. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Kudo, M.; Ikeda, K.; Izumi, N.; Tateishi, R.; Ikeda, M.; Aikata, H.; Kawaguchi, Y.; Wada, Y.; Numata, K.; et al. REFLECT-a phase 3 trial comparing efficacy and safety of lenvatinib to sorafenib for the treatment of unresectable hepatocellular carcinoma: An analysis of Japanese subset. J. Gastroenterol. 2020, 55, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Kawaoka, T.; Amioka, K.; Naruto, K.; Ogawa, Y.; Yoshikawa, Y.; Kikukawa, C.; Kosaka, Y.; Uchikawa, S.; Morio, K.; et al. Efficacy and Safety of Lenvatinib-Transcatheter Arterial Chemoembolization Sequential Therapy for Patients with Intermediate-Stage Hepatocellular Carcinoma. Oncology 2021, 99, 507–517. [Google Scholar] [CrossRef]
- Kuroda, H.; Oikawa, T.; Ninomiya, M.; Fujita, M.; Abe, K.; Okumoto, K.; Katsumi, T.; Sato, W.; Igarashi, G.; Iino, C.; et al. Objective Response by mRECIST to Initial Lenvatinib Therapy Is an Independent Factor Contributing to Deep Response in Hepatocellular Carcinoma Treated with Lenvatinib-Transcatheter Arterial Chemoembolization Sequential Therapy. Liver Cancer 2022, 11, 383–396. [Google Scholar] [CrossRef]
- Peng, Z.; Fan, W.; Zhu, B.; Wang, G.; Sun, J.; Xiao, C.; Huang, F.; Tang, R.; Cheng, Y.; Huang, Z.; et al. Lenvatinib Combined With Transarterial Chemoembolization as First-Line Treatment for Advanced Hepatocellular Carcinoma: A Phase III, Randomized Clinical Trial (LAUNCH). J. Clin. Oncol. 2022, 41, Jco2200392. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Cheng, A.-L.; Vogel, A.; Tovoli, F.; Ueshima, K.; Aikata, H.; et al. Overall survival and objective response in advanced unresectable hepatocellular carcinoma: A subanalysis of the REFLECT study. J. Hepatol. 2022, 78, 133–141. [Google Scholar] [CrossRef]
- He, X.; Hikiba, Y.; Suzuki, Y.; Nakamori, Y.; Kanemaru, Y.; Sugimori, M.; Sato, T.; Nozaki, A.; Chuma, M.; Maeda, S. EGFR inhibition reverses resistance to lenvatinib in hepatocellular carcinoma cells. Sci. Rep. 2022, 12, 8007. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Liu, J.; Wang, Y.; Dong, J. Co-administration of MDR1 and BCRP or EGFR/PI3K inhibitors overcomes lenvatinib resistance in hepatocellular carcinoma. Front. Oncol. 2022, 12, 944537. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Shi, Y.; Lv, Y.; Yuan, S.; Ramirez, C.F.A.; Lieftink, C.; Wang, L.; Wang, S.; Wang, C.; Dias, M.H.; et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature 2021, 595, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Roy, S.; Wang, C.; Goel, A. A Combined Treatment with Berberine and Andrographis Exhibits Enhanced Anti-Cancer Activity through Suppression of DNA Replication in Colorectal Cancer. Pharmaceuticals 2022, 15, 262. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Goel, A. Curcumin and colorectal cancer: An update and current perspective on this natural medicine. Semin. Cancer Biol. 2022, 80, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Toden, S.; Ravindranathan, P.; Han, H.; Goel, A. Curcumin sensitizes pancreatic cancer cells to gemcitabine by attenuating PRC2 subunit EZH2, and the lncRNA PVT1 expression. Carcinogenesis 2017, 38, 1036–1046. [Google Scholar] [CrossRef]
- Okuno, K.; Xu, C.; Pascual-Sabater, S.; Tokunaga, M.; Han, H.; Fillat, C.; Kinugasa, Y.; Goel, A. Berberine Overcomes Gemcitabine-Associated Chemoresistance through Regulation of Rap1/PI3K-Akt Signaling in Pancreatic Ductal Adenocarcinoma. Pharmaceuticals 2022, 15, 1199. [Google Scholar] [CrossRef]
- Okuno, K.; Garg, R.; Yuan, Y.C.; Tokunaga, M.; Kinugasa, Y.; Goel, A. Berberine and Oligomeric Proanthocyanidins Exhibit Synergistic Efficacy Through Regulation of PI3K-Akt Signaling Pathway in Colorectal Cancer. Front. Oncol. 2022, 12, 855860. [Google Scholar] [CrossRef]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem. Pharmacol. 2008, 75, 787–809. [Google Scholar] [CrossRef]
- Goel, A.; Aggarwal, B.B. Curcumin, the golden spice from Indian saffron, is a chemosensitizer and radiosensitizer for tumors and chemoprotector and radioprotector for normal organs. Nutr. Cancer 2010, 62, 919–930. [Google Scholar] [CrossRef]
- Toden, S.; Goel, A. The Holy Grail of Curcumin and its Efficacy in Various Diseases: Is Bioavailability Truly a Big Concern? J. Restor. Med. 2017, 6, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ravindranathan, P.; Pasham, D.; Balaji, U.; Cardenas, J.; Gu, J.; Toden, S.; Goel, A. A combination of curcumin and oligomeric proanthocyanidins offer superior anti-tumorigenic properties in colorectal cancer. Sci. Rep. 2018, 8, 13869. [Google Scholar] [CrossRef] [PubMed]
- Toden, S.; Okugawa, Y.; Jascur, T.; Wodarz, D.; Komarova, N.L.; Buhrmann, C.; Shakibaei, M.; Boland, C.R.; Goel, A. Curcumin mediates chemosensitization to 5-fluorouracil through miRNA-induced suppression of epithelial-to-mesenchymal transition in chemoresistant colorectal cancer. Carcinogenesis 2015, 36, 355–367. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Gomez-Quiroz, L.; Arreguin Camacho, L.O.; Pinna, F.; Lee, Y.H.; Kitade, M.; Domínguez, M.P.; Castven, D.; Breuhahn, K.; Conner, E.A.; et al. Curcumin effectively inhibits oncogenic NF-κB signaling and restrains stemness features in liver cancer. J. Hepatol. 2015, 63, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Zhao, J.; Su, J.; Chen, J.; Cui, X.; Sun, M.; Zhang, X. Curcumin induces mitochondrial apoptosis in human hepatoma cells through BCLAF1-mediated modulation of PI3K/AKT/GSK-3β signaling. Life Sci. 2022, 306, 120804. [Google Scholar] [CrossRef]
- Golonko, A.; Lewandowska, H.; Świsłocka, R.; Jasińska, U.T.; Priebe, W.; Lewandowski, W. Curcumin as tyrosine kinase inhibitor in cancer treatment. Eur. J. Med. Chem. 2019, 181, 111512. [Google Scholar] [CrossRef]
- Chen, P.; Huang, H.P.; Wang, Y.; Jin, J.; Long, W.G.; Chen, K.; Zhao, X.H.; Chen, C.G.; Li, J. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J. Exp. Clin. Cancer Res. 2019, 38, 254. [Google Scholar] [CrossRef]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Roy, S.; Zhao, Y.; Yuan, Y.C.; Goel, A. Metformin and ICG-001 Act Synergistically to Abrogate Cancer Stem Cells-Mediated Chemoresistance in Colorectal Cancer by Promoting Apoptosis and Autophagy. Cancers 2022, 14, 1281. [Google Scholar] [CrossRef]
- Tsui, Y.M.; Chan, L.K.; Ng, I.O. Cancer stemness in hepatocellular carcinoma: Mechanisms and translational potential. Br. J. Cancer 2020, 122, 1428–1440. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyazaki, K.; Morine, Y.; Xu, C.; Nakasu, C.; Wada, Y.; Teraoku, H.; Yamada, S.; Saito, Y.; Ikemoto, T.; Shimada, M.; et al. Curcumin-Mediated Resistance to Lenvatinib via EGFR Signaling Pathway in Hepatocellular Carcinoma. Cells 2023, 12, 612. https://doi.org/10.3390/cells12040612
Miyazaki K, Morine Y, Xu C, Nakasu C, Wada Y, Teraoku H, Yamada S, Saito Y, Ikemoto T, Shimada M, et al. Curcumin-Mediated Resistance to Lenvatinib via EGFR Signaling Pathway in Hepatocellular Carcinoma. Cells. 2023; 12(4):612. https://doi.org/10.3390/cells12040612
Chicago/Turabian StyleMiyazaki, Katsuki, Yuji Morine, Caiming Xu, Chiharu Nakasu, Yuma Wada, Hiroki Teraoku, Shinichiro Yamada, Yu Saito, Tetsuya Ikemoto, Mitsuo Shimada, and et al. 2023. "Curcumin-Mediated Resistance to Lenvatinib via EGFR Signaling Pathway in Hepatocellular Carcinoma" Cells 12, no. 4: 612. https://doi.org/10.3390/cells12040612