
Mitochondrial Lipid Peroxidation Is Responsible for Ferroptosis

 ,
,  ,
, 
Abstract
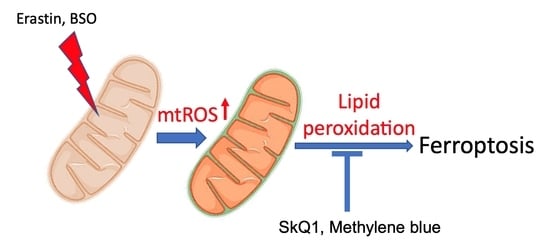
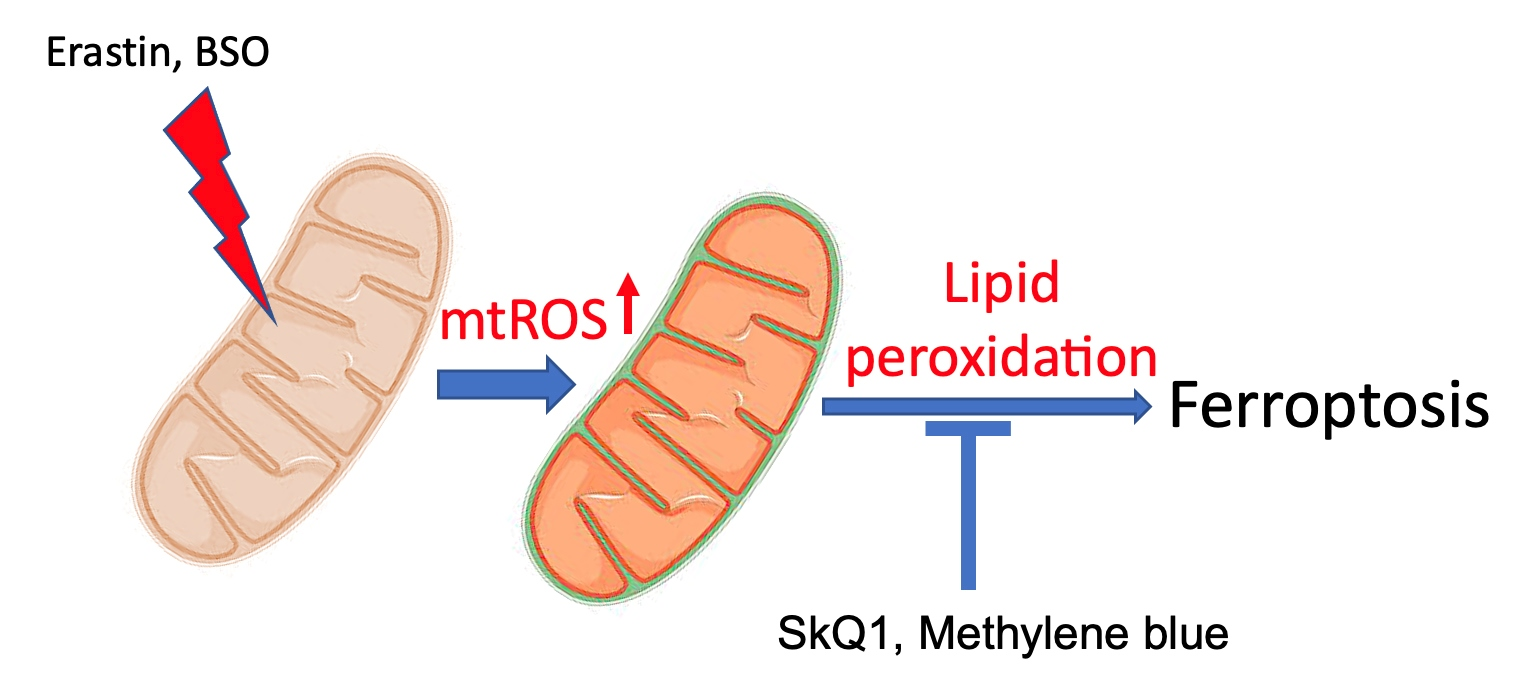{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Chemicals
2.2. Cell Cultures
2.3. Microscopy
2.4. Flow Cytometry
2.5. Isolated Mitochondria
2.6. Statistics
3. Results
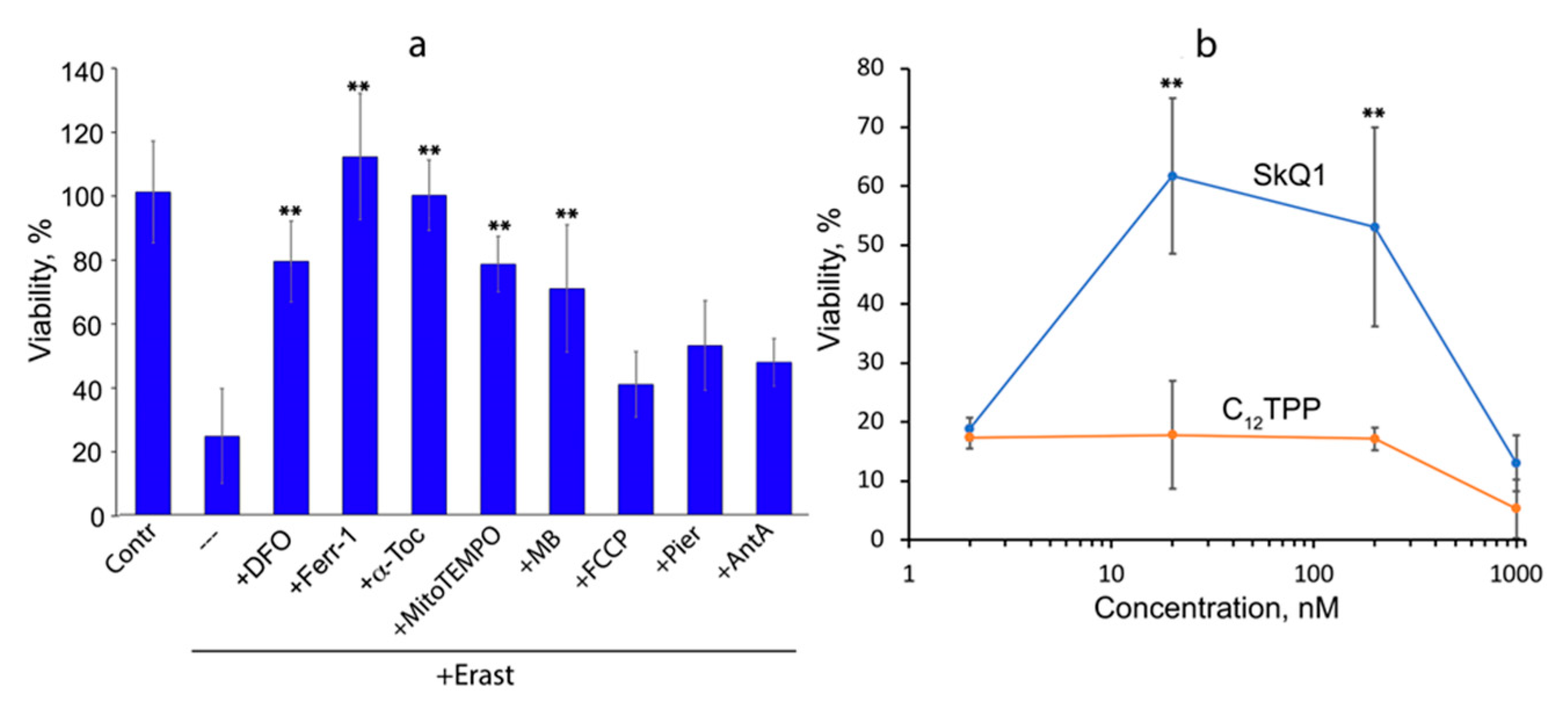
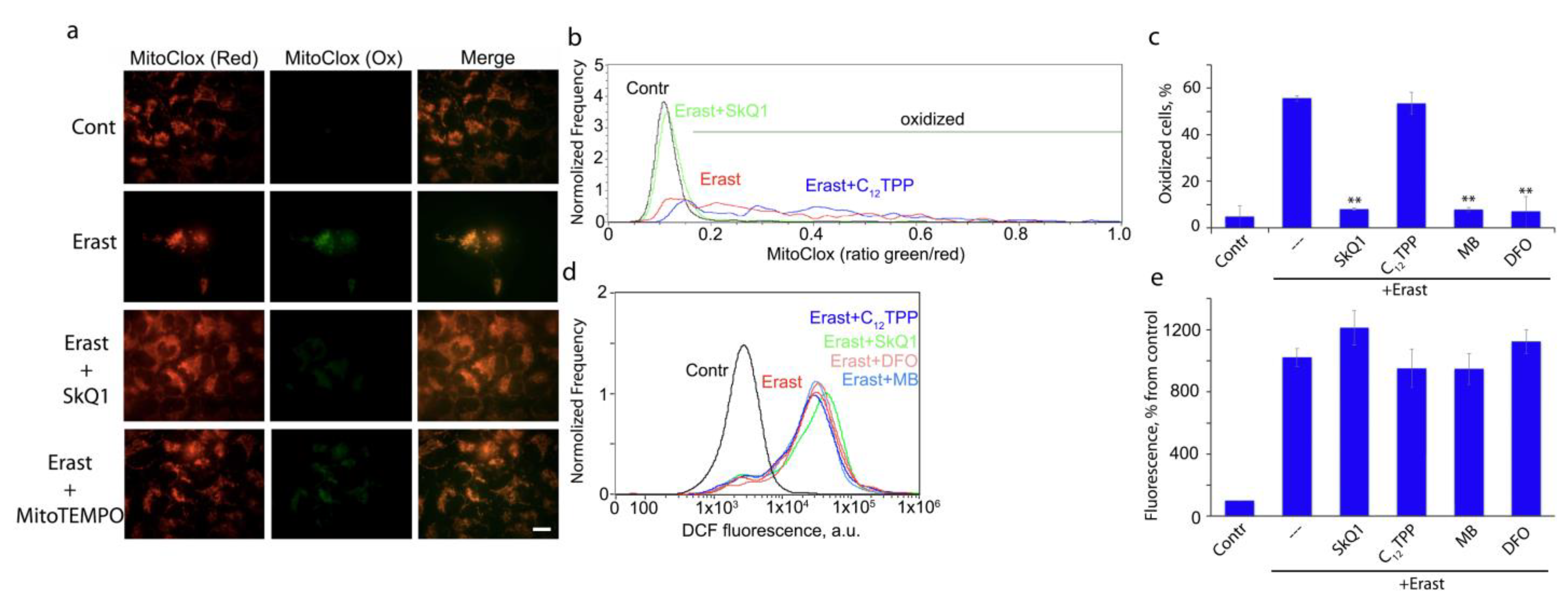
3.1. Mitochondria-Targeted Antioxidant SkQ1 Inhibits Ferroptosis Induced with Erastin
3.2. Ferroptosis Induced by Glutathione Depletion in Fibroblasts Derived from Patient with Leber’s Hereditary Optic Neuropathy (LHON) Depends on mtROS Production
3.3. Peroxidation of Mitochondrial Lipids Is Critical for Ferroptotic Cell Death
3.4. Lipid Peroxidation in Isolated Mitochondria Is Catalyzed by Complex I but Not Complex III
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell. Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363 e353. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Hu, F.; Feng, H.; Linkermann, A.; Min, W.; Stockwell, B.R. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem. Biol. 2018, 13, 1013–1020. [Google Scholar] [CrossRef]
- Basit, F.; van Oppen, L.M.; Schockel, L.; Bossenbroek, H.M.; van Emst-de Vries, S.E.; Hermeling, J.C.; Grefte, S.; Kopitz, C.; Heroult, M.; Hgm Willems, P.; et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death. Dis. 2017, 8, e2716. [Google Scholar] [CrossRef]
- Oh, S.J.; Ikeda, M.; Ide, T.; Hur, K.Y.; Lee, M.S. Mitochondrial event as an ultimate step in ferroptosis. Cell Death. Discov. 2022, 8, 414. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Krainz, T.; Gaschler, M.M.; Lim, C.; Sacher, J.R.; Stockwell, B.R.; Wipf, P. A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS Cent. Sci. 2016, 2, 653–659. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Mao, C.; Kondiparthi, L.; Poyurovsky, M.V.; Olszewski, K.; Gan, B. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc. Natl. Acad. Sci. USA 2022, 119, e2121987119. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Sumbatyan, N.V.; Nesterenko, A.M.; Kholina, E.G.; Voskoboynikova, N.; Steinhoff, H.J.; Mulkidjanian, A.Y.; Chernyak, B.V. MitoCLox: A Novel Mitochondria-Targeted Fluorescent Probe for Tracing Lipid Peroxidation. Oxid. Med. Cell Longev. 2019, 2019, 9710208. [Google Scholar] [CrossRef] [PubMed]
- Lyamzaev, K.G.; Panteleeva, A.A.; Karpukhina, A.A.; Galkin, I.I.; Popova, E.N.; Pletjushkina, O.Y.; Rieger, B.; Busch, K.B.; Mulkidjanian, A.Y.; Chernyak, B.V. Novel Fluorescent Mitochondria-Targeted Probe MitoCLox Reports Lipid Peroxidation in Response to Oxidative Stress In Vivo. Oxid. Med. Cell Longev. 2020, 2020, 3631272. [Google Scholar] [CrossRef] [PubMed]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, D.; Belisle, E.; Severino, D.; Kowaltowski, A.J.; Baptista, M.S. Binding, aggregation and photochemical properties of methylene blue in mitochondrial suspensions. Photochem. Photobiol. 2004, 79, 227–232. [Google Scholar] [CrossRef]
- Atamna, H.; Nguyen, A.; Schultz, C.; Boyle, K.; Newberry, J.; Kato, H.; Ames, B.N. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2008, 22, 703–712. [Google Scholar] [CrossRef]
- Liu, J.; Bandyopadhyay, I.; Zheng, L.; Khdour, O.M.; Hecht, S.M. Antiferroptotic Activity of Phenothiazine Analogues: A Novel Therapeutic Strategy for Oxidative Stress Related Disease. ACS Med. Chem. Lett. 2020, 11, 2165–2173. [Google Scholar] [CrossRef]
- Stack, C.; Jainuddin, S.; Elipenahli, C.; Gerges, M.; Starkova, N.; Starkov, A.A.; Jove, M.; Portero-Otin, M.; Launay, N.; Pujol, A.; et al. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum. Mol. Genet. 2014, 23, 3716–3732. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Jang, S.; Chapa-Dubocq, X.R.; Tyurina, Y.Y.; St Croix, C.M.; Kapralov, A.A.; Tyurin, V.A.; Bayir, H.; Kagan, V.E.; Javadov, S. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox. Biol. 2021, 45, 102021. [Google Scholar] [CrossRef] [PubMed]
- Krylova, T.D.; Sheremet, N.L.; Tabakov, V.Y.; Lyamzaev, K.G.; Itkis, Y.S.; Tsygankova, P.G.; Andreeva, N.A.; Shmelkova, M.S.; Nevinitsyna, T.A.; Kadyshev, V.V.; et al. Three rare pathogenic mtDNA substitutions in LHON patients with low heteroplasmy. Mitochondrion 2020, 50, 139–144. [Google Scholar] [CrossRef]
- Brzheskiy, V.V.; Efimova, E.L.; Vorontsova, T.N.; Alekseev, V.N.; Gusarevich, O.G.; Shaidurova, K.N.; Ryabtseva, A.A.; Andryukhina, O.M.; Kamenskikh, T.G.; Sumarokova, E.S.; et al. Results of a Multicenter, Randomized, Double-Masked, Placebo-Controlled Clinical Study of the Efficacy and Safety of Visomitin Eye Drops in Patients with Dry Eye Syndrome. Adv. Ther. 2015, 32, 1263–1279. [Google Scholar] [CrossRef] [PubMed]
- Shrader, W.D.; Amagata, A.; Barnes, A.; Enns, G.M.; Hinman, A.; Jankowski, O.; Kheifets, V.; Komatsuzaki, R.; Lee, E.; Mollard, P.; et al. alpha-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg. Med. Chem. Lett. 2011, 21, 3693–3698. [Google Scholar] [CrossRef] [PubMed]
- Prime, T.A.; Forkink, M.; Logan, A.; Finichiu, P.G.; McLachlan, J.; Li Pun, P.B.; Koopman, W.J.; Larsen, L.; Latter, M.J.; Smith, R.A.; et al. A ratiometric fluorescent probe for assessing mitochondrial phospholipid peroxidation within living cells. Free Radic. Biol. Med. 2012, 53, 544–553. [Google Scholar] [CrossRef]
- Vinogradov, A.D.; Grivennikova, V.G. Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 863–871. [Google Scholar] [CrossRef]
- Pletjushkina, O.Y.; Lyamzaev, K.G.; Popova, E.N.; Nepryakhina, O.K.; Ivanova, O.Y.; Domnina, L.V.; Chernyak, B.V.; Skulachev, V.P. Effect of oxidative stress on dynamics of mitochondrial reticulum. Biochim. Biophys. Acta 2006, 1757, 518–524. [Google Scholar] [CrossRef]
- Izyumov, D.S.; Domnina, L.V.; Nepryakhina, O.K.; Avetisyan, A.V.; Golyshev, S.A.; Ivanova, O.Y.; Korotetskaya, M.V.; Lyamzaev, K.G.; Pletjushkina, O.Y.; Popova, E.N.; et al. Mitochondria as source of reactive oxygen species under oxidative stress. Study with novel mitochondria-targeted antioxidants--the "Skulachev-ion" derivatives. Biochemistry 2010, 75, 123–129. [Google Scholar] [CrossRef]
- Saretzki, G.; Murphy, M.P.; von Zglinicki, T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell 2003, 2, 141–143. [Google Scholar] [CrossRef]
- Tretter, L.; Horvath, G.; Holgyesi, A.; Essek, F.; Adam-Vizi, V. Enhanced hydrogen peroxide generation accompanies the beneficial bioenergetic effects of methylene blue in isolated brain mitochondria. Free Radic. Biol. Med. 2014, 77, 317–330. [Google Scholar] [CrossRef]
- Boveris, A.; Cadenas, E.; Stoppani, A.O. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem. J. 1976, 156, 435–444. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Sato, H.; Fujii, J. Superoxide produced by mitochondrial complex III plays a pivotal role in the execution of ferroptosis induced by cysteine starvation. Arch. Biochem. Biophys 2021, 700, 108775. [Google Scholar] [CrossRef]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 2021, 218, e20210518. [Google Scholar] [CrossRef]
- Otsu, W.; Ishida, K.; Chinen, N.; Nakamura, S.; Shimazawa, M.; Tsusaki, H.; Hara, H. Cigarette smoke extract and heated tobacco products promote ferritin cleavage and iron accumulation in human corneal epithelial cells. Sci. Rep. 2021, 11, 18555. [Google Scholar] [CrossRef]
- Murphy, C.J.; Oudit, G.Y. Iron-overload cardiomyopathy: Pathophysiology, diagnosis, and treatment. J. Card. Fail. 2010, 16, 888–900. [Google Scholar] [CrossRef]
- Borawski, B.; Malyszko, J. Iron, ferroptosis, and new insights for prevention in acute kidney injury. Adv. Med. Sci. 2020, 65, 361–370. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-Targeted Drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214. [Google Scholar] [CrossRef]
- Bakeeva, L.E.; Barskov, I.V.; Egorov, M.V.; Isaev, N.K.; Kapelko, V.I.; Kazachenko, A.V.; Kirpatovsky, V.I.; Kozlovsky, S.V.; Lakomkin, V.L.; Levina, S.B.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 2. Treatment of some ROS- and age-related diseases (heart arrhythmia, heart infarctions, kidney ischemia, and stroke). Biochemistry 2008, 73, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Chernikov, V.P.; Prusov, A.N.; Kireev, I.I.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mitochondrial Damage and Mitochondria-Targeted Antioxidant Protection in LPS-Induced Acute Kidney Injury. Antioxidants 2019, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Morosanova, M.A.; Pevzner, I.B.; Zorova, L.D.; Manskikh, V.N.; Pulkova, N.V.; Galkina, S.I.; Skulachev, V.P.; Zorov, D.B. Protective effect of mitochondria-targeted antioxidants in an acute bacterial infection. Proc. Natl. Acad. Sci. USA 2013, 110, E3100–E3108. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, V.V.; Pletjushkina, O.Y.; Galkin, I.I.; Zinovkin, R.A.; Chernyak, B.V.; Krysko, D.V.; Bachert, C.; Krysko, O.; Skulachev, V.P.; Popova, E.N. Low concentration of uncouplers of oxidative phosphorylation decreases the TNF-induced endothelial permeability and lethality in mice. Biochim. Biophys Acta Mol. Basis. Dis. 2017, 1863, 968–977. [Google Scholar] [CrossRef]
- Neroev, V.V.; Archipova, M.M.; Bakeeva, L.E.; Fursova, A.; Grigorian, E.N.; Grishanova, A.Y.; Iomdina, E.N.; Ivashchenko Zh, N.; Katargina, L.A.; Khoroshilova-Maslova, I.P.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 4. Age-related eye disease. SkQ1 returns vision to blind animals. Biochemistry 2008, 73, 1317–1328. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyamzaev, K.G.; Panteleeva, A.A.; Simonyan, R.A.; Avetisyan, A.V.; Chernyak, B.V. Mitochondrial Lipid Peroxidation Is Responsible for Ferroptosis. Cells 2023, 12, 611. https://doi.org/10.3390/cells12040611
Lyamzaev KG, Panteleeva AA, Simonyan RA, Avetisyan AV, Chernyak BV. Mitochondrial Lipid Peroxidation Is Responsible for Ferroptosis. Cells. 2023; 12(4):611. https://doi.org/10.3390/cells12040611
Chicago/Turabian StyleLyamzaev, Konstantin G., Alisa A. Panteleeva, Ruben A. Simonyan, Armine V. Avetisyan, and Boris V. Chernyak. 2023. "Mitochondrial Lipid Peroxidation Is Responsible for Ferroptosis" Cells 12, no. 4: 611. https://doi.org/10.3390/cells12040611
APA StyleLyamzaev, K. G., Panteleeva, A. A., Simonyan, R. A., Avetisyan, A. V., & Chernyak, B. V. (2023). Mitochondrial Lipid Peroxidation Is Responsible for Ferroptosis. Cells, 12(4), 611. https://doi.org/10.3390/cells12040611