



Pathogens 2025, 14(10), 1053; https://doi.org/10.3390/pathogens14101053 - 18 Oct 2025
Viewed by 1062
Abstract
►
Show Figures
CWD surveillance and diagnosis are important issues in Europe since its detection in Norway, as some of its strains, like that of classical scrapie, are contagious. In addition, there are concerns as several matters about CWD are not yet known. Although diagnostic methods
[...] Read more.
CWD surveillance and diagnosis are important issues in Europe since its detection in Norway, as some of its strains, like that of classical scrapie, are contagious. In addition, there are concerns as several matters about CWD are not yet known. Although diagnostic methods for the active surveillance in bovine and small ruminants have been able to detect the European CWD strains, a retrospective study on Italian wild red deer (Cervus elaphus) samples was performed to compare the results obtained from rapid screening tests, authorized according to EU Regulation 999/2001, and the RT-QuIC, a highly sensitive method in the detection of prion disease infection. A total of one hundred brainstems and medial retropharyngeal lymph nodes were selected out of those received from the CWD Italian surveillance system. Confirmed CWD-positive and -negative samples were included in the study as controls. All of the samples were first tested with the HerdChek BSE–Scrapie Antigen Test and then using the RT-QuIC. The rapid test was negative in all brainstem and lymph node samples. RT-QuIC analyses showed only one red deer brainstem sample positive for seeding activity, while all lymph nodes were negative, including the one from this case. This positive brainstem sample was then re-extracted and retested using two different recombinant prion protein substrates (Ha90-231; BV23-231) and their different batches from the first analyses. Seeding activity was consistently confirmed across both substrates and extractions, with positive signals detected down to dilutions of 10−4 using rPrP Ha90-231 and as low as 10−6 with rPrP BV23-231. The additional diagnostic investigations performed on this red deer using the alternative rapid test (TeSeE SAP Combi), Western blot, and immunohistochemistry showed negative results both in the brainstem and lymph nodes. This study showed that overall, the results obtained with the HerdChek BSE–Scrapie Antigen Test and RT-QuIC agree except in one case. Our findings highlight the potential of the RT-QuIC method to detect very low levels of PrPSc-associated seeding activity that may escape detection using classical methods. While seeding activity does not always equate to infectivity, only a bioassay will confirm the real disease status of this Italian case. These findings support the integration of RT-QuIC as a powerful complementary tool within existing surveillance frameworks to strengthen early detection and diagnostic accuracy.
Full article
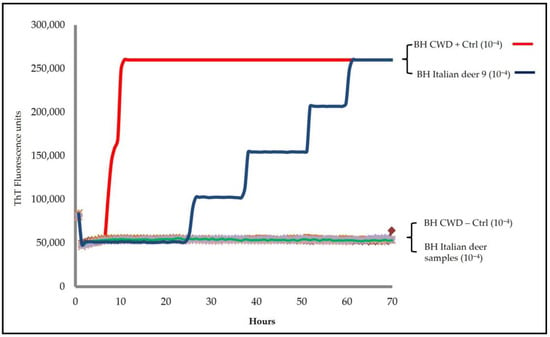
Figure 1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}