
Comparative Analysis of Felixounavirus Genomes Including Two New Members of the Genus That Infect Salmonella Infantis

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
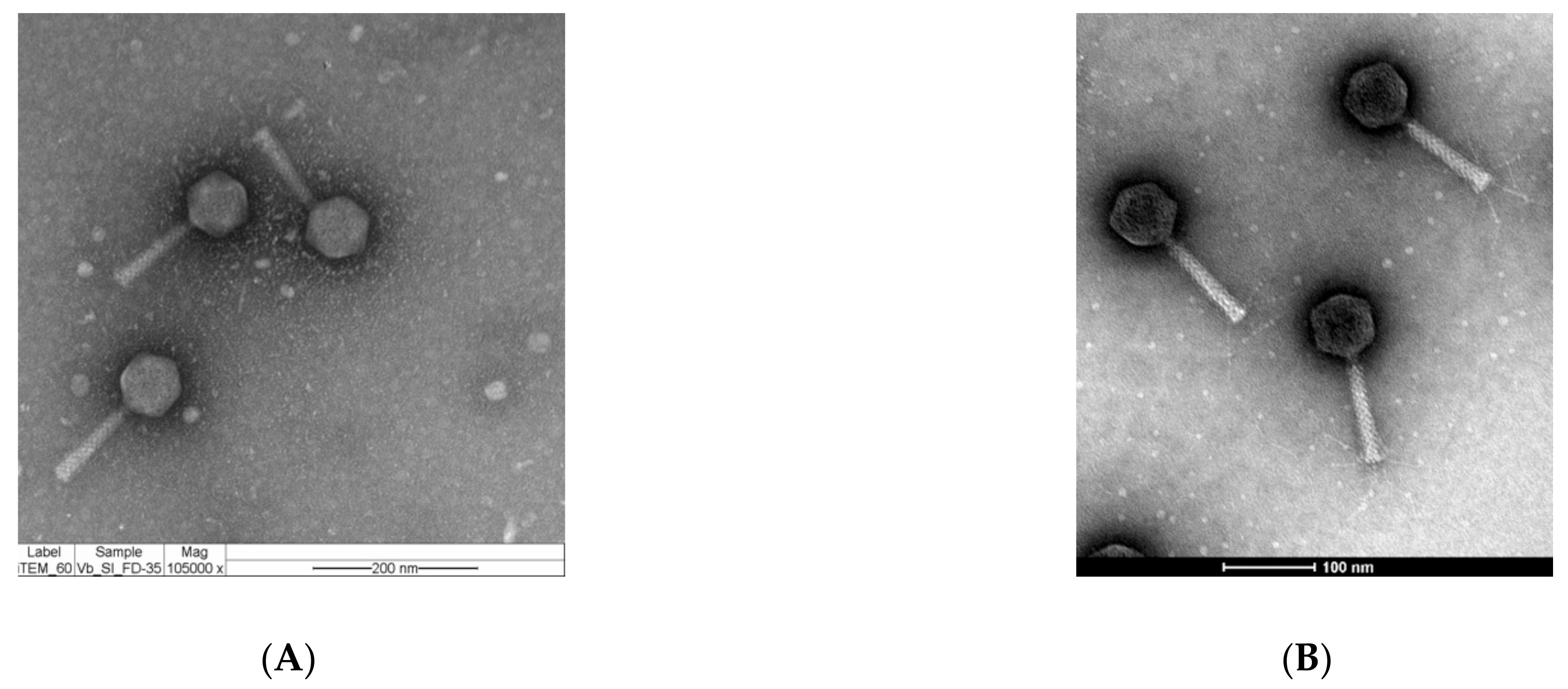
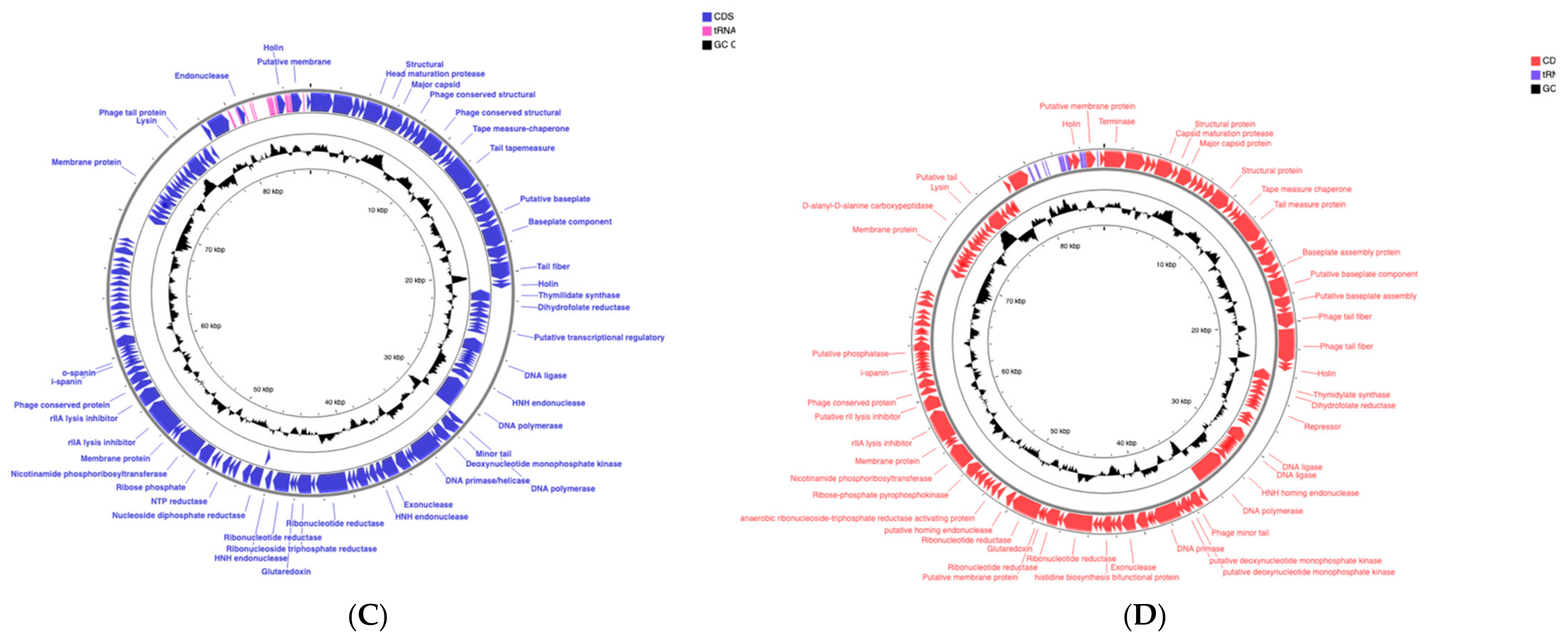
2.1. Phages vB_Si_35FD and vB_Si_DR094 Represent New Members of the Felixounavirus Genus that Infect a Large Number of Salmonella serovars
2.2. Comparative Analysis of Felixounavirus that Infect Salmonella
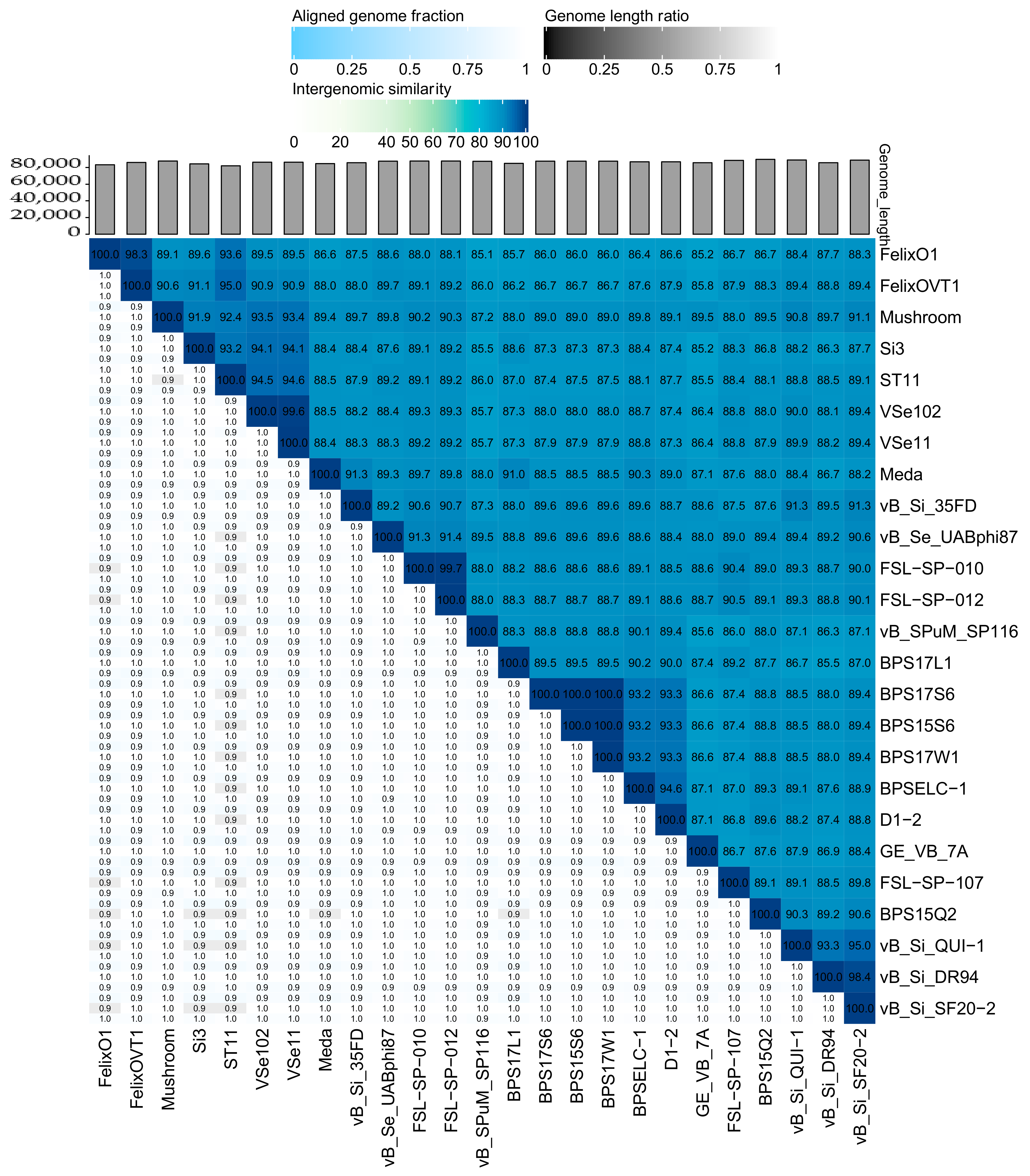
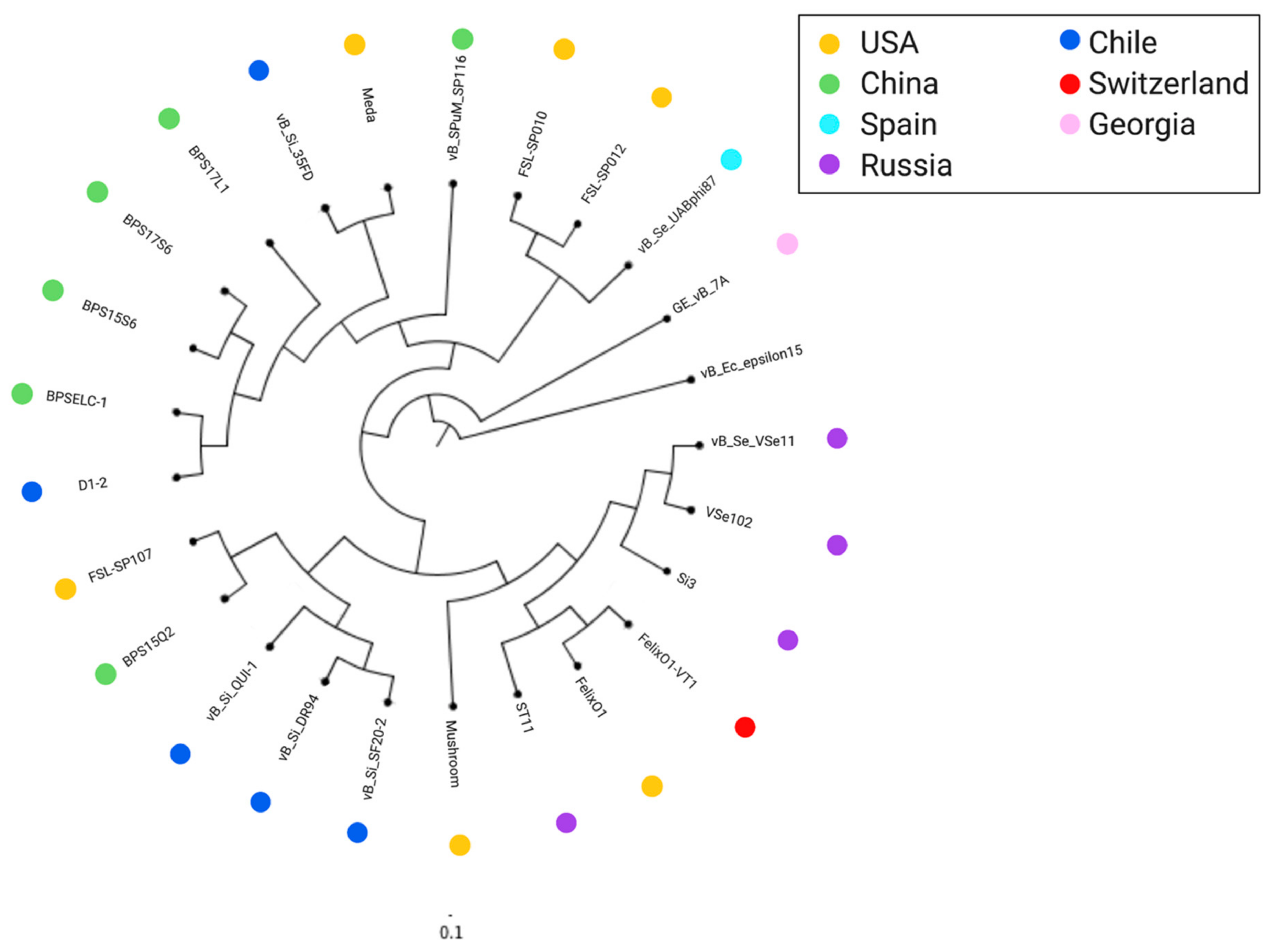
2.2.1. Felixounavirus that Infect Salmonella are Highly Similar on a Global Scale
2.2.2. Similar Genomic Backbone in Felixounaviruses that Infect Salmonella
3. Materials and Methods
3.1. Bacteria and Phage Growth Conditions
3.2. Host Range Characterization Phages vB_Si_35FD and vB_Si_DR94
3.3. Morphological Characterization
3.4. DNA Extraction and Sequencing
3.5. Genome Annotation
3.6. Genome Sequence Accession Number
3.7. Selection of Genomes and Comparative Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hendriksen, R.S.; Vieira, A.R.; Karlsmose, S.; Lo, D.M.A.; Wong, F.; Jensen, A.B.; Wegener, H.C.; Aarestrup, F.M. Global Monitoring of Salmonella Serovar Distribution from the World Health Organization Global Foodborne Infections Network Country Data Bank: Results of Quality Assured Laboratories from 2001 to 2007. Foodborne Pathog. Dis. 2011, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majowicz, S.E.; Musto, J.; Scallan, E.; Angulo, F.J.; Kirk, M.; O’Brien, S.J.; Jones, T.F.; Fazil, A.; Hoekstra, R.M. The Global Burden of Nontyphoidal Salmonella Gastroenteritis. Clin. Infect. Dis. 2010, 50, 882–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharff, R.L. Food Safety Economics. In Food Safety Economics; Springer: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Ohad Gal-Mora, B. Persistent Infection and Long-Term Carriage of Typhoidal and Nontyphoidal Salmonellae. Clin. Microbiol. Rev. 2019, 32, e00088-18. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention (CDC). National Enteric Disease Surveillance: Salmonella Annual Report. 2011. Available online: https://www.cdc.gov/ncezid/dfwed/PDFs/salmonella-annual-report-2011-508c.pdf (accessed on 1 September 2020).
- Antunes, P.; Mourão, J.; Campos, J.; Peixe, L. Salmonellosis: The Role of Poultry Meat. Clin. Microbiol. Infect. 2016, 22, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.P.; Andres, V.; Cheney, T.E.; Martelli, F.; Gosling, R.; Marier, E.; Rabie, A.; Gilson, D.; Davies, R.H. How Do Pig Farms Maintain Low Salmonella Prevalence: A Case-Control Study. Epidemiol. Infect. 2018, 146, 1909–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suttle, C.A. Marine Viruses—Major Players in the Global Ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Twort, F.W. An investigation on the nature of Ultra-Microcopic Viruses. Lancet 1915, 186, 1241–1243. [Google Scholar] [CrossRef] [Green Version]
- D’Hérelle, F. On an invisible microbe antagonistic to dysentery bacilli. Bacteriophage 2011, 1, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Tolstoy, I.; Kropinski, A.M.; Brister, J.R. Bacteriophage Taxonomy: An Evolving Discipline. Bacteriophage Therapy—From Lab to Clinical Practice. Antimicrob. Agents Chemother. 2018, 1693, 649–659. [Google Scholar] [CrossRef]
- Yap, M.L.; Rossmann, M.G. Structure and function of bacteriophage T4. Future Microbiol. 2014, 9, 1319–1327. [Google Scholar] [CrossRef] [Green Version]
- Felix, A.; Callow, B.R. Typing of Paratyphoid B Bacilli by Vi Bacteriophage. Br. Med. J. 1943, 2, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.J.; Carstens, E.B. Ratification Vote on Taxonomic Proposals to the International Committee on Taxonomy of Viruses. Arch. Virol. 2012, 157, 1411–1422. [Google Scholar] [CrossRef] [Green Version]
- Welkos, S.; Schreiber, M.; Baer, H. Identification of Salmonella with the O-1 Bacteriophage. Appl. Microbiol. 1974, 28, 618–622. [Google Scholar] [CrossRef]
- Rivera, D.; Hudson, L.K.; Denes, T.G.; Hamilton-West, C.; Pezoa, D.; Moreno-Switt, A.I. Two Phages of the Genera Felixunavirus Subjected to 12 Hour Challenge on Salmonella Infantis Showed Distinct Genotypic and Phenotypic Changes. Viruses 2019, 11, 586. [Google Scholar] [CrossRef] [Green Version]
- Volozhantsev, N.V.; Denisenko, E.A.; Kislichkina, A.A.; Myakinina, V.P.; Krasilnikova, V.M.; Verevkin, V.V.; Kadnikova, L.A.; Maiskaya, N.V.; Bogun, A.G.; Dyatlov, I.A. Complete Genome Sequences of Two Salmonella Viruses, VSe11 and VSe102 (Family Myoviridae, Subfamily Ounavirinae), with a Very High Degree of Similarity. Genome Announc. 2018, 6, 11–12. [Google Scholar] [CrossRef] [Green Version]
- Neff, J.K.; Xie, Y.; Gill, J.J.; Liu, M. Complete Genome Sequence of Salmonella Enterica Serovar Heidelberg Myophage Meda. Microbiol. Resour. Announc. 2019, 8, e00253-19. [Google Scholar] [CrossRef] [Green Version]
- Rivera, D.; Toledo, V.; Pillo, F.D.I.; Dueñas, F.; Tardone, R.; Hamilton-West, C.; Vongkamjan, K.; Wiedmann, M.; Moreno Switt, A.I. Backyard Farms Represent a Source of Wide Host Range Salmonella Phages That Lysed the Most Common Salmonella Serovars. J. Food Prot. 2018, 81, 272–278. [Google Scholar] [CrossRef] [Green Version]
- Makalatia, K.; Kakabadze, E.; Wagemans, J.; Grdzelishvili, N.; Bakuradze, N.; Natroshvili, G.; Macharashvili, N.; Sedrakyan, A.; Arakelova, K.; Ktsoyan, Z.; et al. Characterization of Salmonella Isolates from Various Geographical Regions of the Caucasus and Their Susceptibility to Bacteriophages. Viruses 2020, 12, 1418. [Google Scholar] [CrossRef]
- Bardina, C.; Spricigo, D.A.; Cortés, P.; Llagostera, M. Significance of the Bacteriophage Treatment Schedule in Reducing Salmonella Colonization of Poultry. Appl. Environ. Microbiol. 2012, 78, 6600–6607. [Google Scholar] [CrossRef] [Green Version]
- Spricigo, D.A.; Bardina, C.; Cortés, P.; Llagostera, M. Use of a Bacteriophage Cocktail to Control Salmonella in Food and the Food Industry. Int. J. Food Microbiol. 2013, 165, 169–174. [Google Scholar] [CrossRef]
- Abedon, S.T.; Yin, J. Chapter 17 Bacteriophage Plaques: Theory and Analysis. Bacteriophages Methods Protoc. 2009, 501, 161–175. [Google Scholar] [CrossRef]
- Vinner, G.K.; Rezaie-Yazdi, Z.; Leppanen, M.; Stapley, A.G.F.; Leaper, M.C.; Malik, D.J. Microencapsulation of Salmonella-Specific Bacteriophage Felix O1 Using Spray-Drying in a Ph-Responsive Formulation and Direct Compression Tableting of Powders into a Solid Oral Dosage Form. Pharmaceuticals 2019, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Niu, Y.D.; Nan, Y.; Stanford, K.; Holley, R.; McAllister, T.; Narváez-Bravo, C. SalmoFresh™ effectiveness in controlling Salmonella on romaine lettuce, mung bean sprouts and seeds. Int. J. Food Microbiol. 2019, 305, 108250. [Google Scholar] [CrossRef]
- Zschach, H.; Joensen, K.G.; Lindhard, B.; Lund, O.; Goderdzishvili, M.; Chkonia, I.; Jgenti, G.; Kvatadze, N.; Alavidze, Z.; Kutter, E.M.; et al. What Can We Learn from a Metagenomic Analysis of a Georgian Bacteriophage Cocktail? Viruses 2015, 7, 6570–6589. [Google Scholar] [CrossRef] [Green Version]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage Diversity, Genomics and Phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef]
- Whichard, J.M.; Weigt, L.A.; Borris, D.J.; Li, L.L.; Zhang, Q.; Kapur, V.; William Pierson, F.; Lingohr, E.J.; She, Y.M.; Kropinski, A.M.; et al. Complete Genomic Sequence of Bacteriophage Felix O1. Viruses 2010, 2, 710–730. [Google Scholar] [CrossRef]
- Morgado, S.; Vicente, A.C. Global In-Silico Scenario of tRNA Genes and Their Organization in Virus Genomes. Viruses 2019, 11, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the Intriguing Presence of TRNAs in Phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herskowitz, I. Control of Gene Expression in Bacteriophage Lambda. Annu. Rev. Genet. 1973, 7, 289–324. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; van Raaij, M. Bacteriophage T4 Long Tail Fiber Domains. Biophys. Rev. 2018, 10, 463–471. [Google Scholar] [CrossRef] [Green Version]
- MacPhee, D.G.; Krishnapillai, V.; Roantree, R.J.; Stocker, B.A.D. Mutations in Salmonella Typhimurium Conferring Resistance to Felix O Phage without Loss of Smooth Character. J. Gen. Microbiol. 1975, 86, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kirtania, P.; Bhattacharya, B.; Das Gupta, S.K. Mycobacteriophage L5Gp56, a Novel Member of the NrdH Family of Redoxins. FEMS Microbiol. Lett. 2014, 357, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Lundin, D.; Torrents, E.; Poole, A.M.; Sjöberg, B.M. RNRdb, a Curated Database of the Universal Enzyme Family Ribonucleotide Reductase, reveals a High Level of Misannotation in Sequences Deposited to Genbank. BMC Genom. 2009, 10, 589. [Google Scholar] [CrossRef] [Green Version]
- Switt, A.I.M.; Orsi, R.H.; Bakker, H.C.D.; Vongkamjan, K.; Altier, C.; Wiedmann, M. Genomic characterization provides new insight into Salmonella phage diversity. BMC Genom. 2013, 14, 481. [Google Scholar] [CrossRef] [Green Version]
- Edgell, D.R.; Gibb, E.A.; Belfort, M. Mobile DNA Elements in T4 and Related Phages. Virol. J. 2010, 7, 290. [Google Scholar] [CrossRef] [Green Version]
- Bonocora, R.P.; Zeng, Q.; Abel, E.V.; Shub, D.A. A Homing Endonuclease and the 50-Nt Ribosomal Bypass Sequence of Phage T4 Constitute a Mobile DNA Cassette. Proc. Natl. Acad. Sci. USA 2011, 108, 16351–16356. [Google Scholar] [CrossRef] [Green Version]
- Young, R. Phage Lysis: Three Steps, Three Choices, One Outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef]
- Kongari, R.; Rajaure, M.; Cahill, J.; Rasche, E.; Mijalis, E.; Berry, J.; Young, R. Phage Spanins: Diversity, Topological Dynamics and Gene Convergence. BMC Bioinform. 2018, 19, 326. [Google Scholar] [CrossRef] [Green Version]
- Cahill, J.; Young, R. Phage Lysis: Multiple Genes for Multiple Barriers. Adv. Virus Res. 2019, 103, 33–70. [Google Scholar] [CrossRef]
- Chen, Y.; Young, R. The Last r Locus Unveiled: T4 RIII Is a Cytoplasmic Antiholin. J. Bacteriol. 2016, 198, 2448–2457. [Google Scholar] [CrossRef] [Green Version]
- Paddison, P.; Abedon, S.T.; Dressman, H.K.; Gailbreath, K.; Tracy, J.; Mosser, E.; Neitzel, J.; Guttman, B.; Kutter, E. The roles of the bacteriophage T4 r genes in lysis inhibition and fine-structure genetics: A new perspective. Genetics 1998, 148, 1539–1550. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage Therapy: Eco-Physiological Pharmacology. Scientifica 2014, 2014, 581639. [Google Scholar] [CrossRef]
- Mahony, J.; McAuliffe, O.; Ross, R.P.; van Sinderen, D. Bacteriophages as Biocontrol Agents of Food Pathogens. Curr. Opin. Biotechnol. 2011, 22, 157–163. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Tolen, T.N.; Xie, Y.; Hernandez, A.C.; Everett, G.F.K. Complete Genome Sequence of Salmonella Enterica Serovar Typhimurium Myophage Mushroom. Genome Announc. 2016, 3, 2011–2012. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.T.; Carvalho, J.; Cunha, M.V.; Serrano, E.; Palmeira, J.D.; Fonseca, C. Temporal and Geographical Research Trends of Antimicrobial Resistance in Wildlife—A Bibliometric Analysis. One Health 2021, 11, 100198. [Google Scholar] [CrossRef]
- Dyatlov, I.A.; Aleshkin, V.A.; Aleshkin, A.V.; Afanas’Ev, S.S.; Svetoch, E.A.; Volozhantsev, N.V.; Vasyliev, D.A.; Zolotukhin, S.N.; Amerkhanova, A.M.; Kiseleva, I.A.; et al. Antibacterial composition, strain of bacteriophage Escherichia coli, used for obtaining thereof patent ru2518303. 2019. Available online: https://patents.google.com/patent/RU2518303C2/en (accessed on 1 July 2021).
- Kakabadze, E.; Makalatia, K.; Grdzelishvili, N.; Bakuradze, N.; Goderdzishvili, M.; Kusradze, I.; Phoba, M.-F.; Lunguya, O.; Lood, C.; Lavigne, R.; et al. Selection of Potential Therapeutic Bacteriophages that Lyse a CTX-M-15 Extended Spectrum β-Lactamase Producing Salmonella enterica Serovar Typhi Strain from the Democratic Republic of the Congo. Viruses 2018, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Shcherbakov, V.P.; Plugina, L.; Shcherbakova, T. Endonuclease VII Is a Key Component of the Mismatch Repair Mechanism in Bacteriophage T4. DNA Repair 2011, 10, 356–362. [Google Scholar] [CrossRef]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host Receptors for Bacteriophage Adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef] [Green Version]
- Linberg, A.A. Studies of a Receptor for Felix 0-1 Phage in Salmonella minnesota. J. Gen. Micriobiol. 1967, 48, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, M.; Wright, A. Variation in the Structure and Bacteriophage-Inactivating Capacity of Salmonella Anatum Lipopolysaccharide as a Function of Growth Temperature. J. Bacteriol. 1979, 137, 746–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azam, A.H.; Tanji, Y. Bacteriophage-Host Arm Race: An Update on the Mechanism of Phage Resistance in Bacteria and Revenge of the Phage with the Perspective for Phage Therapy. Appl. Microbiol. Biotechnol. 2019, 103, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, F.; Rivera, D.; Toledo, V.; Tardone, R.; Hervé-Claude, L.P.; Hamilton-West, C.; Switt, A.I.M. Short Communication: Characterization of Salmonella Phages from Dairy Calves on Farms with History of Diarrhea. J. Dairy Sci. 2017, 100, 2196–2200. [Google Scholar] [CrossRef] [Green Version]
- Parra, B.; Robeson, J. Selection of Polyvalent Bacteriophages Infecting Salmonella Enterica Serovar Choleraesuis. Electron. J. Biotechnol. 2016, 21, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, H.W. Basic Phage Electron Microscopy. Methods Mol. Biol. 2009, 501, 113–126. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A Modular and Extensible Implementation of the RAST Algorithm for Building Custom Annotation Pipelines and Annotating Batches of Genomes. Sci. Rep. 2015, 5, srep08365. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A Comparative Genomics Tool for Circular Genomes. Nucleic Acids Res. 2008, 36 (Suppl. 2), W181–W184. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based Phylogeny and Classification of Prokaryotic Viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [Green Version]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–667. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, 699–710. [Google Scholar] [CrossRef]
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; A Goodwin, L.; et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A Genome Comparison Visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogeneticst; Oxford University Press: New York, NY, USA, 2001. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Accession Number | Genome Size (kb) * | %GC * | CDS * | tRNA * | Isolation Country | Isolation Sample |
|---|---|---|---|---|---|---|---|
| FelixO1 | JF461087.1 | 83.33 | 38.9 | 125 | 18 | Switzerland | - |
| Mushroom | KP143762.1 | 87.71 | 39 | 129 | 22 | USA | IntestiPhage |
| D1-2 | MN481367.1 | 86.88 | 38.7 | 132 | 18 | China | Sewage |
| vB_Si_SF20-2 (DaR-2019b) | MK965970.1 | 88.97 | 39.1 | 131 | 20 | Chile | Poultry feces |
| vB_Si_QUI-1 (DaR-2019a) | MK965969.1 | 89.09 | 39.1 | 129 | 20 | Chile | Poultry feces |
| Meda | MH586731.1 | 84.67 | 38.8 | 131 | 19 | USA | Soil in the cattle holding pen of cattle harvest facility |
| GE_vB_7A | MG969404.1 | 85.78 | 39.0 | 165 | 21 | Georgia | Mtkvari river water |
| BPS17W1 | MG646669.1 | 87.61 | 38.8 | 130 | 19 | China | Sewage samples from hog house |
| BPS17S6 | MG646671.1 | 87.63 | 38.8 | 131 | 19 | China | Sewage samples from layer house |
| BPS17L1 | MG646672.1 | 84.92 | 38.9 | 125 | 21 | China | Sewage samples from slaughterhouse |
| BPS15S6 | MG646670.1 | 87.61 | 38.8 | 130 | 19 | China | Sewage samples from layer house |
| VSe102 | MG251392.1 | 86.37 | 39.0 | 126 | 17 | Russia | Farm sewage |
| VSe11 | MG251391.1 | 86.36 | 39.0 | 126 | 17 | Russia | Sewage |
| ST11 | MF370225.1 | 82.1 | 39.0 | 130 | 19 | Russia | Chicken feces |
| Si3 | KY626162.1 | 84.42 | 39.0 | 125 | 17 | Russia | - |
| BPS15Q2 | KX405003.1 | 89.82 | 38.9 | 132 | 20 | China | Domestic sewage samples |
| vB_SPuM_SP116 | KP010413.1 | 87.51 | 38.8 | 130 | 21 | China | Sewage |
| FelixO1VT | AF320576.1 | 86.16 | 39.0 | 126 | 19 | USA | - |
| BPSELC-1 | MN227145.1 | 86.99 | 38.8 | 129 | 19 | China | Chicken manure |
| FSL-SP-010 | ** KC139526.1-KC139527.1-KC139528.1 | 87.73 | 39.1 | 134 | 18 | USA | Bovine feces |
| FSL-SP-012 | ** KC139543.1-KC139544.1 | 87.81 | 39.0 | 132 | 19 | USA | Bovine feces |
| FSL-SP-107 | ** KC139640.1-KC139638.1 | 88.52 | 39.0 | 136 | 19 | USA | Bovine feces |
| vB_Si_35FD | MZ327261 | 85.81 | 38.9 | 129 | 19 | Chile | Bovine feces |
| vB_Si_DR094 | MZ327262 | 85.7 | 39.2 | 125 | 20 | Chile | Bovine feces |
| vB_Se_ UABphi87 | NC_027360.1 | 87.8 | 38.9 | 129 | 23 | Spain | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barron-Montenegro, R.; García, R.; Dueñas, F.; Rivera, D.; Opazo-Capurro, A.; Erickson, S.; Moreno-Switt, A.I. Comparative Analysis of Felixounavirus Genomes Including Two New Members of the Genus That Infect Salmonella Infantis. Antibiotics 2021, 10, 806. https://doi.org/10.3390/antibiotics10070806
Barron-Montenegro R, García R, Dueñas F, Rivera D, Opazo-Capurro A, Erickson S, Moreno-Switt AI. Comparative Analysis of Felixounavirus Genomes Including Two New Members of the Genus That Infect Salmonella Infantis. Antibiotics. 2021; 10(7):806. https://doi.org/10.3390/antibiotics10070806
Chicago/Turabian StyleBarron-Montenegro, Rocío, Rodrigo García, Fernando Dueñas, Dácil Rivera, Andrés Opazo-Capurro, Stephen Erickson, and Andrea I Moreno-Switt. 2021. "Comparative Analysis of Felixounavirus Genomes Including Two New Members of the Genus That Infect Salmonella Infantis" Antibiotics 10, no. 7: 806. https://doi.org/10.3390/antibiotics10070806