Abstract
Glomerular podocytes are highly differentiated cells that cover glomerular capillaries from the outside and have a characteristic morphology with numerous foot processes. The formation of slit membranes between the foot processes serves as a final filtration barrier for urine filtration from the blood. Podocyte damage causes disruption of the slit membrane, subsequent proteinuria and finally glomerulosclerosis, which is a common pathway in various types of chronic kidney disease (CKD). In recent years, there has been an increase in diabetes, due to rapid lifestyle changes, which is the main cause of CKD. Therefore, understanding the effect of diabetic status on podocytes is of great importance to establish a strategy for preventing CKD progression. In this review, we summarize altered glucose and lipid metabolism in diabetic podocytes and also discuss the reversibility of the changes in podocyte phenotype.
1. Introduction
Chronic kidney disease (CKD), defined as evidence of structural or functional renal impairment for 3 or more months, is generally progressive and irreversible. The global prevalence of diabetes mellitus (DM) has increased over the past few decades, mainly driven by an increase in the prevalence of type 2 diabetes mellitus (T2DM) due to obesity and the metabolic syndrome [1,2]. Microvascular changes within the kidney often lead to chronic kidney disease, an entity referred to as diabetic nephropathy (DN) [3]. DN is the most common cause of CKD and end-stage kidney disease worldwide [4].
It has recently become clear that the initial glomerular injury affects the podocytes as important target cells for the progression of CKD and end-stage kidney disease [5]. Podocytes are highly differentiated epithelial cells that cover the outer layer of the glomerular basement membrane. Podocytes serve as the final barrier to urinary protein loss through the special formation of foot processes and an interposed slit diaphragm. As podocytes are terminally differentiated cells without a capacity for proliferation or replenishment, chronic injury causes phenotypical changes, detachment and apoptosis in podocytes, leading to disruption of the slit membrane and finally glomerulosclerosis [6,7].
Therefore, understanding podocyte phenotype and function in diabetes is necessary for CKD management and for considering future therapeutic targets. This review summarizes podocyte injury in diabetes, focusing on not only the effect of high glucose itself, but also lipotoxicity that is frequently associated with diabetes.
2. Effects of Hyperglycemia on Podocytes in Diabetic Nephropathy (DN)
Hyperglycemia induces a podocytopathy, characterized by cellular hypertrophy, foot process effacement, and podocyte depletion. It is suggested that podocytes may respond to injurious stimuli in different ways, including hypertrophy, dedifferentiation, detachment and depletion, depending on the severity and duration of the injury [8]. Some of the new mechanisms that drive the effects of hyperglycemia on podocytes and possible therapeutic targets are discussed below, divided into chapters by types of podocyte morphological changes, because the function of podocytes primarily depends on their structure and morphology. The key molecules in the impact of hyperglycemia on podocytes in DN are summarized in Table 1.
Glycemic control is the main determinant to prevent the progression to overt DN. Improvement in HbA1c decelerates glomerular filtration rate (GFR) loss and delays the onset of End stage renal diseases (ESRD) in patients with type 1 diabetes mellitis (T1DM) and proteinuria [9]. Moreover, 10 years after pancreas transplantation in type 2 diabetes mellitus (T2DM) under normoglycemia, glomerular lesions were markedly improved, which suggests that the lesions of diabetic nephropathy are reversible and that the kidney can undergo remodeling upon long-term normoglycemia [10]. On the other hand, large clinical trials of diabetes have suggested that even transient treatments or injuries could have a sustained effect on the onset or progression of complications for longer periods, which is known as ‘metabolic memory’ [11,12,13,14]. One of possible mechanisms of the memory is epigenetic alteration. Recent studies indicate that specific DNA methylation of blood or kidney cells in diabetic patients may be associated with development of the nephropathy [15,16,17]. Therefore, better understanding of epigenetic regulation in podocytes is necessary when considering the reversibility or plasticity of podocyte damage following therapeutic approaches in DN. Epigenetic alterations in diabetic podocytes are also described.
2.1. Podocyte Hypertrophy
Previous studies of animal models and humans have established that podocyte hypertrophy is associated with the development of DN [18,19]. Angiotensin Ⅱ has been shown to increase the expression of parathyroid hormone-related protein (PTHrP), Transformin Growth Factor β1 (TGF-β1), and cell cycle regulatory protein-p27Kip, which promotes the aggravation of podocyte hypertrophy in high-glucose conditions [20]. Several studies have suggested that mTORC1 (mechanistic target of rapamycin signaling complex 1), a kinase that senses nutrient availability, was upregulated in podocyte of diabetic mice, and closely associated with the activation of podocyte hypertrophy induced by high glucose [21]. Inoki et al. have reported that abnormal mTORC1 activation caused mislocalization of slit diaphragm proteins and induced endoplasmic reticulum (ER) stress in podocytes, which suggest mTORC1 activation in podocytes is a critical event in inducing DN [22]. Moreover, knockout of Ragulator component p18, recruiting of mTORC1 to lysosomal membranes, attenuated its activation and cell injury under diabetic conditions. This points to mTOR-lysosome recruitment as a potential therapeutic target for the treatment of DN [23].
2.2. Foot Process Effacement
Foot process effacement is a cytoskeletal rearrangement of podocytes reflected by flattening, widening, and retraction of foot processes that signifies podocyte injury and weakening of the integrity of the glomerular filter barrier, thereby leading to albuminuria in DN [24,25]. Dysregulation of nephrin, an essential transmembrane protein in the slit diaphragm complex [26,27], is an important mechanism of foot process effacement. The expression of nephrin is decreased in DN, resulting in aberrant rearrangement of actin and breakdown of the slit diaphragm and foot process effacement [28]. Mislocalization of nephrin has been also observed in DN [29]. Several studies have suggested that protein kinase C α-type (PKCα) was upregulated under hyperglycemia and mediates beta-arrestin2-dependent nephrin endocytosis [30,31]. Protein kinase C and casein kinase substrate in neurons protein 2(PACSIN2) has also been reported as a molecule involved in nephrin endocytosis [29]. Endpcytosis of nephrin is a promising target molecule for podocyte protective therapy in DN.

Table 1.
Key molecules associated with high glucose in podocytes.
Table 1.
Key molecules associated with high glucose in podocytes.
| Key Molecules | Effect on Podocytes | Mechanism | Experimental Model | Expression | Ref. |
|---|---|---|---|---|---|
| mTORC1 | Hypertyophy Foot process effacement GBM thickening podocyte loss | Mislocalization of nephrin, ER stress | db/db mice | increase | [21,22] |
| PKCα | Foot process effacement | Endocytosis of nephrin | STZ mice DN, human | increase | [30,31] |
| Integrinα3β | Podocyte detachment | connecting podocytes with the 7GBM | STZ rats DN, human | decrease | [32,33] |
| NOX4 | Foot process effacement | ROS production | ApoE(−/−) mice | increase | [34] |
| NOX5 | Foot process effacement GBM thickening | ROS production | DN, human | increase | [35] |
| TRPC5 | Foot process effacement Podocyte loss | cytoskeletal rearrangement | Dhal SS rats | increase | [36,37] |
| TRPC6 | Podocyte apoptosis | intracellular Ca(2+) overload, NOX4-derived ROS production | Dhal SS rats | increase | [38] |
| Dnmt1 | Foot process effacement | DNA methylation in the nephrin promoter region | db/db mice | increase | [39] |
| KLF4 | Foot process effacement | DNA methylation in the nephrin promoter region | db/db mice DN, human | decrease | [40,41] |
| KAT5 | Foot process effacement | Impaired DNA repair | db/db mice | decrease | [42] |
mTORC1: mechanistic target of rapamycin complex1, PKCα: Protein kinase Cα, NOX4: nicotinamide adenine dinucleotide phosphate oxdase4,NOX5: nicotinamide adenine dinucleotide phosphate oxdase5, TRPC5: Transient receptor potential cation channel, subfamily C, member 5, TRPC6: Transient receptor potential cation channel, subfamily C, member 6, Dnmt1: DNA (cytosine-5)-methyltransferase 1, KLF4: Krüppel-like transcription factor 4, GBM: glomerular basement membrane, ER: endoplasmic reticulum, ROS: Reactive Oxygen Species, STZ: Streptozocin, DN: diabetic nephropathy, ApoE(−/−): apo E deficiency, Dhal SS: Dhal salt sensitive.
The reduction of nephrin expression by DN results in not only foot process effacement but also insulin signaling alternation, because the cytoplasmic tail of nephrin is necessary for proper insulin signaling [43]. Mice with podocyte-specific deletion of the insulin receptor develop significant albuminuria together with histological features that recapitulate DN in a normoglycemia, which reveals insulin signaling to the podocyte is critical for kidney function [8].
2.3. Podocyte Detachment and Apoptosis
The podocyte and glomerular basement membrane (GBM) are closely connected to prevent proteinuria by sustaining the glomerular filtration barrier. Integrin α3β is an important receptor that can tightly connect podocytes with the GBM. The expression of α3β1 has been shown to be decreased in patients with diabetes and in rats with streptozotocin-induced diabetes, contributing to detachment of podocytes from the GBM [32,33]. Podocyte apoptosis is caused by glomerular hyperfiltration as well as hyperglycemia itself. Several studies have shown that TGF-β induces apoptosis in podocytes by stimulating mitogen-activated protein kinase (MAPK) p38 signaling and the classic effector caspase-3 pathway in DN [44,45]. Podocyte apoptosis under high-glucose conditions is associated with the release of mitochondrial and plasma membrane reactive oxygen species (ROS) that trigger the p38MAPK and nicotinamide adenine dinucleotide phosphate oxidases (NOX) signaling pathways [46,47]. In the rodent kidney, three isoforms of the catalytic subunit of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase are expressed (NOX1, NOX2, and NOX4). Nox4 is the main source of renal ROS in a mouse model of DN [34]. NOX5 has been recently reported to be upregulated in human DN podocytes, and alter filtration barrier function through the production of ROS [35]. ROS has obtained increasing attention in recent years as a therapeutic target for DN, as we know bardoxolone methyl, an oral antioxidant inflammation modulator, improves renal function in patients with advanced CKD and T2DM [48]. Several reports have shown that NOX inhibition in vivo reduces albuminuria and podocytopenia in models of diabetes [49,50]. Furthermore, pharmacological inhibition of NOX1 and -4 reduces albuminuria and slows DN progression in a T2DM model [51]. Transient receptor potential canonical 6 (TRPC6) channel is reported to play a critical role on podocyte ROS. High glucose levels have been shown to induce podocyte apoptosis by stimulating TRPC6 channel-mediated elevation of intracellular calcium in the presence of elevated ROS levels [52]. Recent studies demonstrated that angiotensin II enhances albuminuria by activating TRPC6 channels in podocytes, and this pathway is required the production of ROS [53,54]. Furthermore, it has been reported that NOX4-derived ROS is associated with TRPC5/TRPC6 channels [38]. In regard to TRPC5, TRPC5 is activated by Rac-1, which induces podocyte damage [36]. AC1903, A small-molecule inhibitor of TRPC5, suppressed severe proteinuria and prevented podocyte loss in a rat model of hypertensive proteinuric kidney disease, which indicates TRPC5 inhibitors may be valuable for the treatment of progressive kidney diseases [37].
2.4. Epigenetic Regulation in Diabetic Podocytes
Recently, the link between altered gene expression and epigenetic regulation has attracted much attention in CKD, especially that due to DN. Previously, we showed increased DNA methylation at the nephrin promoter region in podocytes of murine models of DN and patients with DN, which is associated with decreased expression of the transcription factor Kruppel-like factor 4 (KLF4) [40,41]. Zhang et al. also reported a role of DNA methylation in the nephrin promoter region by DNA methyltransferase (DNMT) 1, which may be a possible target for the treatment of DN [39]. Regarding histone modifications, it has been reported that hyperglycemia causes direct modification of histone acetylation status [55]. Interestingly, Rizotte et al. demonstrated that altered histone acetylation and methylation in diabetic podocytes were sustained even after correction of glycemic control to the normal range by continuous insulin administration [56]. This result indicates the possibility that persistent epigenetic changes in podocytes may contribute to ‘metabolic memory’, which is the persistent effect of transient treatments or injuries on disease progression. These results indicate the possibility that podocyte phenotype may be recovered if altered epigenetic marks could be reversed. Previously we have reported that podocyte phenotype could be recovered by transient restoration of KLF4 expression in podocytes using the Tet-on system, with reversed DNA methylation status [41]. Up to now, it is still unclear the intensity and duration of treatment which is necessary to cause epigenetic recovery in podocytes. Further study is needed to investigate the reversibility and plasticity of the podocyte epigenome in each stage of DN.
2.5. DNA Damage and Hyperglycemia
We have discussed epigenetic modifications and the development of CKD, but the process of formation of such epigenetic changes has remained unclear. We have recently focused on the involvement of DNA damage repair in DNA methylation changes in podocytes, since they are terminally differentiated cells without DNA replication. A DNA repair factor lysine acetyltransferase 5 (KAT5) was found to be essential for the maintenance of the podocyte genomic integrity and hyperglycemia was shown to decrease the expression of KAT5 as well as to increase DNA damage in podocytes. Therefore, both of these induce DNA methylation changes in podocytes, which may be involved in the pathogenesis of diabetic nephropathy. [42]. This provides a novel possibility linking podocyte DNA damage to epigenetic changes in metabolic diseases [57]. In humans, it is feasible to evaluate podocyte DNA damage and the expression of DNA methylation modulators using urine-derived cells in patients with diabetes and/or hypertension [58]. Thus, podocyte DNA damage and DNA methylation may be a hopeful target of a diagnostic marker as well as a novel therapy for conquering against DN. When considering epigenetic regulation as a target for DN treatment, such as DNMT1, the problem of side effects always arises. Because DNA damage repair system is specific to cell types [57], factors associated with DNA damage repair may be a hopeful therapeutic target regulating epigenetic status. Future studies are necessary to investigate how to induce epigenetic changes only in the target cells.
3. Effects of Lipotoxicity on Podocytes in DN
Hyperglycemia induced metabolic alterations play critical roles in disease initiation, but a cluster of factors, including dyslipidemia and hypertension, could play a role in inducing onset and progression of DN [59]. Evidence has been accumulated to suggest that dyslipidemia is one of the risk factors for progression and regression of diabetic nephropathy [60,61,62,63]. Reduced plasma triglyceride levels in T2DM patients with treatment of fenofibrate result in reduction of albuminuria [64]. Improvement of elevation of serum low-density lipoprotein (LDL)-cholesterol with treatment was associated with an improvement in annual changes in estimated glomerular filtration rate (eGFR) [65]. These observations support the notion of lipid-lowering therapies could provide beneficial effects on dyslipidemia-mediated kidney injury.
Birnkkoetter et al. reported that in mice, podocytes rely primarily on anaerobic glycolysis to maintain glomerular filtration barrier and are relatively insensitive to defect in mitochondrial biogenesis during ischemia damage [66]. Instead, as lipid accumulation is commonly observed in patients with CKD, podocytes are rather sensitive to cellular lipid-mediated glomerular injury [67]. One evolving area of research has focused on the role of lipotoxicity in podocyte damage in DN. Lipotoxicity, which is a disruptive process caused by lipid accumulation in non-adipose tissue, resulting in cell damage and cell death, is closely associated with the pathogenesis of these diseases. Intracellular lipid accumulation causes insulin resistance, the production of reactive oxygen species (ROS) and endoplasmic reticulum stress, all of which could cause renal damage. Several molecules have been reported to be involved in the effects of lipid accumulation on podocytes as described below Table 2 summarizes the key molecules involved in cholesterol and free fatty acids accumulation separately.
Table 2.
Key molecules associated with lipotoxicity in podocytes.
3.1. Cholesterol Accumulation and Podocytes
The accumulation of cholesterol in podocytes has been shown in diabetic nephropathy (DN) to be involved in the development of glomerular sclerosis [77,78,79]. In addition to DN, clinical studies have shown a similar accumulation of cholesterol in patients with atherosclerosis and focal segmental glomerulosclerosis (FSGS) [68,80,81]. ATP-binding cassette transporters, such as ATP-binding cassette subfamily A member 1 (ABCA1) and ABC subfamily G member 1 (ABCG1), have important roles in cholesterol efflux to high-density lipoprotein (HDL) acceptors and have recently been noted for their association with CKD. The expression of ABCA1 is reduced in type 1 diabetic mice [69]. Similarly, in type 2 diabetic mice, the downregulation of ABCA1 and ABCG1 has been reported, leading to lipid accumulation [68]. Recently, Ducasa et al. have reported that podocyte-specific deletion of ABCA1 rendered mice susceptible to DKD and the accumulation of mitochondrial cardiolipin, and in mice with DN, an increase in cardiolipin oxidation was observed and a cardiolipin peroxidase inhibitor treatment reversed DKD progression, with improvements in podocyte number [82]. The involvement of the renin-angiotensin system (RAS) in cholesterol metabolism in podocytes has been extensively studied. Angiotensin Ⅱ has been reported to induce the accumulation of cholesterol in mouse podocytes, which was related to the expression of genes including ABCA1 and ABCG1 [83]. In addition, we have also reported that in a mouse model of hyperlipidemia, treatment with high-dose angiotensin receptor blockers (ARBs) reduced cholesterol accumulation in the glomerulus and improved proteinuria. The suggested mechanism might involve reduced expression of biglycan, a lipid-retaining proteoglycan, and ACAT1, which converts cholesterol to ester, resulting in a relative increase in free cholesterol for lipid release [70]. Although an important role of RAS in CKD development and progression is widely recognized, it would be of great interest to understand the effect of activated RAS on CKD, focusing on lipid accumulation.
3.2. Free Fatty Acids (FFAs) and Triglycerides
Triglycerides (TGs) are lipids that form lipid droplets, and free fatty acids (FFAs) accumulate intracellularly as TGs. Increased uptake of FFAs in podocytes has been observed in DN [79].
CD36 is a multifunctional transmembrane glycoprotein that acts as a transporter for FFA uptake in the kidney, where it plays a main role in FFA uptake in tubular epithelial cells, podocytes and mesangial cells, and is also important in the development of CKD [84]. CD36 is elevated in both mouse models and in humans in the presence of kidney damage [85]. CD36 has been shown to be associated with lipid accumulation in patients with diabetes [71,72]. In addition to DN and in Alport model mice, lipid accumulation with elevated CD36 has been recognized [73]. In podocytes, CD36 mediated FFA uptake increases ROS, leading to apoptosis [86,87,88]. In addition to CD36, other transporters for FFA uptake include fatty acid transport protein 4 (FATP4) and fatty acid-binding protein (FABP). FATP4 is upregulated in podocytes in a diabetic model and is involved in TG accumulation and cell damage [75]. H-FABP has been reported to be expressed specifically in podocytes and is associated with proteinuria in a mouse model of diabetes and in patients with obesity-related nephropathy [76].
3.3. Ketone Bodies and Mechanistic Target of Rapamycin Signaling Complex 1 (mTORC1)
Ketone bodies are produced from fatty acid degradation in the liver in the state of glucose depletion, such as fasting, and are known to be an important alternative energy source that is quickly utilized in place of glucose in other tissues. In recent years, ketone bodies have also attracted attention for their role as mediators of nutrient signals in cells [89]. One of the nutrient-sensing signals is mTORC1, which is upregulated in podocytes and tubular cells in mouse models of diabetes and in patients with DM [22,90]. Administration of ketones to a mouse model of diabetes suppressed mTORC1, decreased proteinuria, and prevented podocyte damage. Sodium glucose cotransporter 2 (SGLT2) is known to increase the blood levels of ketones, and the renoprotective effects of SGLT2 have been reported to be associated with elevated ketones and inhibition of mTORC1 [91]. In addition to DN, ketone administration produces similar changes in 5/6 nephrectomized mice and polycystic kidney disease (PKD) model mice, which indicates that time-restricted feeding and ketogenic diets that promote ketosis attenuate mTORC1 signaling and inhibit PKD progression [92].
4. Summary
This review summarizes podocyte damage caused by high glucose and lipotoxicity in diabetes as shown in Figure 1. Recent advances in research techniques such as single-cell analysis and epigenetic analysis, are revealing the biology of podocytes in detail. As a characteristic feature of glomerular podocytes, their damage is directly linked to kidney dysfunction and renal prognosis. Epigenetic alteration in podocytes may be important to discuss the reversibility of podocyte phenotype following therapeutic intervention. Understanding metabolic alterations in diabetic podocytes is important to investigate novel strategies for the treatment of ever-increasing DN.
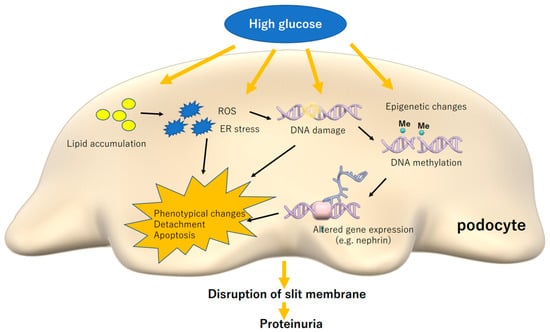
Figure 1.
High glucose and induced lipotoxicity causes stress reactions in podocytes, which can cause phenotypical changes, detachment and apoptosis, leading to disruption of slit membrane and proteinuria. ROS: reactive oxygen species, ER: endoplasmic reticulum.
Author Contributions
R.N. and K.H. wrote the manuscript. H.I. supervised the project and contributed to the manuscript development. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated during this study are included in this published article.
Acknowledgments
K.H. was supported by Grants for Scientific Research (19K08688) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan; Keio University Research Grants for Life Science and Medicine; Naito Memorial Foundation, Takeda Science Foundation, Tokyo, Japan.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yun, H.R.; Kim, H.; Park, J.T.; Chang, T.I.; Yoo, T.-H.; Kang, S.-W.; Choi, K.H.; Sung, S.; Kim, S.W.; Lee, J.; et al. Obesity, Metabolic Abnormality, and Progression of CKD. Am. J. Kidney Dis. 2018, 72, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Tervaert, T.W.C.; Mooyaart, A.L.; Amann, K.; Cohen, A.H.; Cook, H.T.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; De Heer, E.; et al. Pathologic Classification of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Koye, D.N.; Shaw, J.E.; Reid, C.M.; Atkins, R.C.; Reutens, A.T.; Magliano, D.J. Incidence of chronic kidney disease among people with diabetes: A systematic review of observa-tional studies. Diabet. Med. 2017, 34, 887–901. [Google Scholar] [CrossRef]
- Assady, S.; Wanner, N.; Skorecki, K.; Huber, T.B. New Insights into Podocyte Biology in Glomerular Health and Disease. J. Am. Soc. Nephrol. 2017, 28, 1707–1715. [Google Scholar] [CrossRef]
- Asanuma, K.; Mundel, P. The role of podocytes in glomerular pathobiology. Clin. Exp. Nephrol. 2003, 7, 255–259. [Google Scholar] [CrossRef]
- Mundel, P.; Kriz, W. Structure and function of podocytes: An update. Brain Struct. Funct. 1995, 192, 385–397. [Google Scholar] [CrossRef]
- Welsh, G.I.; Hale, L.J.; Eremina, V.; Jeansson, M.; Maezawa, Y.; Lennon, R.; Pons, D.A.; Owen, R.J.; Satchell, S.C.; Miles, M.J.; et al. Insulin Signaling to the Glomerular Podocyte Is Critical for Normal Kidney Function. Cell Metab. 2010, 12, 329–340. [Google Scholar] [CrossRef]
- Skupien, J.; Warram, J.H.; Smiles, A.; Galecki, A.; Stanton, R.C.; Krolewski, A.S. Improved Glycemic Control and Risk of ESRD in Patients with Type 1 Diabetes and Proteinuria. J. Am. Soc. Nephrol. 2014, 25, 2916–2925. [Google Scholar] [CrossRef]
- Fioretto, P.; Barzon, I.; Mauer, M. Is diabetic nephropathy reversible? Diabetes Res. Clin. Pract. 2014, 104, 323–328. [Google Scholar] [CrossRef]
- Nathan, D.M.; Lachin, J.; Cleary, P.; Orchard, T.; Brillon, D.J.; Backlund, J.-Y.; O’Leary, D.H.; Genuth, S.; Trial, D.C.A.C. Epidemiology of Diabetes Interventions and Complications Research Group Intensive Diabetes Therapy and Carotid Intima–Media Thickness in Type 1 Diabetes Mellitus. N. Engl. J. Med. 2003, 348, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.-Y.C.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group Intensive Diabetes Treatment and Cardiovascular Disease in Patients with Type 1 Diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Lachin, J.M.; Genuth, S.; Cleary, P.; Davis, M.D.; Nathan, D.M. Retinopathy and Nephropathy in Patients with Type 1 Diabetes Four Years after a Trial of Intensive Therapy. N. Engl. J. Med. 2000, 342, 381–389. [Google Scholar] [CrossRef] [PubMed]
- de Boer, I.H.; Sun, W.; Cleary, P.A.; Lachin, J.M.; Molitch, M.E.; Steffes, M.W.; Zinman, B. Intensive diabetes therapy and glomerular filtration rate in type 1 diabetes. N. Engl. J. Med. 2011, 365, 2366–2376. [Google Scholar] [PubMed]
- Chen, Z.; Miao, F.; Braffett, B.H.; Lachin, J.M.; Zhang, L.; Wu, X.; Roshandel, D.; Carless, M.; Li, X.A.; Tompkins, J.D.; et al. DNA methylation mediates development of HbA1c-associated complications in type 1 diabetes. Nat. Metab. 2020, 2, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gluck, C.; Qiu, C.; Han, S.Y.; Palmer, M.; Park, J.; Ko, Y.-A.; Guan, Y.; Sheng, X.; Hanson, R.L.; Huang, J.; et al. Kidney cytosine methylation changes improve renal function decline estimation in patients with diabetic kidney disease. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Park, J.; Guan, Y.; Sheng, X.; Gluck, C.; Seasock, M.J.; Hakimi, A.A.; Qiu, C.; Pullman, J.; Verma, A.; Li, H.; et al. Functional methylome analysis of human diabetic kidney disease. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Lin, J.S.; Susztak, K. Podocytes: The Weakest Link in Diabetic Kidney Disease? Curr. Diab. Rep. 2016, 16, 45. [Google Scholar] [CrossRef]
- Herbach, N.; Schairer, I.; Blutke, A.; Kautz, S.; Siebert, A.; Göke, B.; Wolf, E.; Wanke, R. Diabetic kidney lesions of GIPRdn transgenic mice: Podocyte hypertrophy and thickening of the GBM precede glomerular hypertrophy and glomerulosclerosis. Am. J. Physiol. Physiol. 2009, 296, F819–F829. [Google Scholar] [CrossRef]
- Romero, M.; Ortega, A.; Izquierdo, A.; López-Luna, P.; Bosch, R.J. Parathyroid hormone-related protein induces hypertrophy in podocytes via TGF-beta(1) and p27(Kip1): Implications for diabetic nephropathy. Nephrol. Dial. Transplant. 2010, 25, 2447–2457. [Google Scholar] [CrossRef]
- Lu, M.K.; Gong, X.G.; Guan, K.L. mTOR in podocyte function: Is rapamycin good for diabetic nephropathy? Cell Cycle 2011, 10, 3415–3416. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Rüegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, J.; Yoshida, S.; Nada, S.; Okada, M.; Inoki, K. Role of Ragulator in the Regulation of Mechanistic Target of Rapamycin Signaling in Podocytes and Glomerular Function. J. Am. Soc. Nephrol. 2016, 27, 3653–3665. [Google Scholar] [CrossRef] [PubMed]
- Mundel, P.; Shankland, S.J. Podocyte biology and response to injury. J. Am. Soc. Nephrol. 2002, 13, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Maezawa, Y.; Yokote, K.; Joh, K.; Kobayashi, K.; Kawamura, H.; Nishimura, M.; Roberts, A.B.; Saito, Y.; Mori, S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem. Biophys. Res. Commun. 2003, 305, 1002–1007. [Google Scholar] [CrossRef]
- Li, X.; Chuang, P.Y.; D’Agati, V.D.; Dai, Y.; Yacoub, R.; Fu, J.; Xuezhu, L.; Taku, O.; Premsrirut, P.K.; Holzman, L.B.; et al. Nephrin Preserves Podocyte Viability and Glomerular Structure and Function in Adult Kidneys. J. Am. Soc. Nephrol. 2015, 26, 2361–2377. [Google Scholar] [CrossRef]
- Zhu, J.; Sun, N.; Aoudjit, L.; Li, H.; Kawachi, H.; Lemay, S.; Takano, T. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 2008, 73, 556–566. [Google Scholar] [CrossRef]
- Doublier, S.; Salvidio, G.; Lupia, E.; Ruotsalainen, V.; Verzola, D.; Deferrari, G.; Camussi, G. Nephrin expression is reduced in human diabetic nephropathy: Evidence for a distinct role for glycated albumin and angiotensin II. Diabetes 2003, 52, 1023–1030. [Google Scholar] [CrossRef]
- Dumont, V.; Tolvanen, T.A.; Kuusela, S.; Wang, H.; Nyman, T.A.; Lindfors, S.; Tienari, J.; Nisen, H.; Suetsugu, S.; Plomann, M.; et al. PACSIN2 accelerates nephrin trafficking and is up-regulated in diabetic kidney disease. FASEB J. 2017, 31, 3978–3990. [Google Scholar] [CrossRef]
- Tossidou, I.; Teng, B.; Menne, J.; Shushakova, N.; Park, J.-K.; Becker, J.U.; Modde, F.; Leitges, M.; Haller, H.; Schiffer, M. Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocy-tosis. PLoS ONE 2010, 5, e10185. [Google Scholar] [CrossRef]
- Quack, I.; Woznowski, M.; Potthoff, S.A.; Palmer, R.; Königshausen, E.; Sivritas, S.; Schiffer, M.; Stegbauer, J.; Vonend, O.; Rump, L.C.; et al. PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia. J. Biol. Chem. 2011, 286, 12959–12970. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gui, D.; Chen, Y.; Mou, L.; Liu, Y.; Huang, J. Astragaloside IV improves high glucose-induced podocyte adhesion dysfunction via alpha3beta1 integrin upregulation and integrin-linked kinase inhibition. Biochem. Pharmacol. 2008, 76, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Chen, C.-A.; Guh, J.-Y.; Chang, J.-M.; Shin, S.-J.; Lai, Y.-H. Altering expression of α3β1 integrin on podocytes of human and rats with diabetes. Life Sci. 2000, 67, 2345–2353. [Google Scholar] [CrossRef]
- Jha, J.C.; Gray, S.P.; Barit, D.; Okabe, J.; El-Osta, A.; Namikoshi, T.; Thallas-Bonke, V.; Wingler, K.; Szyndralewiez, C.; Heitz, F.; et al. Genetic Targeting or Pharmacologic Inhibition of NADPH Oxidase Nox4 Provides Renoprotection in Long-Term Diabetic Nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1237–1254. [Google Scholar] [CrossRef] [PubMed]
- Holterman, C.E.; Thibodeau, J.-F.; Towaij, C.; Gutsol, A.; Montezano, A.C.; Parks, R.J.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.J. Nephropathy and Elevated BP in Mice with Podocyte-Specific NADPH Oxidase 5 Expression. J. Am. Soc. Nephrol. 2013, 25, 784–797. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Artomov, M.; Brähler, S.; Stander, M.C.; Shamsan, G.; Sampson, M.G.; White, J.M.; Kretzler, M.; Miner, J.H.; Jain, S.; et al. A role for genetic susceptibility in sporadic focal segmental glomerulosclerosis. J. Clin. Investig. 2016, 126, 1603. [Google Scholar] [CrossRef]
- Zhou, Y.; Castonguay, P.; Sidhom, E.-H.; Clark, A.R.; Dvela-Levitt, M.; Kim, S.; Sieber, J.; Wieder, N.; Jung, J.Y.; Andreeva, S.; et al. A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science 2017, 358, 1332–1336. [Google Scholar] [CrossRef]
- Ilatovskaya, D.V.; Blass, G.; Palygin, O.; Levchenko, V.; Pavlov, T.S.; Grzybowski, M.N.; Winsor, K.; Shuyskiy, L.S.; Geurts, A.M.; Cowley, A.W.; et al. A NOX4/TRPC6 Pathway in Podocyte Calcium Regulation and Renal Damage in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 1917–1927. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Q.; Liu, S.; Chen, Y.; Li, R.; Lin, T.; Yu, C.; Zhang, H.; Huang, Z.; Zhao, X.; et al. DNA methyltransferase 1 may be a therapy target for attenuating diabetic nephropathy and po-docyte injury. Kidney Int. 2017, 92, 140–153. [Google Scholar] [CrossRef]
- Hayashi, K.; Sasamura, H.; Nakamura, M.; Sakamaki, Y.; Azegami, T.; Oguchi, H.; Tokuyama, H.; Wakino, S.; Hayashi, K.; Itoh, H. Renin-angiotensin blockade resets podocyte epigenome through Kruppel-like Factor 4 and at-tenuates proteinuria. Kidney Int. 2015, 88, 745–753. [Google Scholar] [CrossRef]
- Hayashi, K.; Sasamura, H.; Nakamura, M.; Azegami, T.; Oguchi, H.; Sakamaki, Y.; Itoh, H. KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates pro-teinuria. J. Clin. Investig. 2014, 124, 2523–2537. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, A.; Hayashi, K.; Abe, T.; Kaneko, M.; Yokoi, H.; Azegami, T.; Nakamura, M.; Yoshimoto, N.; Kanda, T.; Sakamaki, Y.; et al. Decreased KAT5 Expression Impairs DNA Repair and Induces Altered DNA Methylation in Kidney Podocytes. Cell Rep. 2019, 26, 1318–1332.e4. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, M.; Bitzer, M.; Roberts, I.S.; Kopp, J.B.; ten Dijke, P.; Mundel, P.; Böttinger, E.P. Apoptosis in podocytes induced by TGF-beta and Smad7. J. Clin. Investig. 2001, 108, 807–816. [Google Scholar] [CrossRef]
- Li, J.H.; Huang, X.R.; Zhu, H.-J.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2004, 18, 176–178. [Google Scholar] [CrossRef]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Böttinger, E.P. Affiliations Glucose-Induced Reactive Oxygen Species Cause Apoptosis of Podocytes and Podocyte Depletion at the Onset of Diabetic Nephropathy. Diabetes 2005, 55, 225–233. [Google Scholar] [CrossRef]
- Eid, A.A.; Gorin, Y.; Fagg, B.M.; Maalouf, R.; Barnes, J.L.; Block, K.; Abboud, H.E. Mechanisms of Podocyte Injury in Diabetes: Role of Cytochrome P450 and NADPH Oxidases. Diabetes 2009, 58, 1201–1211. [Google Scholar] [CrossRef]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone Methyl and Kidney Function in CKD with Type 2 Diabetes. New Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef]
- Asaba, K.; Tojo, A.; Onozato, M.L.; Goto, A.; Quinn, M.T.; Fujita, T.; Wilcox, C.S. Effects of NADPH oxidase inhibitor in diabetic nephropathy. Kidney Int. 2005, 67, 1890–1898. [Google Scholar] [CrossRef]
- Thallas-Bonke, V.; Thorpe, S.R.; Coughlan, M.T.; Fukami, K.; Yap, F.Y.; Sourris, K.C.; Penfold, S.A.; Bach, L.A.; Cooper, M.E.; Forbes, J.M. Inhibition of NADPH Oxidase Prevents Advanced Glycation End Product-Mediated Damage in Diabetic Nephropathy Through a Protein Kinase C- -Dependent Pathway. Diabetes 2007, 57, 460–469. [Google Scholar] [CrossRef]
- Sedeek, M.; Gutsol, A.; Montezano, A.C.; Burger, D.; Cat, A.N.D.; Kennedy, C.R.J.; Burns, K.D.; Cooper, M.E.; Jandeleit-Dahm, K.; Page, P.; et al. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of Type 2 diabetes. Clin. Sci. 2013, 124, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.-C.; Song, X.; Lu, X.-Y.; Li, D.T.; Eaton, D.C.; Shen, B.-Z.; Li, X.-Q.; Ma, H.-P. High glucose induces podocyte apoptosis by stimulating TRPC6 via elevation of reactive oxygen species. Biochim. Biophys. Acta BBA Bioenerg. 2013, 1833, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, R.; van der Vlag, J.; Baltissen, M.P.A.; Verkaart, S.A.J.; Wetzels, J.F.M.; Berden, J.H.M.; Hoenderop, J.G.J.; Nijenhuis, T. Glucose specifically regulates TRPC6 expression in the podocyte in an AngII-dependent manner. Am. J. Pathol. 2014, 184, 1715–1726. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Palygin, O.; Chubinskiy-Nadezhdin, V.; Negulyaev, Y.A.; Ma, R.; Birnbaumer, L.; Staruschenko, A. Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glo-meruli. Kidney Int. 2014, 86, 506–514. [Google Scholar] [CrossRef]
- De Marinis, Y.; Cai, M.; Bompada, P.; Atac, D.G.; Kotova, O.; Johansson, M.E.; Garcia-Vaz, E.; Gomez, M.F.; Laakso, M.; Groop, L. Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int. 2016, 89, 342–353. [Google Scholar] [CrossRef]
- Lizotte, F.; Denhez, B.; Guay, A.; Gévry, N.; Côté, A.M.; Geraldes, P. Persistent Insulin Resistance in Podocytes Caused by Epigenetic Changes of SHP-1 in Diabetes. Diabetes 2016, 65, 3705–3717. [Google Scholar] [CrossRef]
- Hayashi, K.; Hishikawa, A.; Itoh, H. DNA Damage Repair and DNA Methylation in the Kidney. Am. J. Nephrol. 2019, 50, 81–91. [Google Scholar] [CrossRef]
- Hishikawa, A.; Hayashi, K.; Yoshimoto, N.; Nakamichi, R.; Homma, K.; Itoh, H. DNA damage and expression of DNA methylation modulators in urine-derived cells of patients with hypertension and diabetes. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Russo, G.; Piscitelli, P.; Giandalia, A.; Viazzi, F.; Pontremoli, R.; Fioretto, P.; De Cosmo, S. Atherogenic dyslipidemia and diabetic nephropathy. J. Nephrol. 2020, 33, 1–8. [Google Scholar] [CrossRef]
- O’Seaghdha, C.M.; Hwang, S.-J.; Upadhyay, A.; Meigs, J.B.; Fox, C.S. Predictors of Incident Albuminuria in the Framingham Offspring Cohort. Am. J. Kidney Dis. 2010, 56, 852–860. [Google Scholar] [CrossRef]
- Ninomiya, T.; Kiyohara, Y.; Kubo, M.; Yonemoto, K.; Tanizaki, Y.; Doi, Y.; Hirakata, H.; Iida, M. Metabolic Syndrome and CKD in a General Japanese Population: The Hisayama Study. Am. J. Kidney Dis. 2006, 48, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Schaeffner, E.S.; Kurth, T.; Curhan, G.C.; Glynn, R.J.; Rexrode, K.; Baigent, C.; Buring, J.E.; Gaziano, J.M. Cholesterol and the risk of renal dysfunction in apparently healthy men. J. Am. Soc. Nephrol. 2003, 14, 2084–2091. [Google Scholar] [PubMed]
- De Koning, L.; Anand, S.S. Adherence to a Mediterranean diet and survival in a Greek population. Engl. J. Med. 2003, 348, 2599–2608. [Google Scholar]
- Keech, A.C.; Simes, R.J.; Barter, P.J.; Best, J.D.; Scott, R.A.P.; Taskinen, M.R.; Forder, P.M.; Pillai, A.; Davis, T.M.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.N.; Hitman, G.A.; Neil, H.A.W.; Livingstone, S.J.; Charlton-Menys, V.; DeMicco, D.A.; Fuller, J.H. Effects of atorvastatin on kidney outcomes and cardiovascular disease in patients with dia-betes: An analysis from the Collaborative Atorvastatin Diabetes Study (CARDS). Am. J. Kidney Dis. 2009, 54, 810–819. [Google Scholar] [CrossRef]
- Brinkkoetter, P.T.; Bork, T.; Salou, S.; Liang, W.; Mizi, A.; Özel, C.; Koehler, S.; Hagmann, H.H.; Ising, C.; Kuczkowski, A.; et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mito-chondrial Metabolism and Dynamics. Cell Rep. 2019, 27, 1551–1566. [Google Scholar] [CrossRef]
- Merscher, S.; Pedigo, C.E.; Mendez, A.J. Metabolism, Energetics, and Lipid Biology in the Podocyte-Cellular Cholesterol-Mediated Glomerular Injury. Front. Endocrinol. 2014, 5, 169. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef]
- Tang, C.; Kanter, J.E.; Bornfeldt, K.E.; Leboeuf, R.C.; Oram, J.F. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. J. Lipid Res. 2009, 51, 1719–1728. [Google Scholar] [CrossRef]
- Hayashi, K.; Sasamura, H.; Azegami, T.; Itoh, H. Regression of atherosclerosis in apolipoprotein E-deficient mice is feasible using high-dose an-giotensin receptor blocker, candesartan. J. Atheroscler. Thromb. 2012, 19, 736–746. [Google Scholar]
- Afshinnia, F.; Rajendiran, T.M.; Soni, T.; Byun, J.; Wernisch, S.; Sas, K.M.; Hawkins, J.; Bellovich, K.; Gipson, D.; Michailidis, G.; et al. Impaired beta-Oxidation and Altered Complex Lipid Fatty Acid Partitioning with Advancing CKD. J. Am. Soc. Nephrol. 2018, 29, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Chen, Y.; Huang, W.; Viterna, J.; Liu, J.; Westfall, K.; Tian, J.; Bartlett, D.J.; Tang, W.H.W.; Xie, Z.-J.; et al. CD36 and Na/K-ATPase-α1 form a proinflammatory signaling loop in kidney. Hypertension 2012, 61, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Romani, P.; Brian, I.; Santinon, G.; Pocaterra, A.; Audano, M.; Pedretti, S.; Mathieu, S.; Forcato, M.; Bicciato, S.; Manneville, J.-B.; et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat. Cell Biol. 2019, 21, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Maimaitiyiming, H.; Zhou, Q.; Norman, H.; Zhou, C.; Wang, S. Interaction of thrombospondin1 and CD36 contributes to obesity-associated podocytopathy. Biochim. Biophys. Acta BBA Bioenerg. 2015, 1852, 1323–1333. [Google Scholar] [CrossRef]
- Falkevall, A.; Mehlem, A.; Palombo, I.; Sahlgren, B.H.; Ebarasi, L.; He, L.; Ytterberg, A.J.; Olauson, H.; Axelsson, J.; Sundelin, B.; et al. Reducing VEGF-B Signaling Ameliorates Renal Lipotoxicity and Protects against Diabetic Kidney Disease. Cell Metab. 2017, 25, 713–726. [Google Scholar] [CrossRef]
- Chen, H.-M.; Zheng, C.-X.; Gao, Q.; Ge, Y.-C.; Liu, P. Heart-Type Fatty Acid Binding Protein Is Associated with Proteinuria in Obesity. PLoS ONE 2012, 7, e45691. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, Z.; Proctor, G.; Moskowitz, S.; Liebman, S.E.; Rogers, T.; Lucia, M.S.; Li, J.; Levi, M. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulo-sclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J. Biol. Chem. 2005, 280, 32317–32325. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, T.; Li, J.; Proctor, G.; McManaman, J.L.; Lucia, S.; Chua, S.; Levi, M. Regulation of Renal Lipid Metabolism, Lipid Accumulation, and Glomerulosclerosis in FVBdb/db Mice with Type 2 Diabetes. Diabetes 2005, 54, 2328–2335. [Google Scholar] [CrossRef]
- Nosadini, R.; Tonolo, G. Role of oxidized low density lipoproteins and free fatty acids in the pathogenesis of glomerulopathy and tubulointerstitial lesions in type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 79–85. [Google Scholar] [CrossRef]
- Moorhead, J.; El-Nahas, M.; Chan, M.; Varghese, Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet 1982, 320, 1309–1311. [Google Scholar] [CrossRef]
- Lee, H.S.; Kruth, H.S. Accumulation of cholesterol in the lesions of focal segmental glomerulosclerosis. Nephrology 2003, 8, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Ducasa, G.M.; Mitrofanova, A.; Mallela, S.K.; Liu, X.; Molina, J.; Sloan, A.; Pedigo, C.E.; Ge, M.; Santos, J.V.; Hernandez, Y.; et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Investig. 2019, 129, 3387–3400. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Q.; Yang, J.; Ma, Y.; Ding, G. Angiotensin II induces cholesterol accumulation and injury in podocytes. Sci. Rep. 2017, 7, 10672. [Google Scholar] [CrossRef] [PubMed]
- Armesilla, A.L.; Vega, M.A. Structural organization of the gene for human CD36 glycoprotein. J. Biol. Chem. 1994, 269, 18985–18991. [Google Scholar]
- Susztak, K.; Ciccone, E.; McCue, P.; Sharma, K.; Bottinger, E.P. Multiple Metabolic Hits Converge on CD36 as Novel Mediator of Tubular Epithelial Apoptosis in Diabetic Nephropathy. PLoS Med. 2005, 2, e45. [Google Scholar] [CrossRef]
- Hua, W.; Huang, H.-Z.; Tan, L.-T.; Wan, J.-M.; Gui, H.-B.; Zhao, L.; Ruan, X.-Z.; Chen, X.-M.; Du, X.-G. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef]
- Baranova, I.N.; Vishnyakova, T.G.; Bocharov, A.V.; Kurlander, R.; Chen, Z.; Kimelman, M.L.; Remaley, A.T.; Csako, G.; Thomas, F.; Eggerman, T.L.; et al. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J. Biol. Chem. 2005, 280, 8031–8040. [Google Scholar] [CrossRef]
- Bige, N.; Shweke, N.; Benhassine, S.; Jouanneau, C.; Vandermeersch, S.; Dussaule, J.-C.; Chatziantoniou, C.; Ronco, P.; Boffa, J.-J. Thrombospondin-1 plays a profibrotic and pro-inflammatory role during ureteric obstruction. Kidney Int. 2012, 81, 1226–1238. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Gödel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mór, A.; Lindenmeyer, M.T.; Rastaldi, M.-P.; Hartleben, G.; et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Investig. 2011, 121, 2197–2209. [Google Scholar] [CrossRef]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 2020, 32, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.A.; Kruger, S.L.; Broderick, C.; Amarlkhagva, T.; Agrawal, S.; Dodam, J.R.; Mrug, M.; Lyons, L.A.; Weimbs, T. Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease. Cell Metab. 2019, 30, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).