Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe COVID-19



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Single-Cell Data Retrieval and Bioinformatics
2.2. Gene Set Enrichment and Modeling of Gene Interactions Networks
2.3. RNA-Seq Analysis of PBMCs from COVID-19 Patients
2.4. Statistical Analyses
3. Results
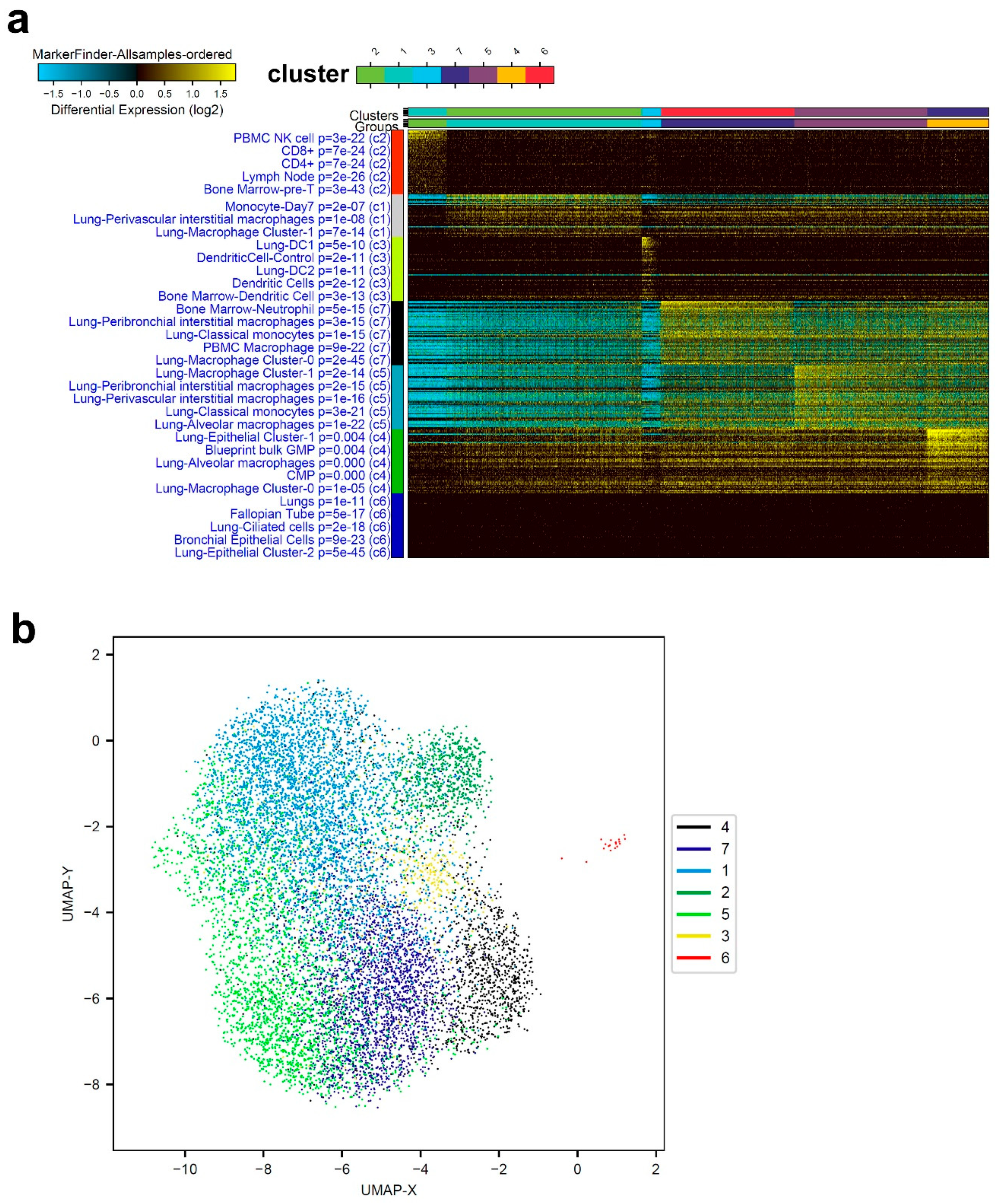
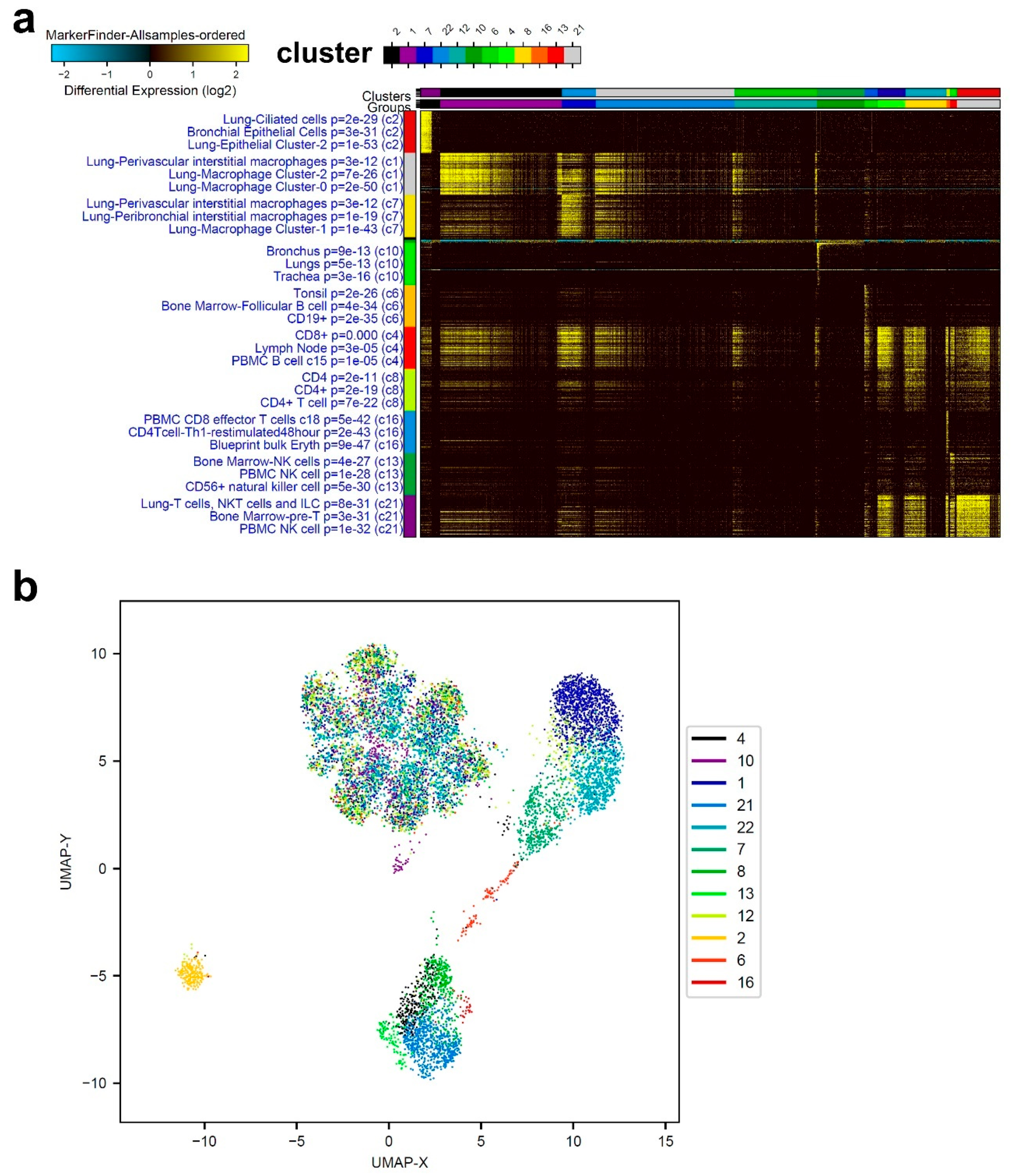
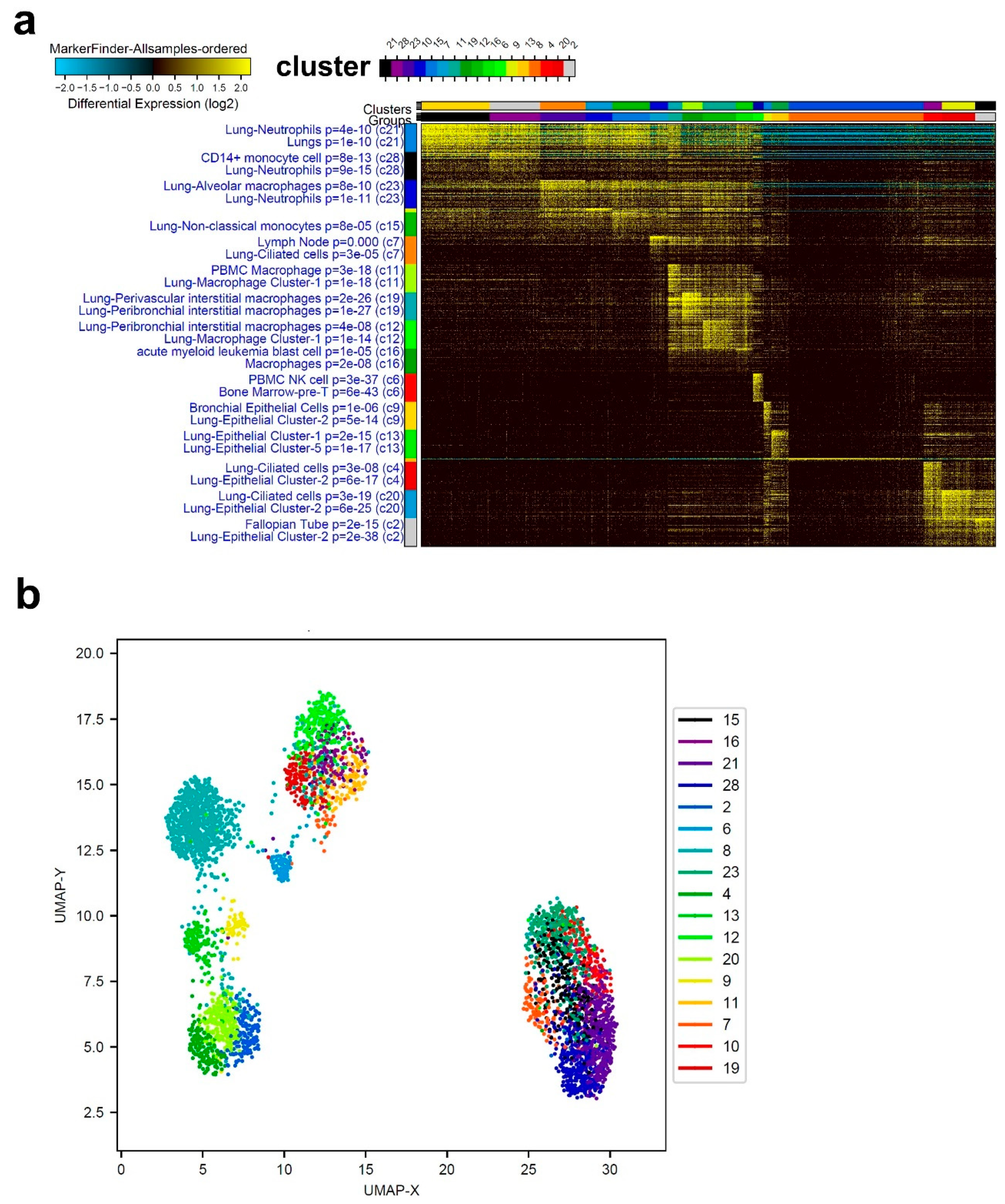
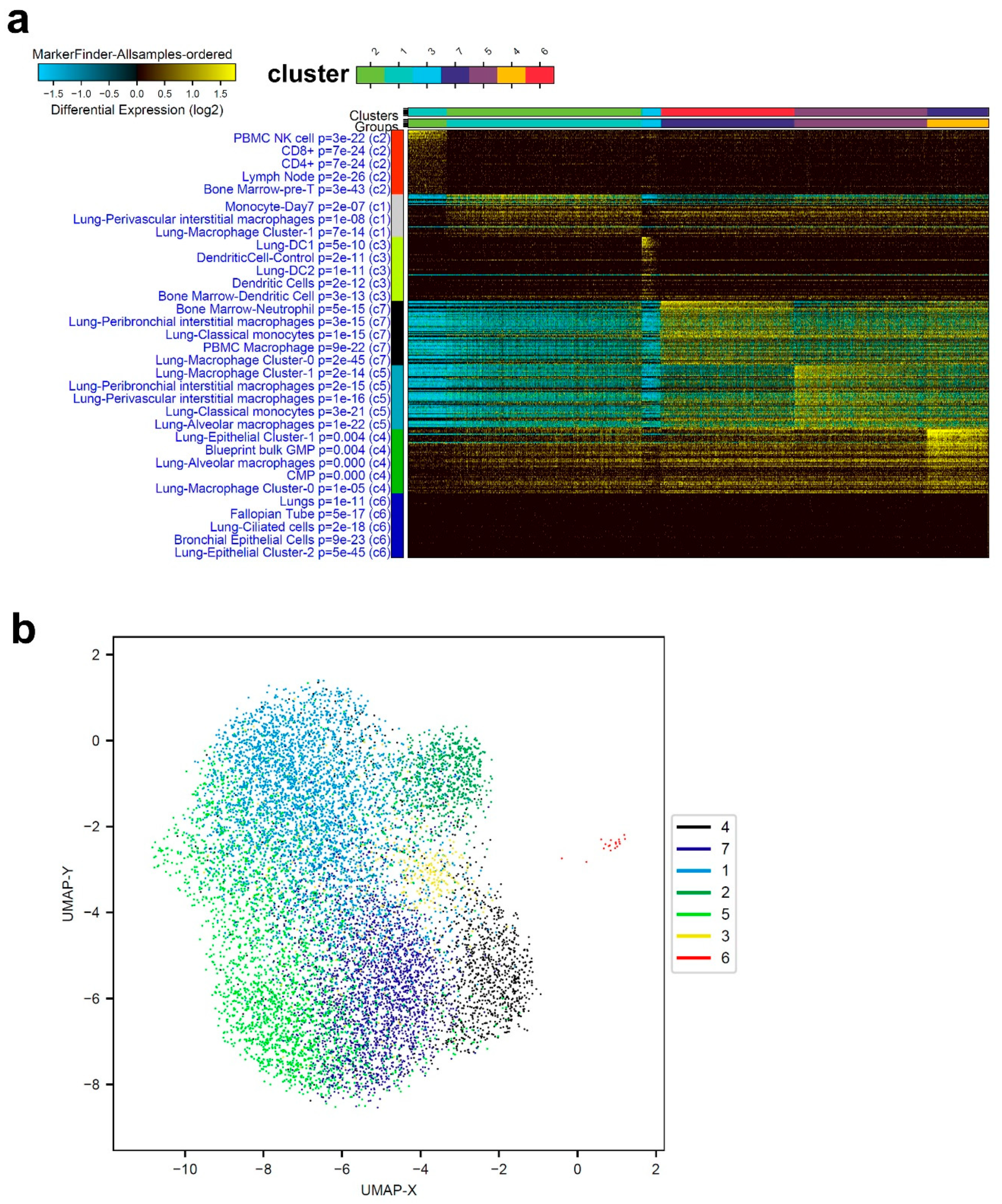
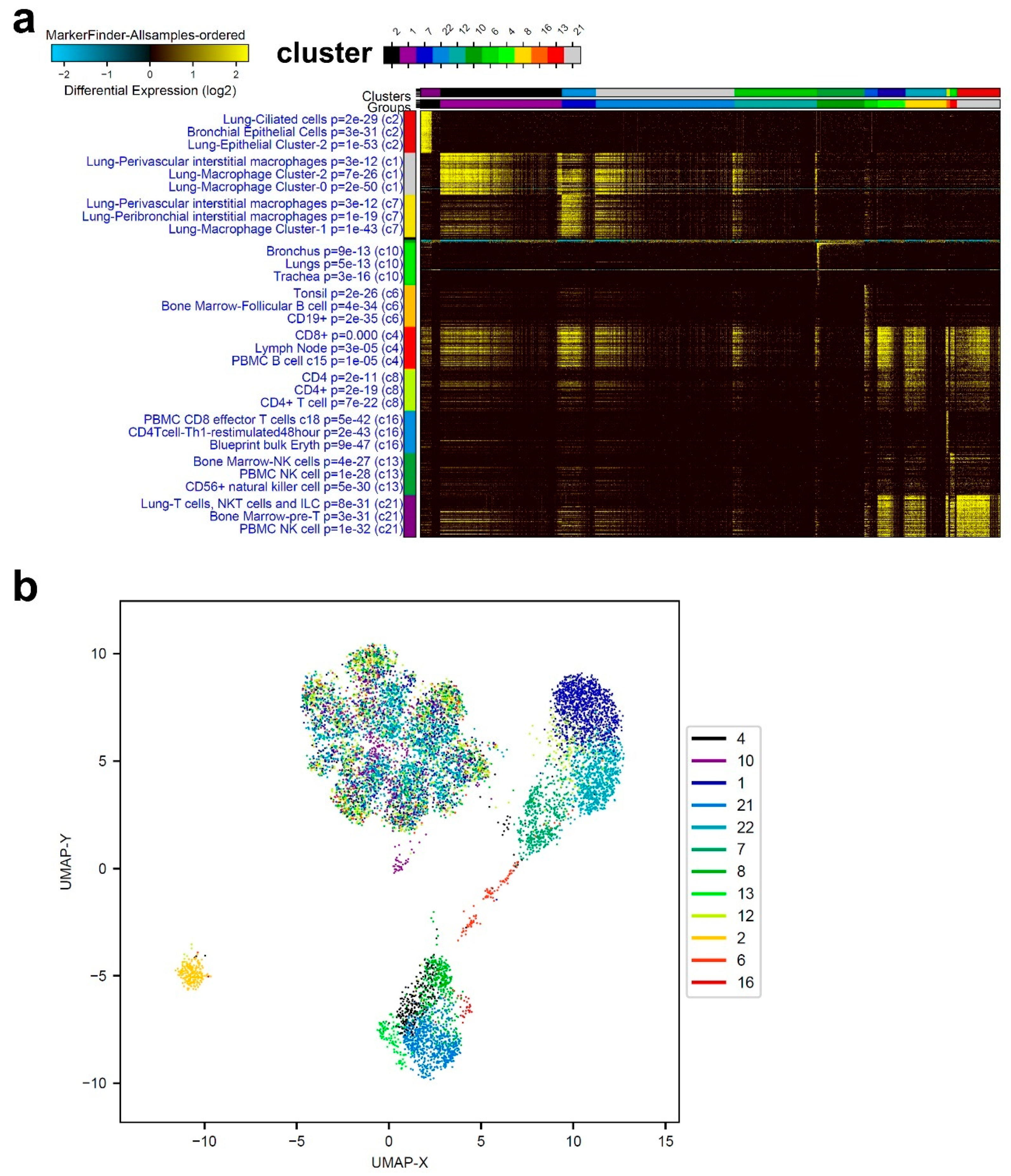
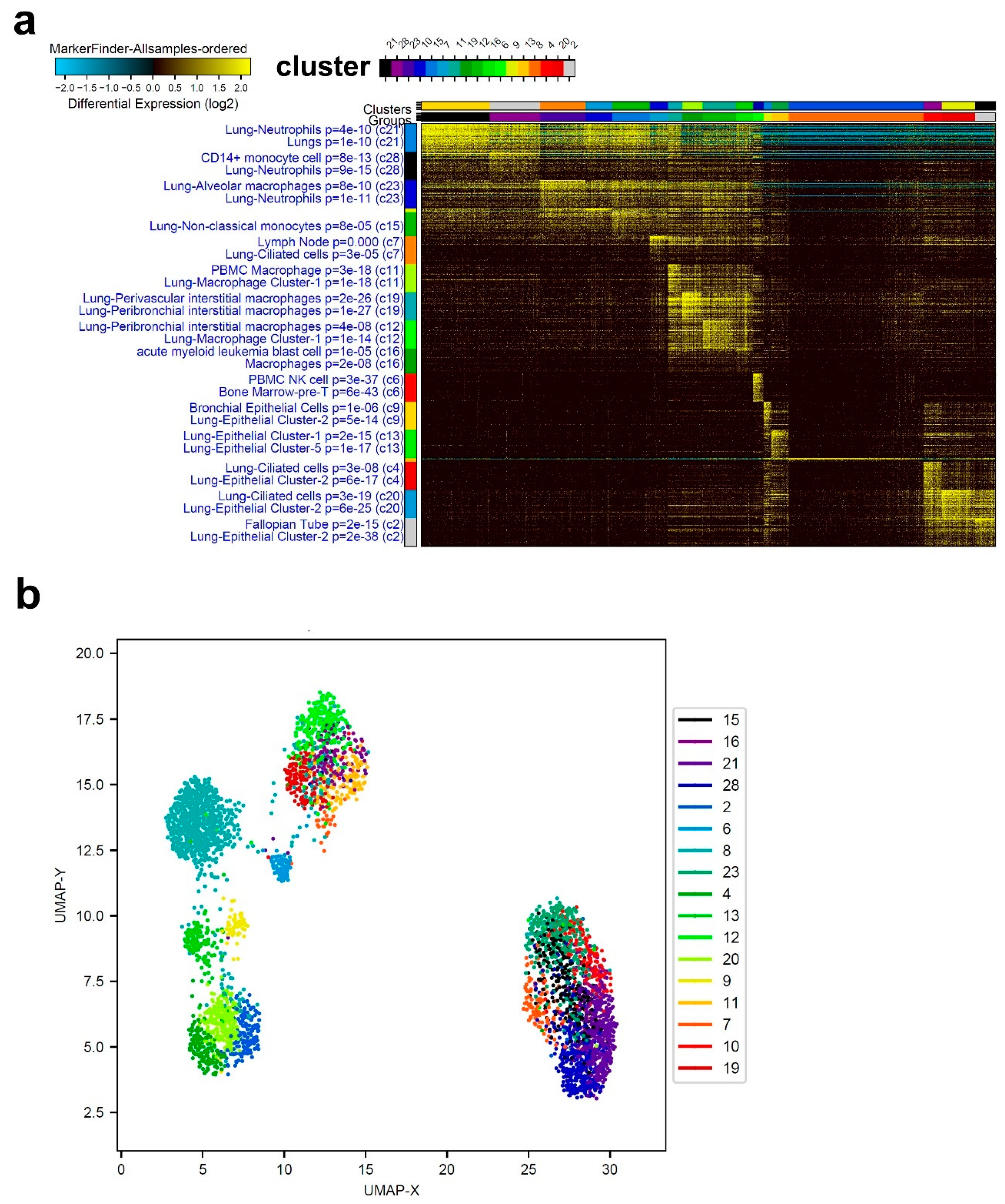
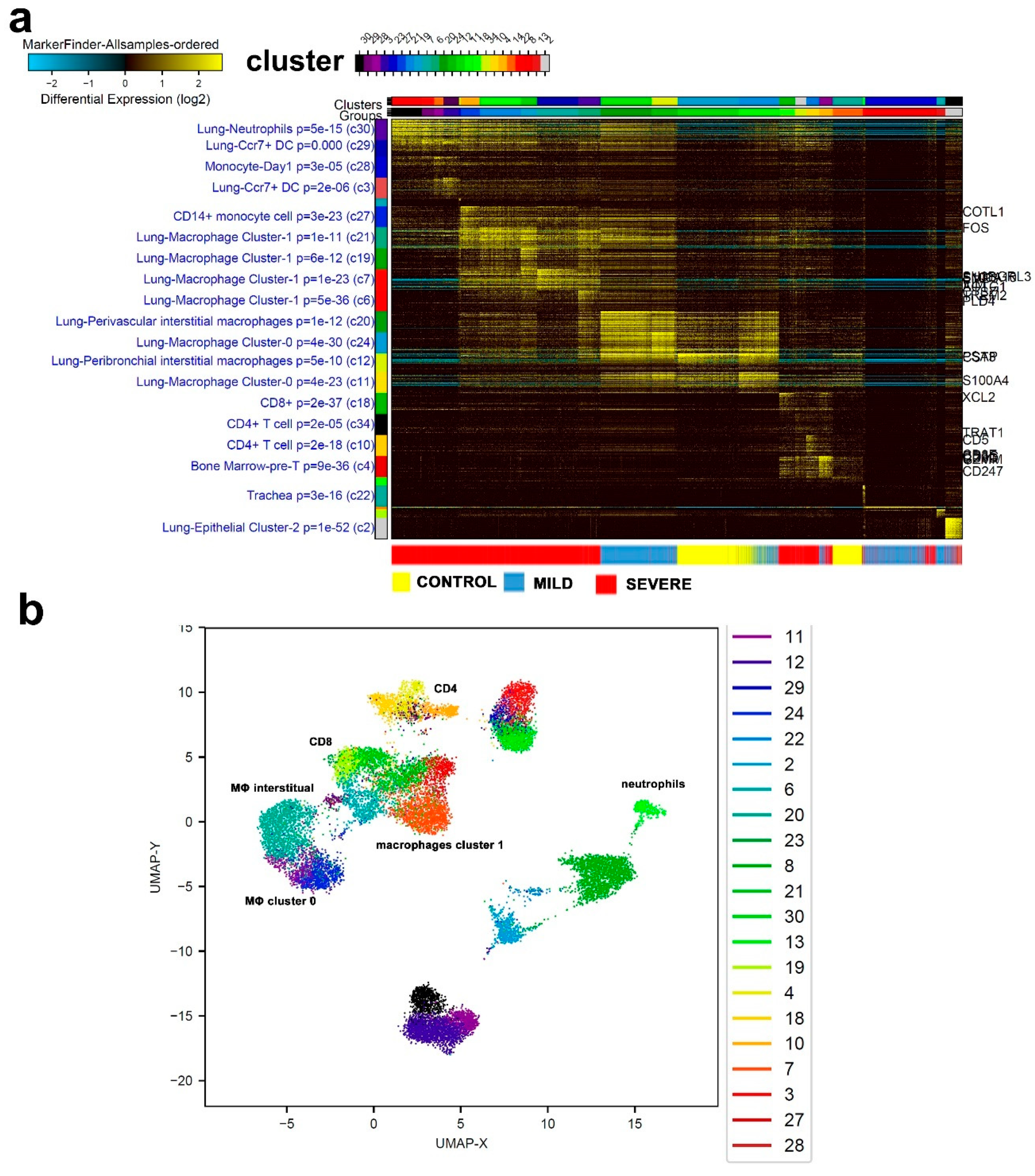
3.1. Single-Cell Transcriptome Analysis of BAL Revealed Variable Cellular Composition in Severe and Mild COVID-19 Patients Compared to Healthy Subjects
3.2. Combination Analysis of Single-Cell Transcriptomes of BAL from Severe- and Mild-COVID-19 Patients Compared to Healthy Subjects
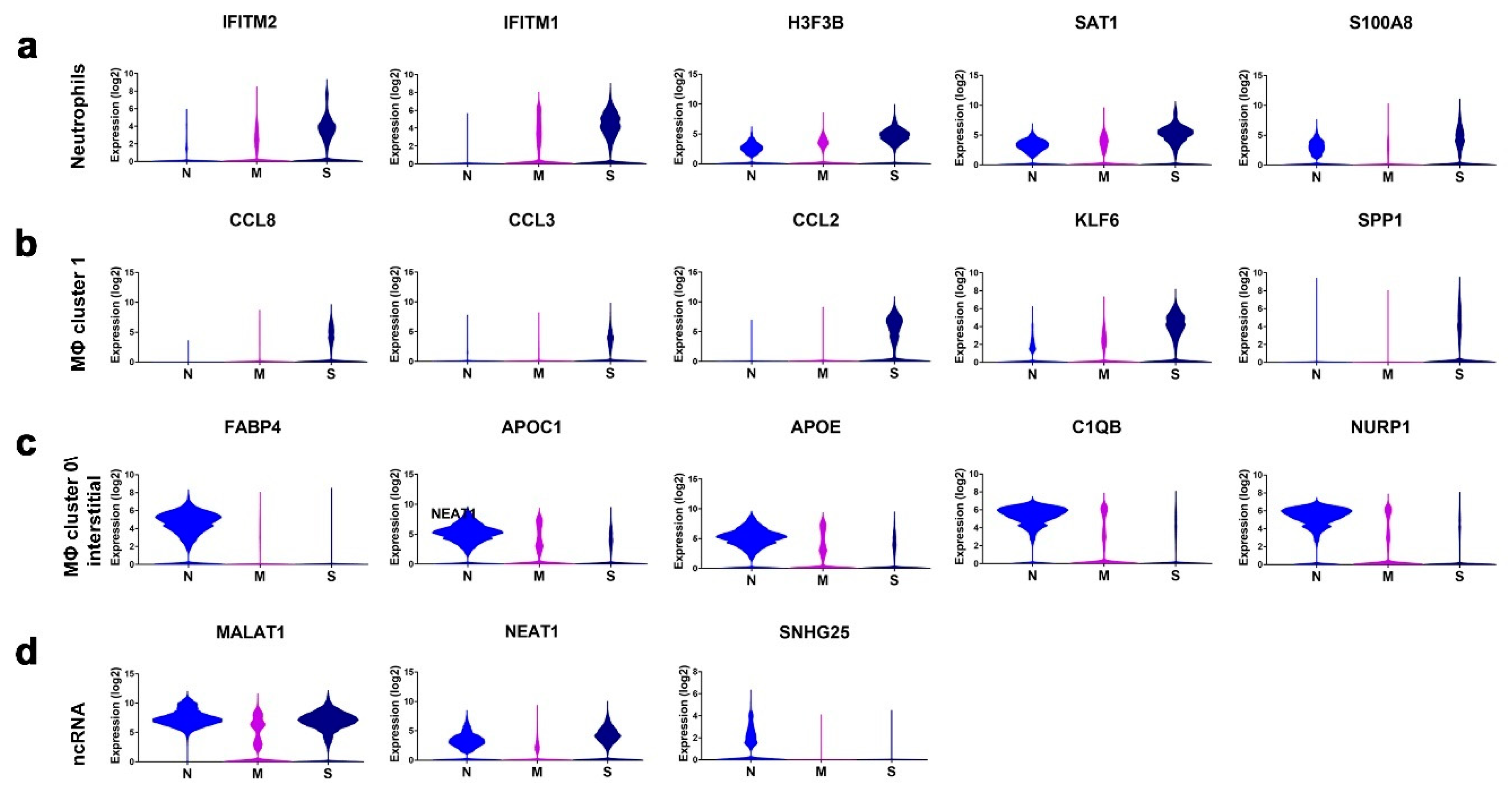
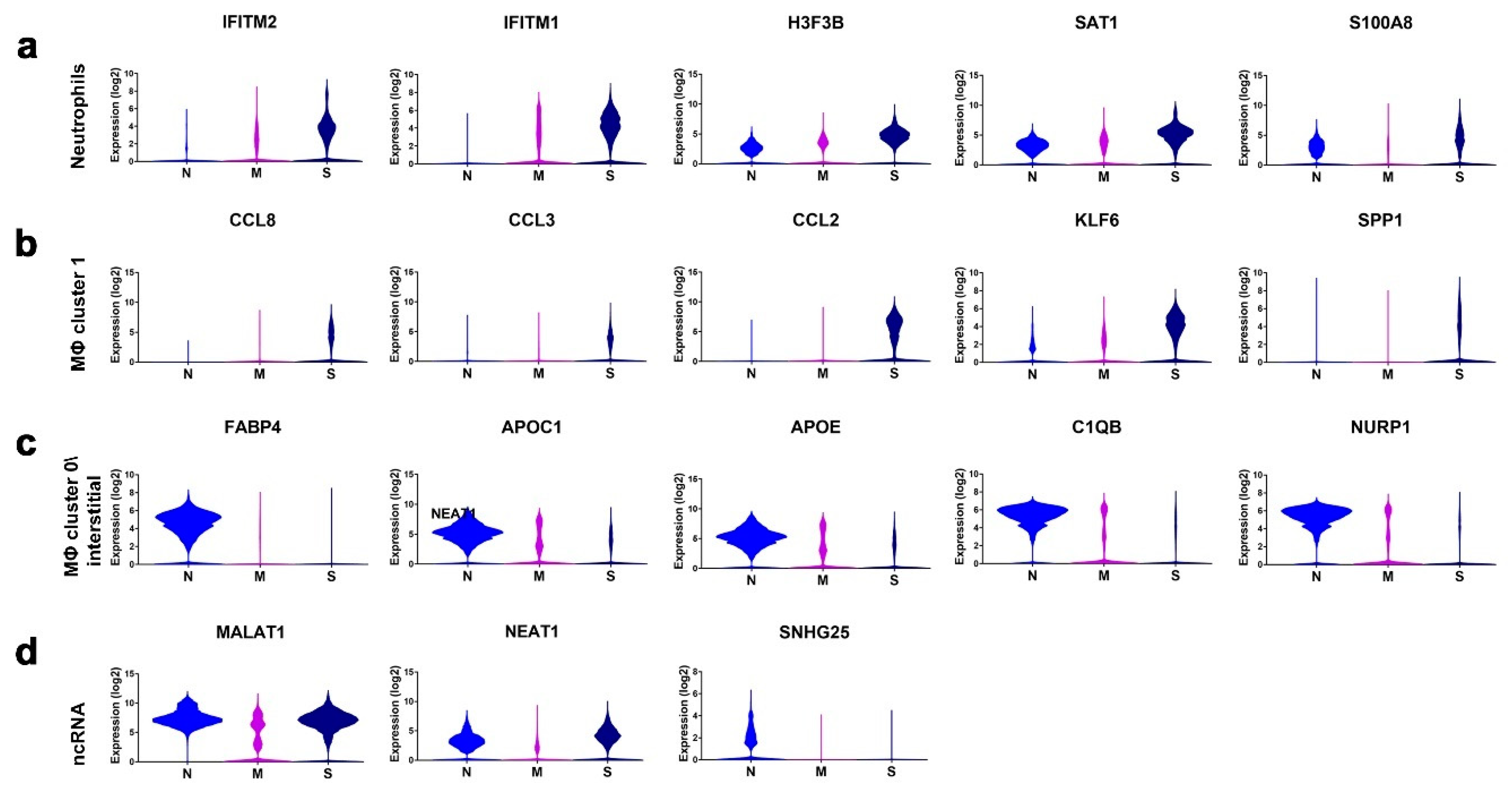
3.3. Enriched Gene Markers in Patients with Mild or Severe COVID-19 Compared to Healthy Subjects
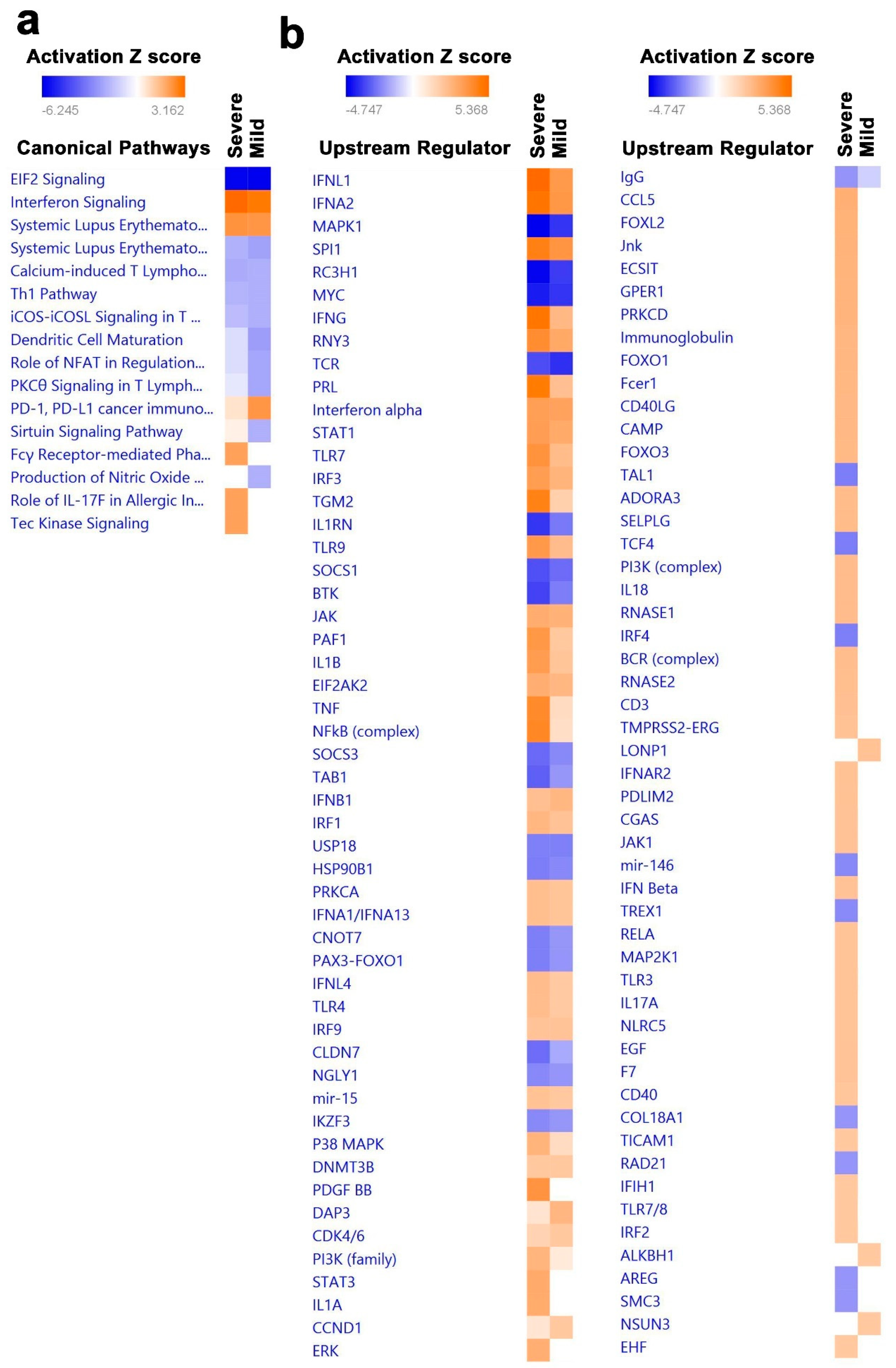
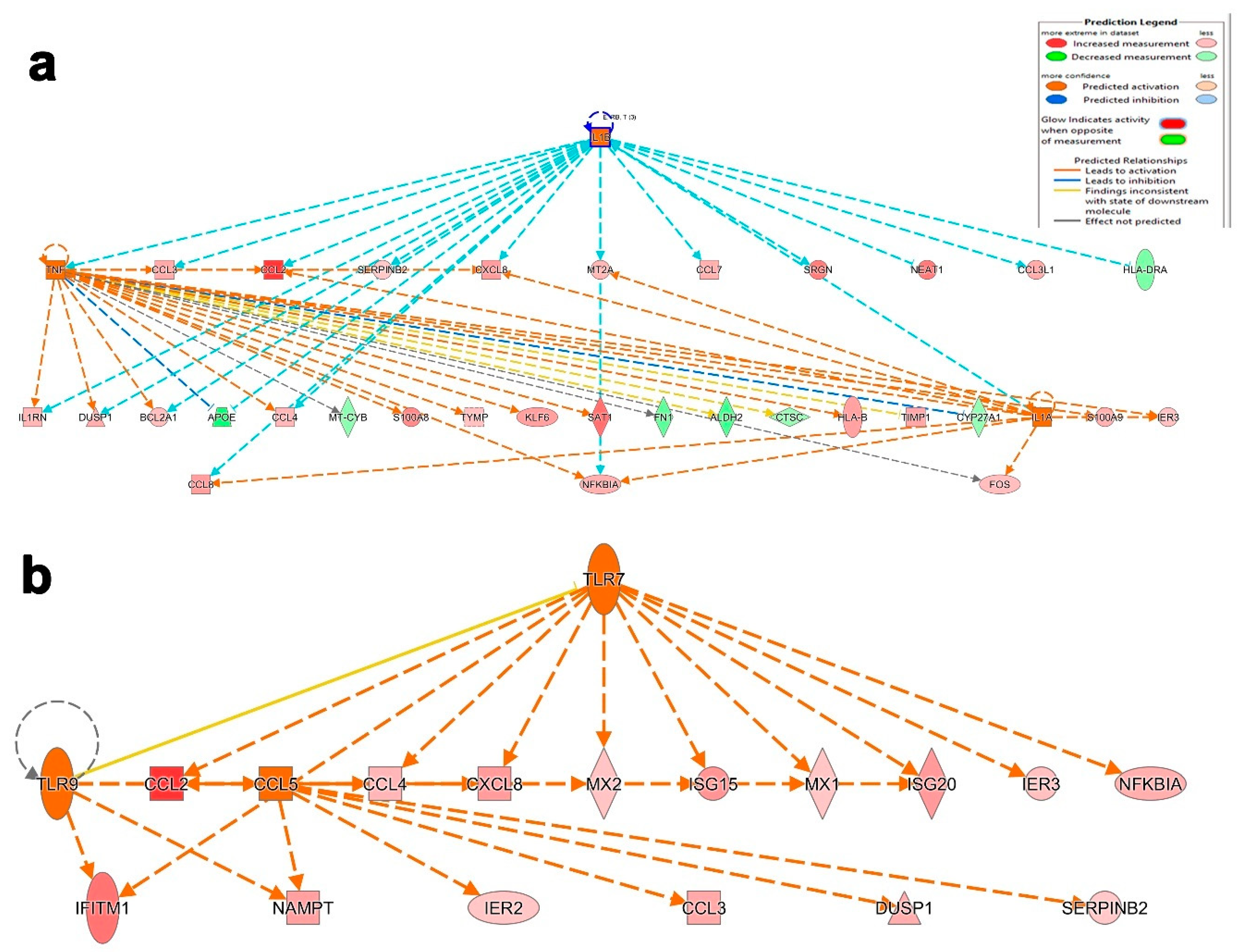
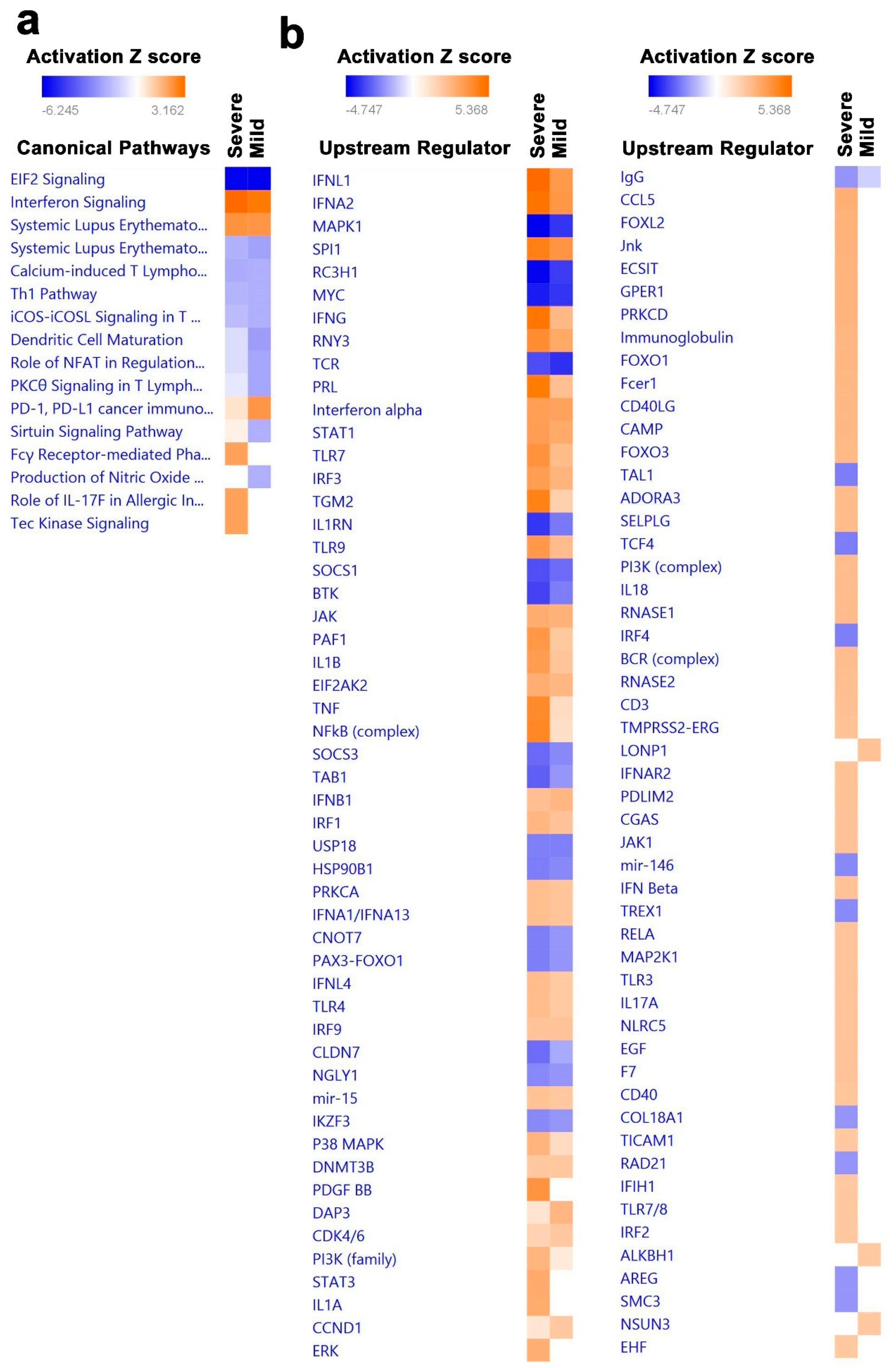
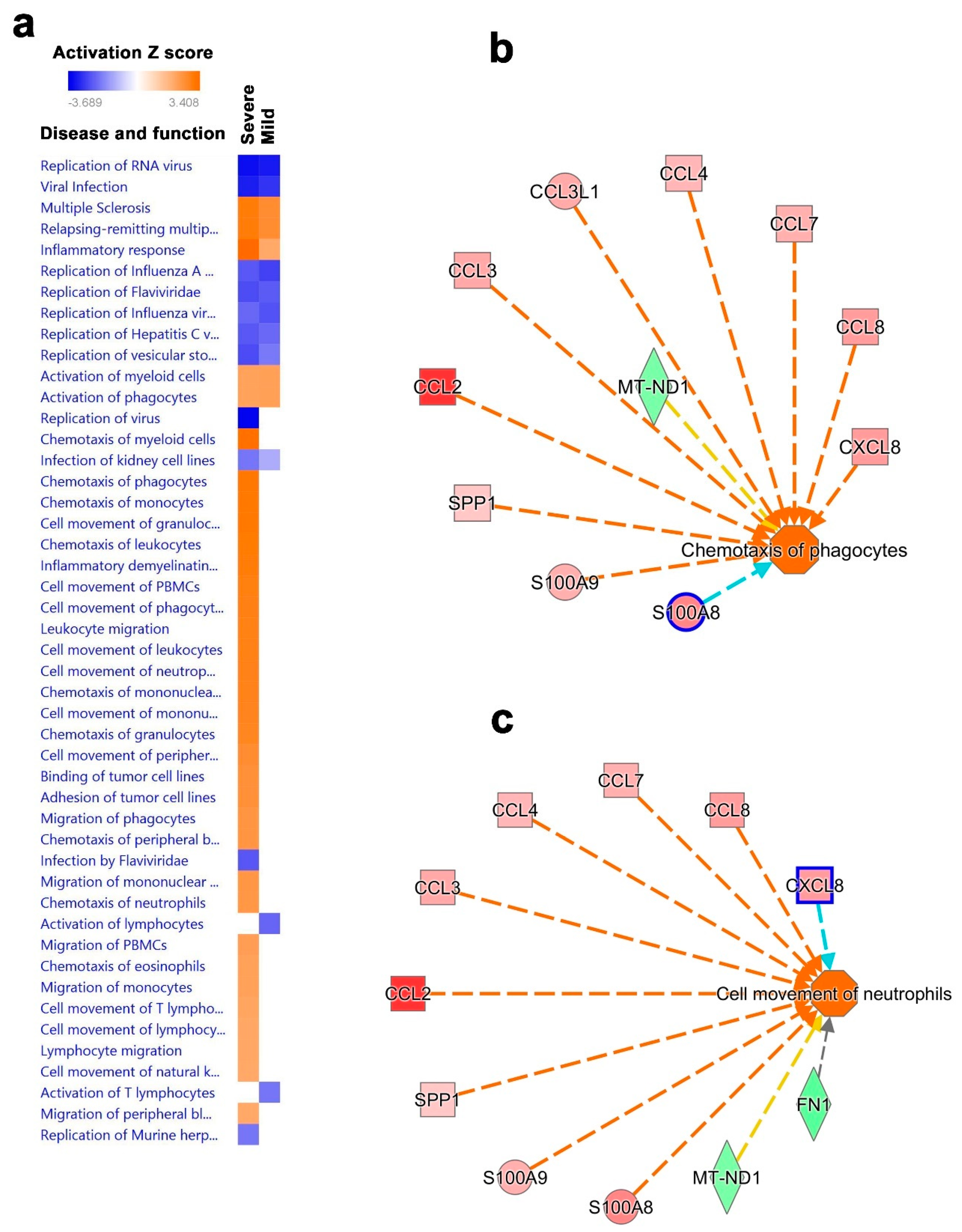
3.4. Canonical and Upstream Regulator Pathway Analyses Highlighted Activation of Innate Immune Response in BAL Cells from Severe COVID-19
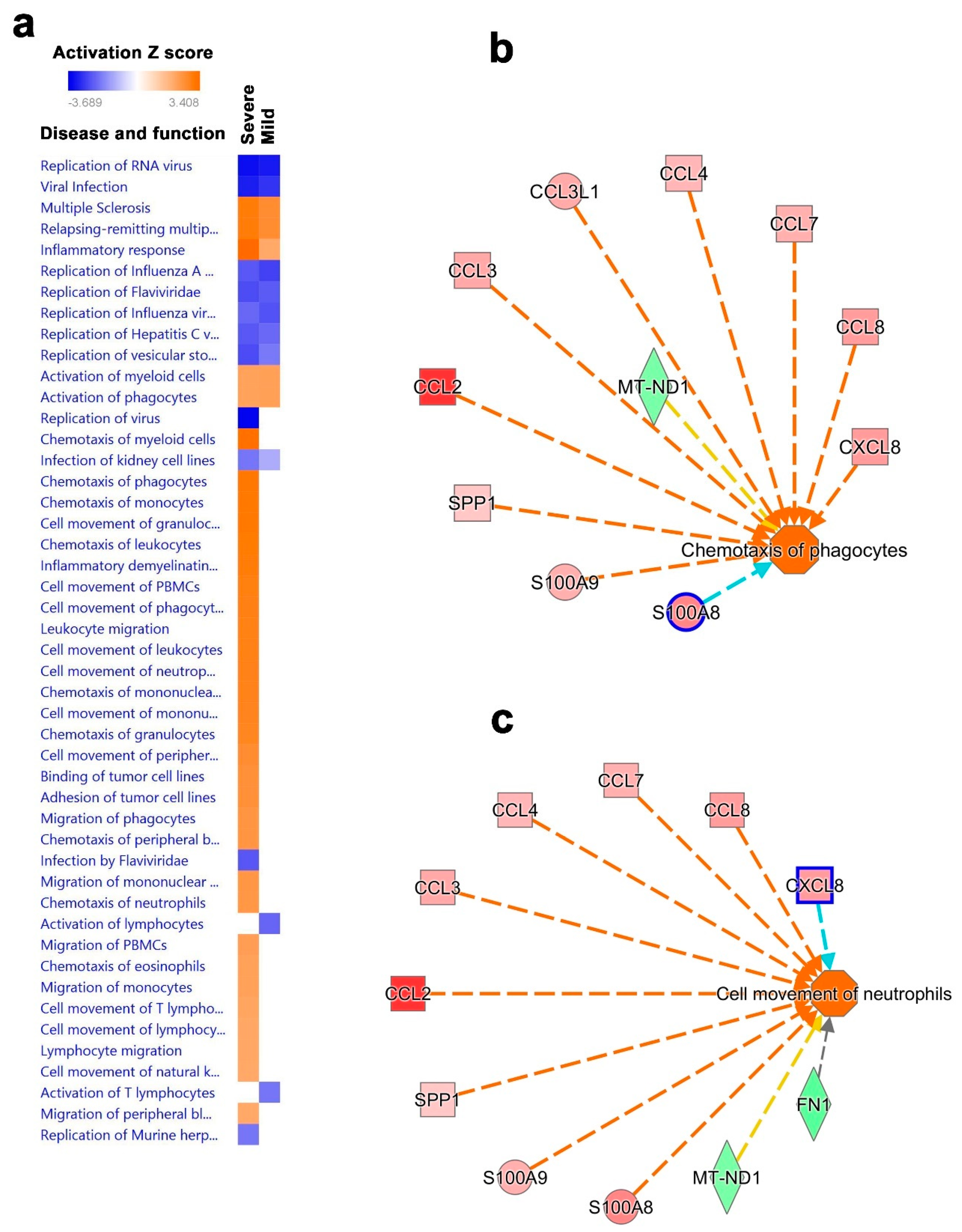
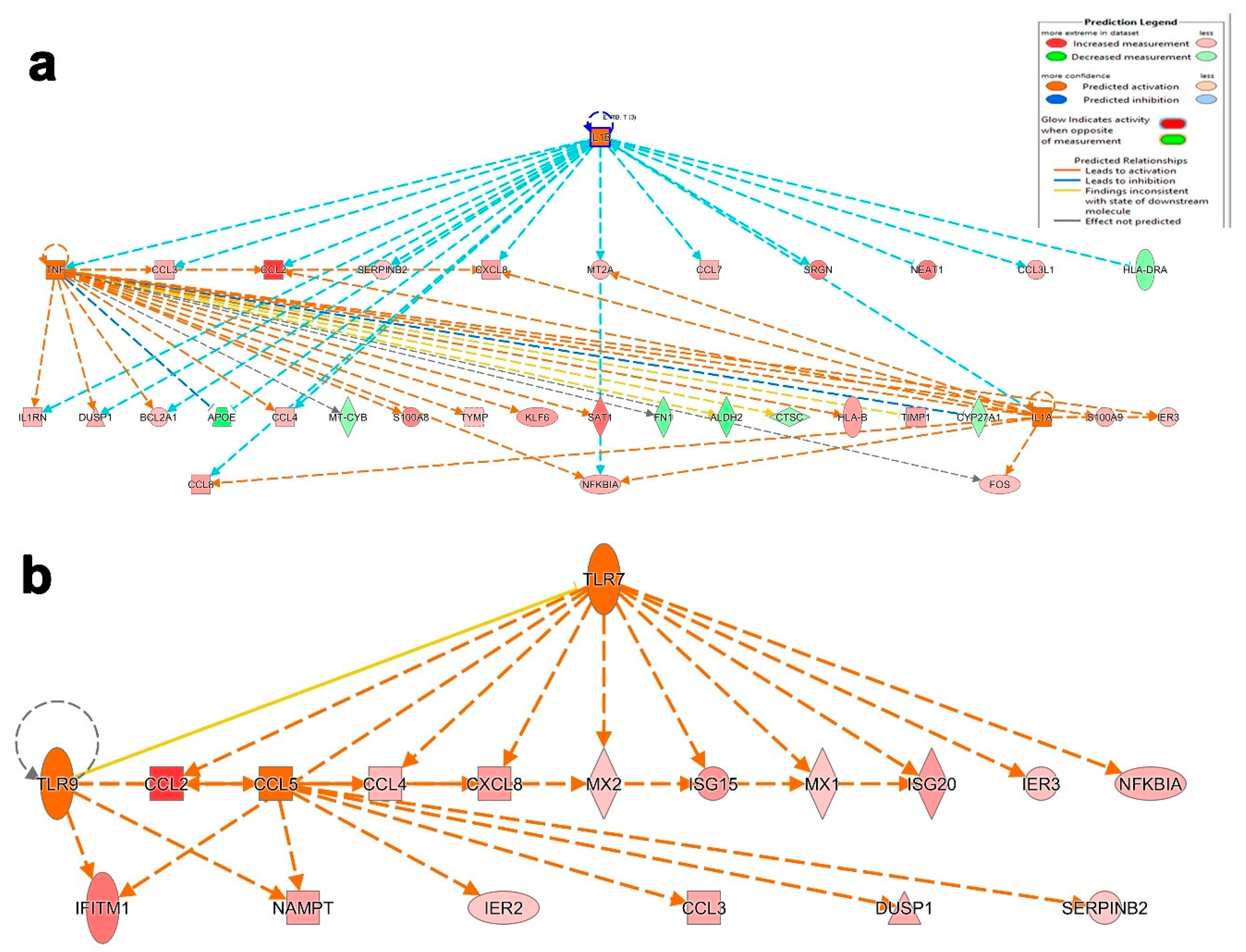
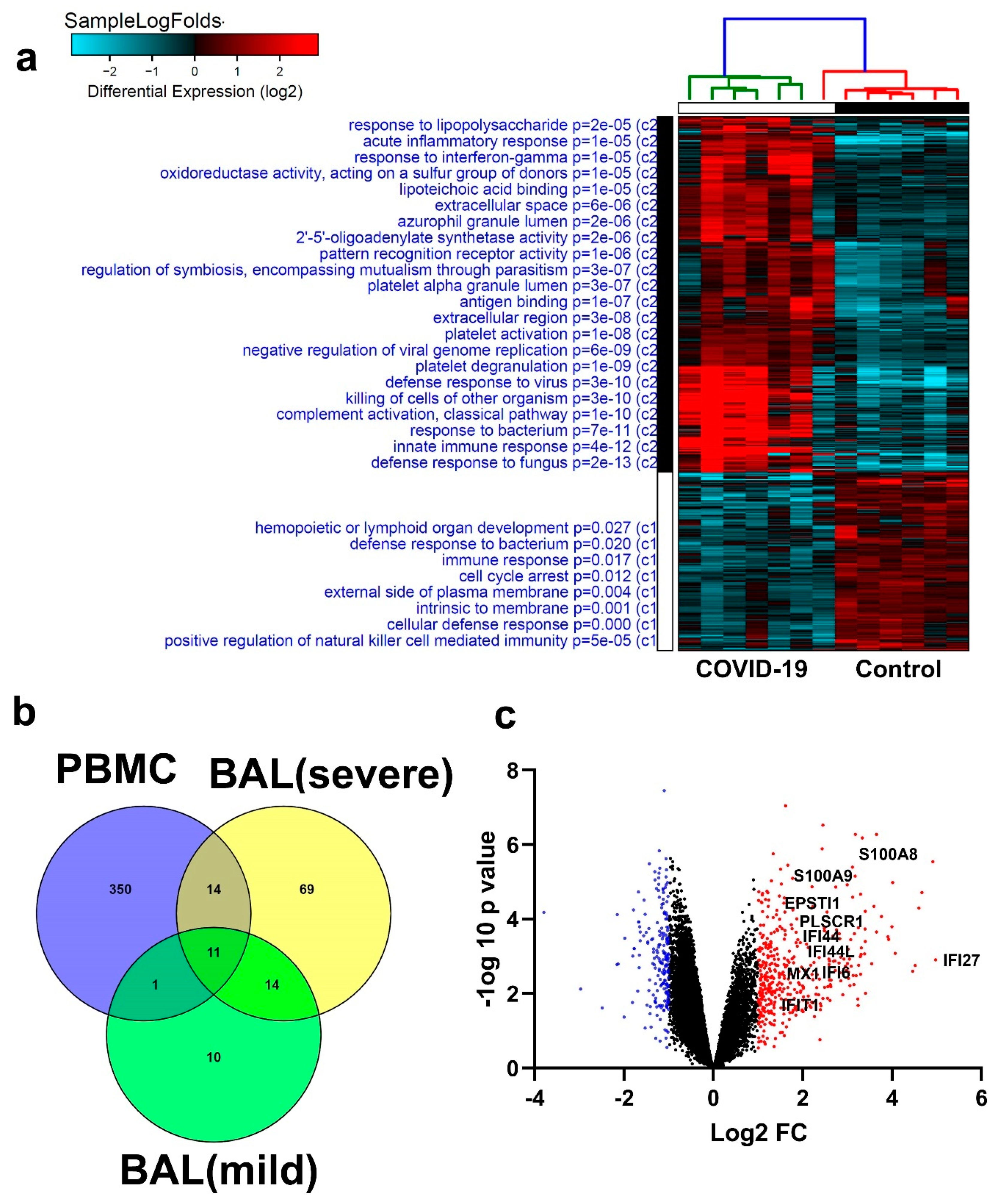
3.5. Disease and Functional Analysis of Differentially Expressed Genes in BAL from Severe- and Mild- COVID-19 Patients Indicated the Activation of Innate Immune Responses
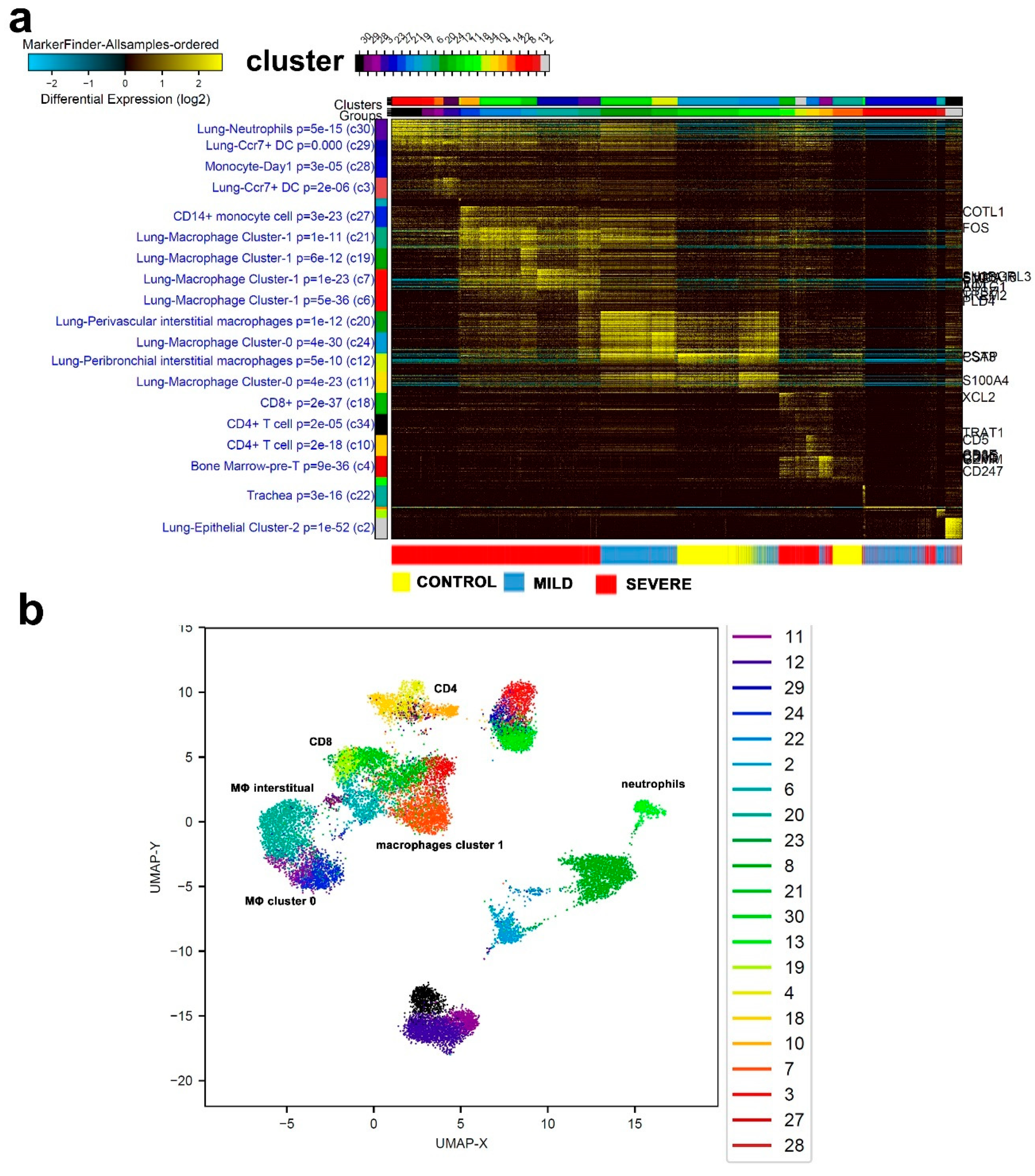
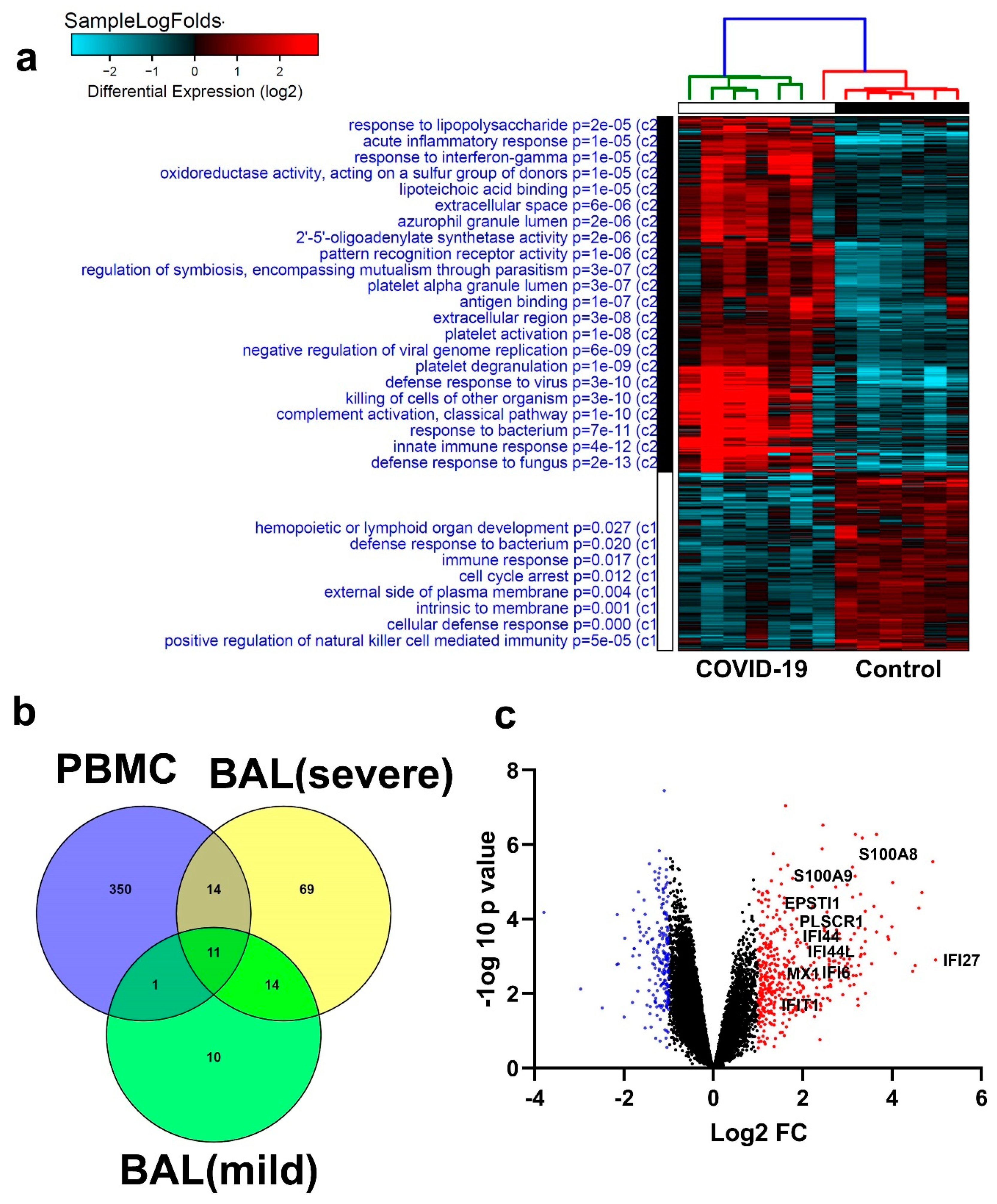
3.6. Similarities in Transcriptome Data from BAL and PBMCs from COVID-19 Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Chen, L.; Peng, G.; Zhou, W.; Pennell, C.A.; Mansky, L.M.; Geraghty, R.J.; Li, F. A Virus-Binding Hot Spot on Human Angiotensin-Converting Enzyme 2 Is Critical for Binding of Two Different Coronaviruses. J. Virol. 2011, 85, 5331–5337. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE2 Receptor Expression and Severe Acute Respiratory Syndrome Coronavirus Infection Depend on Differentiation of Human Airway Epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, C.A.; Ghani, A.C.; Leung, G.M.; Hedley, A.J.; Fraser, C.; Riley, S.; Abu-Raddad, L.J.; Ho, L.-M.; Thach, T.-Q.; Chau, P.; et al. Epidemiological determinants of spread of causal agent of severe acute respiratory syndrome in Hong Kong. Lancet 2003, 361, 1761–1766. [Google Scholar] [CrossRef] [Green Version]
- Cai, A.; McClafferty, B.; Benson, J.; Ramgobin, D.; Kalayanamitra, R.; Shahid, Z.; Groff, A.; Aggarwal, C.S.; Patel, R.; Polimera, H.; et al. COVID-19: Catastrophic Cause of Acute Lung Injury. J. South Dak. Med. 2020, 73, 252–260. [Google Scholar]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Dandekar, A.A.; Perlman, S. Immunopathogenesis of coronavirus infections: Implications for SARS. Nat. Rev. Immunol. 2005, 5, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wang, W.; Yin, J.; Ouyang, Y.; Pang, L.; Feng, Y.; Qiao, L.; Guo, X.; Shi, H.; Jin, R.; et al. The inhibition of IL-2/IL-2R gives rise to CD8(+) T cell and lymphocyte decrease through JAK1-STAT5 in critical patients with COVID-19 pneumonia. Cell Death Dis. 2020, 11, 429. [Google Scholar] [CrossRef] [PubMed]
- Pujhari, S.; Paul, S.; Ahluwalia, J.; Rasgon, J.L. Clotting disorder in severe acute respiratory syndrome coronavirus 2. Rev. Med Virol. 2020, 2177. [Google Scholar] [CrossRef]
- Wang, J.; Li, Q.; Yin, Y. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front. Immunol. 2020, 11, 2063. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Venkatasubramanian, M.; Chetal, K.; Schnell, D.J.; Atluri, G.; Salomonis, N. Resolving single-cell heterogeneity from hundreds of thousands of cells through sequential hybrid clustering and NMF. Bioinformatics 2020, 36, 3773–3780. [Google Scholar] [CrossRef] [Green Version]
- Calvano, S.E.; Xiao, W.; Richards, D.R.; Felciano, R.M.; Baker, H.V.; Cho, R.J.; Chen, R.O.; Brownstein, B.H.; Cobb, J.P.; Tschoeke, S.K.; et al. A network-based analysis of systemic inflammation in humans. Nature 2005, 437, 1032–1037. [Google Scholar] [CrossRef]
- Vishnubalaji, R.; Nair, V.S.; Ouararhni, K.; Elkord, E.; Alajez, N.M. Integrated Transcriptome and Pathway Analyses Revealed Multiple Activated Pathways in Breast Cancer. Front. Oncol. 2019, 9, 910. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Elango, R.; Alsaleh, K.A.; Vishnubalaji, R.; Manikandan, M.; Ali, A.M.; El-Aziz, N.A.; AlTheyab, A.; Al-Rikabi, A.; Alfayez, M.; Aldahmash, A.; et al. MicroRNA Expression Profiling on Paired Primary and Lymph Node Metastatic Breast Cancer Revealed Distinct microRNA Profile Associated With LNM. Front. Oncol. 2020, 10, 756. [Google Scholar] [CrossRef]
- Shaath, H.; Toor, S.M.; Nair, V.S.; Elkord, E.; Alajez, N.M. Transcriptomic Analyses Revealed Systemic Alterations in Gene Expression in Circulation and Tumor Microenvironment of Colorectal Cancer Patients. Cancers 2019, 11, 1994. [Google Scholar] [CrossRef] [Green Version]
- Emig, D.; Salomonis, N.; Baumbach, J.; Lengauer, T.; Conklin, B.R.; Albrecht, M. AltAnalyze and DomainGraph: Analyzing and visualizing exon expression data. Nucleic Acids Res. 2010, 38, W755–W762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, F.W.; Jones, J.M. Diagnosing Bacterial Respiratory Infection by Bronchoalveolar Lavage. J. Infect. Dis. 1987, 155, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Zhong, G.; Huang, I.-C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [Green Version]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; Van Der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishnubalaji, R.; Shaath, H.; Alajez, N.M. Protein Coding and Long Noncoding RNA (lncRNA) Transcriptional Landscape in SARS-CoV-2 Infected Bronchial Epithelial Cells Highlight a Role for Interferon and Inflammatory Response. Genes 2020, 11, 760. [Google Scholar] [CrossRef]
- Pinto, B.G.G.; Oliveira, A.E.R.; Singh, Y.; Jimenez, L.; Gonçalves, A.N.A.; Ogava, R.L.T.; Creighton, R.; Peron, J.P.S.; Nakaya, H.I. ACE2 Expression Is Increased in the Lungs of Patients with Comorbidities Associated with Severe COVID-19. J. Infect. Dis. 2020, 222, 556–563. [Google Scholar] [CrossRef]
- Roessner, A.; Smolle, M.; Hayback, J. Giant cell tumor of bone: Morphology, molecular pathogenesis, and differential diagnosis. Pathologe 2020, 41, 134–142. [Google Scholar]
- Peng, X.; Wei, F.; Hu, X. Long noncoding RNA DLGAP1-AS1 promotes cell proliferation in hepatocellular carcinoma via sequestering miR-486-5p. J. Cell. Biochem. 2019, 121, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Papp, E.; Hallberg, D.; Konecny, G.E.; Bruhm, D.C.; Adleff, V.; Noë, M.; Kagiampakis, I.; Palsgrove, D.; Conklin, D.; Kinose, Y.; et al. Integrated Genomic, Epigenomic, and Expression Analyses of Ovarian Cancer Cell Lines. Cell Rep. 2018, 25, 2617–2633. [Google Scholar] [CrossRef] [Green Version]
- Klupp, F.; Schuler, S.; Kahlert, C.; Halama, N.; Franz, C.; Mayer, P.; Schmidt, T.; Ulrich, A. Evaluation of the inflammatory markers CCL8, CXCL5, and LIF in patients with anastomotic leakage after colorectal cancer surgery. Int. J. Color. Dis. 2020, 35, 1221–1230. [Google Scholar] [CrossRef] [Green Version]
- Koshimune, S.; Kosaka, M.; Mizuno, N.; Yamamoto, H.; Miyamoto, T.; Ebisui, K.; Toyooka, S.; Ohtsuka, A. Prognostic value of OCT4A and SPP1C transcript variant co-expression in early-stage lung adenocarcinoma. BMC Cancer 2020, 20, 521. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.N.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teuwen, L.-A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; A Schuepbach, R.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef]
- Nile, S.H.; Nile, A.; Qiu, J.; Li, L.; Jia, X.; Kai, G. COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020, 53, 66–70. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Palomino, D.C.T.; Marti, L.C. Chemokines and immunity. Einstein (São Paulo) 2015, 13, 469–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felgenhauer, U.; Schoen, A.; Gad, H.H.; Hartmann, R.; Schaubmar, A.R.; Failing, K.; Drosten, C.; Weber, F. Inhibition of SARS–CoV-2 by type I and type III interferons. J. Biol. Chem. 2020, 295, 13958–13964. [Google Scholar] [CrossRef] [PubMed]
- Major, J.; Crotta, S.; Llorian, M.; McCabe, T.M.; Gad, H.H.; Priestnall, S.L.; Hartmann, R.; Wack, A. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 2020, eabc2061. [Google Scholar] [CrossRef]
- Chakraborty, S.; Edwards, K.; Buzzanco, A.S.; Memoli, M.J.; Sherwood, R.; Mallajosyula, V.; Xie, M.M.; Gonzalez, J.; Buffone, C.; Kathale, N.; et al. Symptomatic SARS-CoV-2 infections display specific IgG Fc structures. medRxiv 2020. [Google Scholar] [CrossRef]
- Bournazos, S.; Wang, T.T.; Ravetch, J.V. The Role and Function of Fcgamma Receptors on Myeloid Cells. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Li, J.; Han, Z.; Chen, Z.; Zhang, Q. Silencing of lncRNA MALAT1 Prevents Inflammatory Injury after Lung Transplant Ischemia-Reperfusion by Downregulation of IL-8 via p300. Mol. Ther. Nucleic Acids 2019, 18, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Chen, C.-Y.; Yedavalli, V.S.R.K.; Jeang, K.-T. NEAT1 Long Noncoding RNA and Paraspeckle Bodies Modulate HIV-1 Posttranscriptional Expression. mBio 2013, 4, e00596-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaath, H.; Alajez, N.M. Computational and Transcriptome Analyses Revealed Preferential Induction of Chemotaxis and Lipid Synthesis by SARS-CoV-2. Biology 2020, 9, 260. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaath, H.; Vishnubalaji, R.; Elkord, E.; Alajez, N.M. Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe COVID-19. Cells 2020, 9, 2374. https://doi.org/10.3390/cells9112374
Shaath H, Vishnubalaji R, Elkord E, Alajez NM. Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe COVID-19. Cells. 2020; 9(11):2374. https://doi.org/10.3390/cells9112374
Chicago/Turabian StyleShaath, Hibah, Radhakrishnan Vishnubalaji, Eyad Elkord, and Nehad M. Alajez. 2020. "Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe COVID-19" Cells 9, no. 11: 2374. https://doi.org/10.3390/cells9112374