Breaking Molecular Symmetry through Biocatalytic Reactions to Gain Access to Valuable Chiral Synthons


Institute of Biomolecular Chemistry, National Research Council of Italy, Via Paolo Gaifami 18, 95126 Catania, Italy
*
Author to whom correspondence should be addressed.
Symmetry 2020, 12(9), 1454; https://doi.org/10.3390/sym12091454
Submission received: 12 August 2020
/
Revised: 22 August 2020
/
Accepted: 1 September 2020
/
Published: 4 September 2020
(This article belongs to the Special Issue Chiral Molecules: Properties, Synthesis and Analysis)
Abstract
:In this review the recent reports of biocatalytic reactions applied to the desymmetrization of meso-compounds or symmetric prochiral molecules are summarized. The survey of literature from 2015 up to date reveals that lipases are still the most used enzymes for this goal, due to their large substrate tolerance, stability in different reaction conditions and commercial availability. However, a growing interest is focused on the use of other purified enzymes or microbial whole cells to expand the portfolio of exploitable reactions and the molecular diversity of substrates to be transformed. Biocatalyzed desymmetrization is nowadays recognized as a reliable and efficient approach for the preparation of pharmaceuticals or natural bioactive compounds and many processes have been scaled up for multigram preparative purposes, also in continuous-flow conditions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Chirality is an important molecular feature that gains particular significance in the case of biologically active compounds since the activity is often dependent from the correct stereochemistry fitting with the target sites in living systems [1,2]. This concept is nowadays so widely accepted that pharmacological properties of chiral drugs are studied on both enantiomers [3] in order to highlight, if there are, enhancement of the activity or adverse effects related with a single enantiomer [4]. The increased demand of enantiomerically pure drugs or intermediates, together with the huge advances in total synthesis of natural compounds, have promoted the development of synthetic methodologies aimed to the preparation of a variety of compounds in non-racemic form [5].
In this context, the desymmetrization strategy has attracted great interest [6,7] for the possibility to generate multiple stereocentres starting from relatively simple symmetric prochiral molecules or meso-compounds, without the limitation in the yield connected with the classical kinetic resolution of racemates, that gives at maximum 50% of the desired enantiomer.
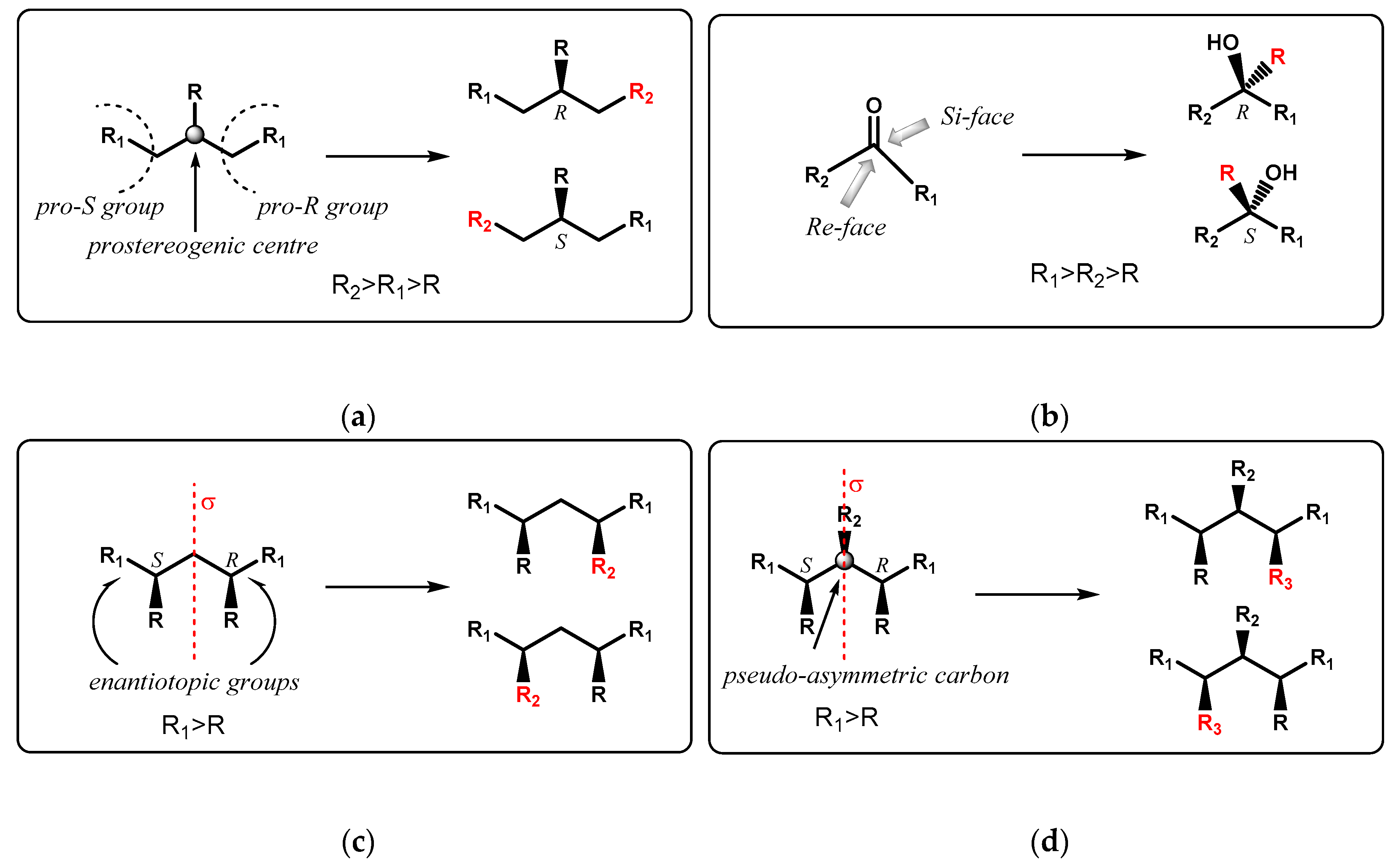
It is worth mentioning that a molecule is prochiral when it contains a tetrahedral atom bonded to two identical functional groups, whose selective modification (or substitution) following an enantiotopic differentiation gives rise to enantiomers (Figure 1a). Are prochiral also those molecules containing a trigonal system (as an alkene or a ketone) that can be made chiral by the addition of a new atom or group with enantiofacial differentiation (Figure 1b). Desymmetrization of prochiral molecules, hence, leads to the creation of a stereogenic centre in place of the pre-existing pro-stereogenic one.
meso-Compounds, on the other hand, are achiral molecules due to the presence of improper symmetry element(s), although they containing pair(s) of chiral centres with opposite configuration (Figure 1c), and possibly also additional stereogenic carbon(s) lying on the symmetry element (Figure 1d), that become “explicited” following desymmetrization since no longer equivalent.
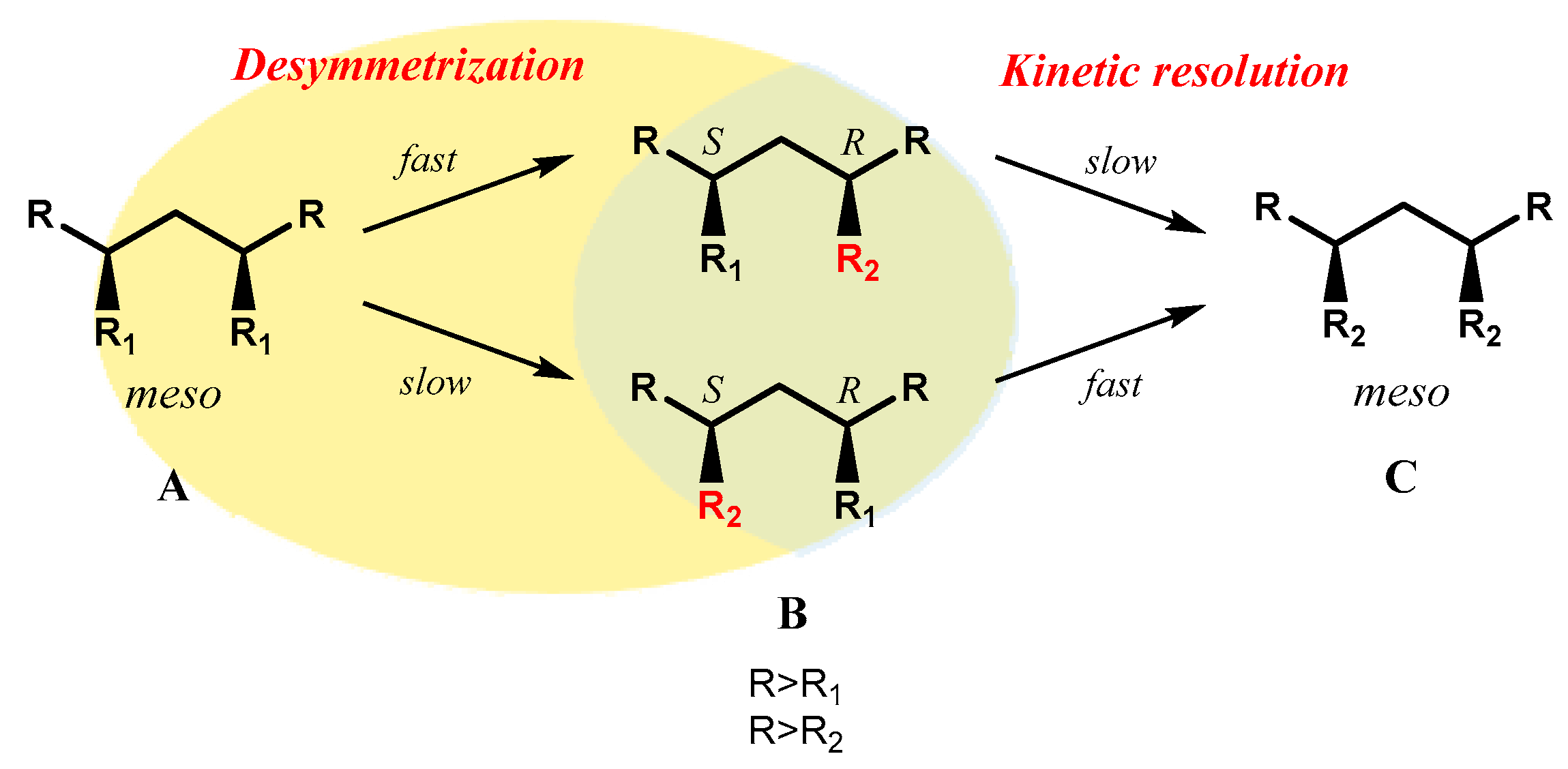
According the IUPAC definition [8], a desymmetrization consists of a modification resulting in the loss of one or more symmetry elements, such as those which preclude chirality (mirror plane, centre of inversion, rotation-reflection axis), so that a prochiral molecular entity is converted into a chiral one. For an effective desymmetrization, hence, two enantiotopic functional groups of the substrate need to be selectively differentiated through the appropriate choice of chiral reagent or catalysts. Furthermore, products in high optical yield at the expense of chemical yield can be obtained even with a moderately selective catalyst, exploiting the synergy between desymmetrization and a subsequent kinetic resolution step, in a Horeau-type amplification of the optical yield [9].
Indeed, when a desymmetrization of a substrate A is carried out in the presence of a catalyst with a given stereopreference, for example R or pro-R, a R-enantioenriched mono-functionalized product B is firstly obtained, but the over-reaction of this compound to the corresponding bis-functionalized derivative C (a symmetrical and achiral by-product) will tend to proceed in a kinetic resolution fashion through the differentiation of the two distinct enantiomers of B, preferentially consuming the minor enantiomer of B, which still has the catalyst-matched R-enantiotopic position intact and suitable to be further transformed. If this is the case, the overall process ultimately results in the improvement of optical purity of the desymmetrization product B (Scheme 1).
An impressive amount of work has been carried out on desymmetrization reactions catalyzed by chiral metal-ligand complexes or chiral organocatalysts as well as by purified enzymes or microbial whole cells and recent reviews are present in literature [10,11,12]. Effective desymmetrization protocols have been developed for substrates bearing a variety of functional groups, also including all-carbon quaternary stereocentres [13], and identified as key steps in the synthesis of several chiral natural compounds or drugs.
Due to the high stereoselectivity of enzymes, biocatalysis offers a valuable strategy in the synthesis of pharmaceuticals and desymmetrization processes [14,15,16], also taking advantages in the use of mild reaction conditions without the need of harsh reagents or special laboratory equipment, in the commercial availability of many enzymes and in the possibility to improve the catalytic performances of enzymes (or microorganisms) by direct evolution techniques [17]. In this review we have selected the most recent (from 2015 up to date) reports of biocatalyzed desymmetrizations of meso and prochiral molecules useful as intermediates in the synthesis of biologically active compounds, limiting the examples of molecules containing trigonal systems to the cases in which the enantiofacial differentiation is coupled with enantiotopic discrimination, as occurs in symmetric diketones.
2. Desymmetrization of Meso-Compounds
2.1. Lipase-Catalyzed Reactions
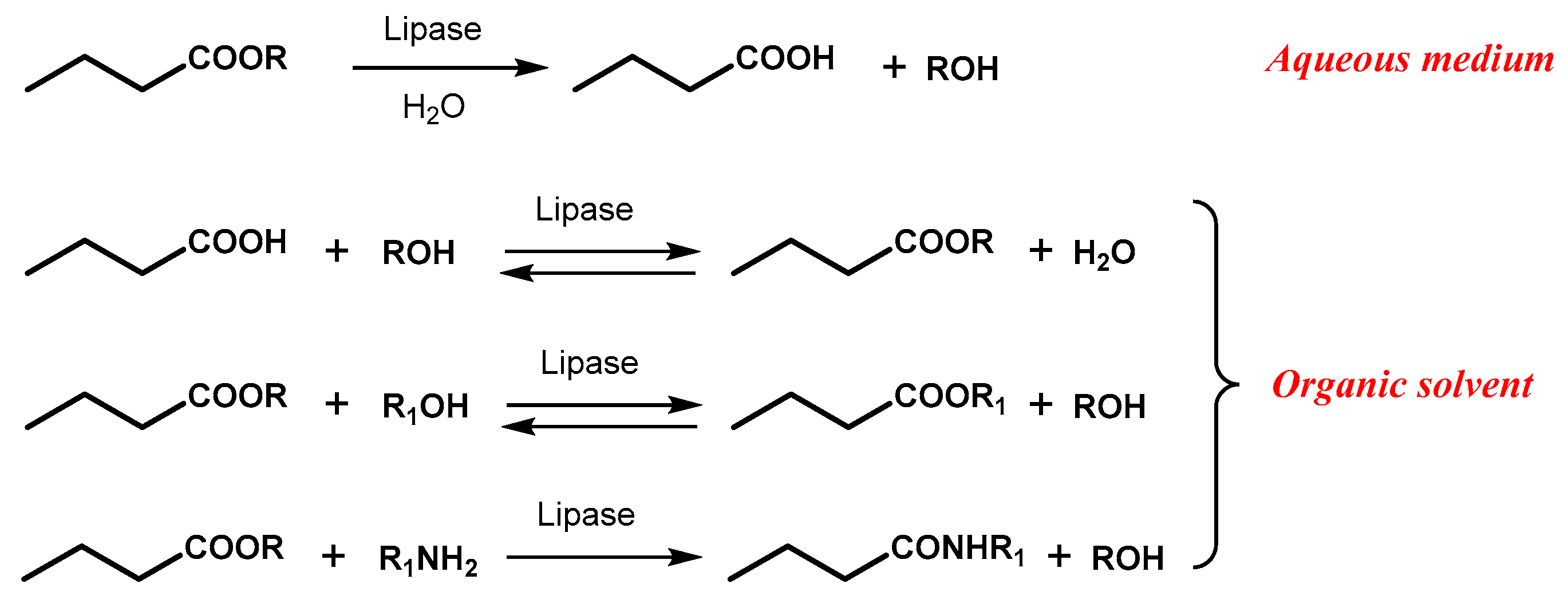
Among the different enzymes used in biocatalytic transformations, lipases are the most popular ones for their wide substrate tolerance, which allows to a notable variety of molecular scaffolds to be accommodated and stereoselectively recognized in the enzymatic active site resulting in efficient kinetic resolution or desymmetrization processes [18,19,20]. Lipases belong to the class of hydrolases and their physiological activity resides in the hydrolysis of fatty acid esters; on this basis most of the applications of these enzyme deal with the hydrolysis of suitable esters of carboxylic acids in buffered aqueous medium. However, lipases are also able to display their activity in organic solvents and this peculiar feature has opened access to the possibility to better solubilize hydrophobic substrates and carry out either direct esterification or transesterification reactions (inhibited in water medium) through the proper combination of an alcohol and a carboxylic acid or derivative (Scheme 2).
Due to its reversibility, the direct esterification often demands an efficient removal of the water produced during the reaction in order to achieve high conversion of the substrate, whereas the use of vinyl esters as acyl donors is usually the best choice to make irreversible the transesterification reactions, thanks to the formation of acetaldehyde [18].
Furthermore, the polarity of the organic medium as well as its water content markedly influence the conformational properties of the enzyme, eventually resulting in changes of its stability, activity and stereoselectivity which can be therefore modulated by “medium engineering” [21,22,23]. The use of lipases in organic solvents gives great advantage when the opposite enantiomers of a target chiral compound are required, since a single enzyme can be used in two complementary reactions, as hydrolysis and esterification or two transesterification reactions, on related substrates without the need of two enzymes with opposite stereopreference. Indeed, if a diester of a meso-diol is hydrolyzed in aqueous medium by a lipase with R-stereopreference a monoester having the free alcohol group located on R-stereocentre will be obtained, but using the meso-diol as substrate in a reaction in the presence of the same lipase and a suitable acyl-donor in organic solvent, the opposite enantiomer is expected to be formed following the preferential esterification of the R-alcohol group. (Scheme 3).
Immobilization of lipases on different inert supports, in addition to facilitating the recovery of the catalyst from aqueous or organic solutions, also leads in many instances to enhanced stability of the enzymes [24,25,26], allowing them to be recycled and/or used in flow-reactors to increase the productivity and sustainability of the biotransformations. Several lipases from bacteria, yeasts, fungi and porcine pancreas are commercially available in free or immobilized forms and in some cases the observed stereoselectivity has been rationalized with the aid of crystallographic and molecular modelling studies [27,28,29].
Selective lipase-catalyzed reactions have been widely applied to the desymmetrization of a variety of meso-diols [12] to give products with defined chirality on two (or more) stereogenic centres, useful as intermediates in the synthesis of natural compounds or drugs.
Chiral 4-hydroxy-2-cyclopentenyl acetate (1), an important building block for the synthesis of prostaglandins, some antibiotics and natural cyclopentanoids, has been enzymatically produced by selective acylation of the cis-2,4-dihydroxycyclopentene (2) or hydrolysis of the corresponding diacetate 3. In the original procedures developed by Theil’s group [30], porcine pancreas lipase (PPL) or pancreatin, a crude preparation of the pancreatic enzymes, were employed for both reactions, but also immobilized lipase from Mucor miehei (Chirazyme®) [31] and Pseudomonas fluorescens lipase (PFL) immobilized on superparamagnetic nanoparticles [32] were found effective in the preparation of optically active 1 (Figure 2).
Recently, both the enantiomers of 1 have been prepared by using two enzymes with opposite stereopreference. By using a recombinant pig liver esterase (PLE), an enzyme acting on the esters of short chain acids with the same hydrolytic function of lipases, the desymmetrization of diacetate 3 was carried out in multigram scale to afford (1S,4R)-1 in good optical yield. Coupling of the process with enantioselective crystallization allowed to obtain the target monoacetate in 99% ee (Figure 3a) at expense of the chemical yield (40%) [33].
On the other hand, the transesterification reaction of 3 with MeOH in the presence of immobilized lipase from Candida antarctica (Novozym® 435) gave, after careful choice of the solvent, (1R,4S)-1 in excellent chemical and optical yields (95%, 99% ee). In the optimized conditions the formation of diol 2 was not observed and the reaction was carried out on a 10 g scale, reusing the enzyme up to 10 cycles without appreciable changes in its activity (Figure 3b) [34].
In the course of a study on diastereoselective Passerini reaction, Riva et al. prepared both enantiomers of monoester 4 exploiting the desymmetrizations of diol 5 and its dibutyrate ester 6 through complementary reactions of acylation and hydrolysis, respectively [35]. In comparison with previous works [36,37], the acylation of 5 with vinyl butyrate was optimized using lipase from Burkholderia cepacia (Amano PS) immobilized on Celite in a 4:1 i-Pr2O:CH2Cl2 mixture and the addition of powdered 3Å molecular sieves was found beneficial for the reproducibility in preparative scale.
Prolonging the reaction time (about 20 h) the amount of formed 6 increased, but (2R,5S)-4 could be obtained in enantiopure form (72% isolated yield) thanks to kinetic amplification (Figure 4).
The same lipase Amano PS also promoted the selective acetylation of diol 7 to give monoacetate (1S,2R)-8 and the hydrolysis of diacetate 9 to afford the opposite enantiomer (1R,2S)-8, in both cases with satisfactory chemical yield and optical purity [38]. Swern oxidation of the free alcohol group present in both enantiomers of 8 gave the corresponding chiral aldehydes, which were used as substrates in Ugi or Passerini multicomponent reactions followed by cyclization to trans or cis-fused bicyclic pyrrolidines, to afford 6 out of all 8 possible stereoisomers of the peptidomimetic derivative 10. Starting from (1R,2S)-8, the same strategy was also applied to the synthesis of the antiviral drug telaprevir, which was obtained in 8 step and 26% overall yield, in a valuable alternative to other reported processes employing the more expensive monoamine oxidase or chemical chiral catalysts (Figure 5).
In the retrosynthetic analysis for two yohimbine alkaloids, the desymmetrization of diacetate 11 was identified as a crucial step to install the right stereochemistry in the pentacyclic indole skeleton of the target molecules. In their work [39], Ghosh and Sarkar modified the reported procedure of hydrolysis of 11 in the presence of PPL with the use of a solution of NaHCO3 instead of NaOH as neutralizing agent of the acetic acid released in the reaction. In this way the competing non-enzymatic hydrolysis of 11, that results in the erosion of the enantiomeric purity of monoacetate 12, was minimized and it was possible to obtain (1R,2S)-12 in reproducible yield (>80%) and optical purity >95% ee, even in a 50 g preparative scale. With this optimized protocol in hand, the same authors also succeeded in converting the optically active 12 into the 6,5 fused ring (3aS,4S,7aR)-4H-furo [2,3-b] pyran-4-ol, which was then incorporated in the structure of an analogue of the HIV-protease inhibitor daunavir (Figure 6).
The desymmetrization of 1,3,5-cyclohexanetriols, originally investigated by Wirz and coworkers [40], is an important strategy to control of the stereochemistry of ring A in the synthesis of vitamin D3 analogues. On the basis of literature data, Garcia et al. [41] selectively acetylated cis,cis-O-protected diol 13 with lipase from Pseudomonas cepacia immobilized on diatomaceous earth (Amano IM) and reported higher reaction rate and selectivity of the enzyme in toluene or TBME compared to THF. The monoacetate 14 with (1R,3S,5S)-configuration was obtained as exclusive product and then manipulated to prepare both 1,3-epimers of vitamin nor-D3 (Figure 7).
In a screening of lipases for the desymmetrization of myo-inositol derivative 15 [42], it was found that only lipases from Rhizomucor miehei (Lipozyme RM-IM) and Thermomyces lanuginosus (Lipozyme TL-IM), both in immobilized form, were active in catalyzing the formation of monoacetate 16 with the same excellent stereoselectivity. In comparison with ethyl acetate, the use of vinyl acetate as solvent/acylating agent led to higher conversion of the substrate and the reaction rate increased in the presence of a co-solvent in 1:1 ratio. In the assay for enzyme reuse, it was found that Lipozyme TL-IM maintained its activity and selectivity over seven cycles (Figure 8).
The stereoselective hydrolysis of diester 17, catalyzed by lipase from Candida rugosa (Lipase AYS), has proven to be a robust route for the preparation in multigram-scale of the chiral monoester (1S,3R)-18 [43], a key synthon for the insertion of the suitable chirality in 19, an inhibitor of human influenza virus replication, whose optimized synthesis was carried out in just 5 steps and 35% overall yield from 18 (Figure 9).
2.2. Reactions Catalyzed by other Enzymes than Lipases
Beside the hydrolysis/transesterification reactions catalyzed by lipases, reactions in the presence of oxidoreductases are an important group of biotransformations with significant potential in organic synthesis and in the preparation of chiral compounds [44,45,46]. In this group of enzymes, alcohol dehydrogenases (ADHs) have been widely investigated [47,48,49,50] due to their ready availability from horse liver [51] or baker’s yeast [52]. ADHs catalyze the reversible oxidation of alcohols to the corresponding carbonyl compounds, through the transfer of a hydride anion to the oxidized form of a nicotinamide cofactor, NAD+ or NADP+, which is converted into its reduced form, NADH or NADPH. The cofactor is then regenerated [53,54] for the subsequent cycles of oxidation through the conversion of an ancillary substrate by a specific NAD(P)H oxidase. Although a variety of purified ADHs can be today produced from engineered microorganisms or recombinant techniques [55,56], the need of a coupled two-enzyme system can be in some instances advantageously overcome by using microbial whole cells containing both the required enzymes [57,58].
For their high stereospecificity, ADHs can be exploited in the desymmetrization of diols through the selective oxidation of one alcohol group following the enzymatic enantiotopic differentiation. This strategy has been recently applied for the production (R)-acetoin 21 from 2,3-butanediol 20 in the presence of a specific ADH from Corynebacterium glutamicum, obtained from expression in Escherichia coli of the corresponding gene and subsequent purification [59]. In the search of an effective regeneration system for the expensive cofactor NAD+, different oxidases were produced and tested; the oxidase from Lactococcus lactis gave the best reaction performances and the overall process led to 21 in good chemical and optical yields (Figure 10).
The selective conversion of diol 2 into monoalcohol (R)-22 was carried out in the presence of a crude lysate of cells from Ralstonia sp. (RasADH) expressing a NADP(H)-dependent ADH and it was also found that an efficient cofactor regeneration is provided by the reduction of cyclohexanone by the same enzyme. Chiral 21 was then used as a building block in a three-component coupling reaction to generate the antiglaucoma drug travoprost, an analogue of prostaglandin PGF2a, in high enantiomeric purity [60] (Figure 11).
The biotransformation of diols 23a–c to the corresponding γ-lactones 24a–c, probably proceeding through a lactol intermediate as previously reported for the oxidation of other diols with the same microorganism [61], was investigated in the presence of whole cells of Rhodococcus erythropolis DSM 44354 as biocatalyst [62].
The microorganism was cultured in a mineral medium containing ethanol as sole source of carbon and no additional enzyme or cofactor were required for the biooxidation of the substrates. The reaction outcome was found to be dependent by the structure of the starting diol and the selectivity in the desymmetrization increased going from 23a to 23c, although the observed chemical yield followed a different order (Figure 12).
Amidases are an important group of hydrolytic enzymes able to convert amides in the corresponding free carboxylic acids with release of ammonia. Ubiquitously diffused in living organisms, they play an important role in the detoxification of xenobiotics and in many microorganisms they are often coupled to nitrile hydratases, concurring to convert nitriles into carboxylic acids [63,64]. In the context of broadening the scope of biocatalysis in sustainable organic synthesis, the use of these enzymes is attracting great interest since the chemical hydrolysis of nitriles and amides often requires harsh conditions, such as the use of strong acids and elevated temperatures, not compatible with the presence of other labile groups, it is not selective, so generating mixtures of products, and produces large amount of waste with consequent environmental concerns.
Although they have been known for long time, the application of amidases in enantioselective biotransformations is rather limited, mostly to the resolution of racemates, and work in this field has been recently reviewed [65]. Extensive investigation of the desymmetrization of dicarboxamides has been carried out by Ao’s group using whole cells of R. erythropolis AJ270, a microorganism that exhibits catalytic nitrile-hydrolyzing activity, and in their recent work a systematic study was carried out on a series of cyclopentane- and cyclohexane-derivatives [66]. Compounds 25a–c were hydrolyzed in high reaction rate and excellent pro-S-stereoselectivity, independently from the size of the carbocycle, to give the corresponding monoamides 26a–c, in which the enzymatically generated carboxylic acid group was chemically alkylated by benzyl bromide to facilitate the isolation and detection of the products (Figure 13).
The biocatalytic procedure was also applied to derivatives 27a–c, containing an additional stereogenic centre lying in the symmetry plane of the molecules, and a marked lowering of the reaction rate was evidenced, probably related with the steric hindrance effects exerted by the substituent in the 2-position of the ring. However, the efficiency of the biocatalyst in the stereorecognition was maintained and, interestingly, in spite of the opposite configuration at C-2 in compounds 27a and 27b the same S-configuration was assigned to the obtained products 28a and 28b, suggesting that the hydroxyl group may not participate in the chiral recognition with amidase. The presence of a rigid cyclic backbone in the substrate was shown essential for the enantiocontrol of the reaction, as indirectly deduced by the reaction of the prochiral glutaramide 29 that afforded 30 in quite low optical purity. Indeed, in 29 the two amide groups are located on a flexible acyclic skeleton and may adapt in comparable rate to the active site of the enzyme (Figure 14).
A marked influence of steric effects on the reactivity of amidase in whole cells of R. erythropolis AJ270 was also revealed in the desymmetrization of bis-amides 31a–e and 33a–b, bearing an all-carbon quaternary stereogenic centre in the C-2 position of cyclopentane ring [67].
When the large C-2 substituent was in the trans-configuration with respect to the reactive amide groups, complete conversion of the substrates to monoamides 32a–e was achieved within 2 days, but the opposite configuration of C-2 led to detrimental effect on the reaction rate, even though the S-stereopreference in the formation of 34a–b was maintained at excellent level. The described biotransformation represents an important approach to enantiopure densely functionalized cyclopentanes, that are not readily available by other routes and can be useful intermediates in the synthesis of natural compounds (Figure 15).
3. Desymmetrization of Prochiral Compounds
3.1. Lipase-Catalyzed Reactions
The recent reports of lipase-catalyzed desymmetrizations of prochiral compounds deal in most cases with substrates belonging to the class of 2-substituted propane-1,3-diols, which are important precursors in the synthesis of biologically active compounds.
As an example, in their search for synthetic access to chiral 2-piperidones, whose skeleton is embedded in many active alkaloids, Judeh and coworkers identified alcohol 36 as an important synthon and investigated the possibility to prepare it by desymmetrization of diol 35 [68]. Following a screening of enzymes, lipase from P. fluorescence was the most stereoselective toward the formation of (R)-36 and it was observed that the amount of the diacetylated product 37 decreased in polar aprotic solvents such as CH3CN. The opposite enantiomer, (S)-36 was obtained through the complementary reaction of hydrolysis of diacetate 37 with the same lipase and the presence of an organic co-solvent (10% v/v) was essential for high enantioselectivity, probably in consequence of a decreased conformational mobility of the enzyme compared to the aqueous medium. However, the hydrolysis of 37 proceeded with concomitant formation of diol 35 and, in the best conditions, gave (S)-36 in 51% isolated yield (Figure 16).
The desymmetrization of diacetate 38, previously reported by Nanda et al. [69], was recently exploited for the construction of the fatty acid unit in the N-terminus of natural cyclodepsipeptides callipeltin A and homophymine B, which possess antiviral, antifungal and antitumoral activities. In their work [70], Tokairin and Konno carried out the hydrolysis of 38 at low temperature in the presence of a commercially available lipase (Amano PS) instead of the original enzyme PS-D (lipase from B. cepacia immobilized on diatomite) and the optically pure monoacetate (R)-39 was converted into (2R,3R,4R)-3-hydroxy-2,4,6- trimethylheptanoic acid in 40% yield over 2 steps (Figure 17).
In a study on the lipase-catalyzed transesterification of pyrimidine acyclonucleoside 40 in organic solvent it was found that the reaction in the presence of Amano PS lipase was not selective for the formation of monoester (R)-41a, but proceeded to the corresponding diester so that the choice of the optimal reaction time was essential to maximize the yield of the target product.
From a reaction in THF, (R)-41a was isolated in 65% yield and its optical purity (95% ee) clearly resulted from the occurrence of a kinetic amplification process during the desymmetrization of 40 [71]. In a subsequent work [72], the same authors screened a series of ionic liquids in different ratios to TBME as reaction medium and they found that the discriminating ability of lipase increased in the presence of hydrophobic 1-butyl-3-methylimidazolium hexafluorophosphate salt ([BMIM][PF6]). In addition, the formation of the diester product was markedly suppressed and further improvement in the enantioselectivity was achieved changing vinyl acetate with vinyl butyrate as acyl donor, leading to the isolation of (R)-41b in excellent chemical and optical yield (Figure 18).
In the de novo synthesis of EFdA, a potent inhibitor of nucleoside reverse transcriptase with high potential in antiretroviral therapy, the desymmetrization of glycerol derivative 42 was retrosynthetically recognized as a key step for the installation of the fully substituted carbinol stereocentre within the molecule. Among the different tested enzymes, lipase A from C. antarctica (CAL-A), was able to catalyze the formation of (R)-43 in very good optical purity without significant over-reaction to the corresponding free triol. However, the enzyme selectivity was found to be dependent by pH and optimal performances were observed in the pH range 4.5–5.6 in the presence of methanol as co-solvent [73] (Figure 19).
3.2. Reactions Catalyzed by Other Enzymes than Lipases
Although oxidation of alcohols catalyzed by purified oxidase or microorganisms is recognized as an important tool in sustainable biotransformations [74], the application of this strategy to the desymmetrization of prochiral diols is rather limited. A systematic study on the desymmetrization of 2-alkyl propane-1,3-diols 44a–g was recently carried out by Brenna et al. [75], that employed Acetobacter aceti MIM 2000/28 as biocatalyst taking advantage from the natural ability of this microorganism to work in acidic medium and tolerate high concentrations of carboxylic acids. It is known that this type of bacteria oxidizes alcohols through a two-step transformation, the first of which involves an alcohol dehydrogenase to give rise to an aldehyde intermediate, then transformed into the final carboxylic acid by means of an aldehyde dehydrogenase.
As could be expected, the biocatalyzed oxidation of substrates 44a–g to the chiral hydroxyacids 45a–g proceeded with different selectivity and reaction rate, depending on the nature of the substituent in the C-2 position of the molecule. With sterically demanding substituents, as iso-butyl (a branched alkyl chain) and phenyl groups, the substrates were recovered unreacted, indicating that the access to the catalytic sites of both the involved enzymes is fully inhibited. Satisfactory results in terms of enantioselectivity were observed with substrates 44a, 44f and 44g, the latter one obtained in moderate chemical yield, but an inversion of the absolute configuration of the corresponding acids was observed going from 45a to 45f. These substrates could be considered the extreme points for that concerns the difference in the steric hindrance between the C-2 substituent and the reactive hydroxymethyl group, a factor that seems to drive the selectivity of the microbial oxidation and could also explain the results observed for the intermediate cases of diols 44b–e (Figure 20).
In a further development, this oxidation reaction was adapted for a continuous flow synthesis of the angiotensin converting enzyme (ACE) inhibitor captopril starting from diol 44a [76]. The A. aceti catalyst was immobilized as alginate-entrapped dried cells and in this form was used to pack a glass column reactor inside which a solution of 44a in acetate buffer and an oxygen stream were let to flow. Due to the high local concentration of the biocatalyst in the reactor, 95% conversion of the substrate was reached in 10 min. Following immobilization, the enantioselectivity of the biocatalyst was maintained and 45a with 96–97% ee was obtained. Furthermore, the alginate beads showed good stability over the time, giving the same substrate conversion for about 10 h of continuous work. At the end of the reaction the product 45a was separated from the unreacted substrate 44a by flowing through an anionic resin, from which it was released by elution with HCl. Even the subsequent reaction steps were optimized for in-flow conditions and coupled with in-line separation of co-products, by-products, and excess reagents to ultimately give captopril in 50% global yield (Figure 21).
In the course of their intensive study on biocatalytic reactions catalyzed by whole cells of the amidase-containing bacterium R. erythropolis AJ270, Ao and coworkers investigated the desymmetrization of a series of prochiral 3-aryl and 3-arylmethylglutaramides [77] and put in evidence a marked effect of the “remote” aromatic substituent on the catalytic activity and selectivity.
Indeed, in the group of 3-arylglutaramides 46, which all required long reaction times (72–168 h) for sufficient substrate conversion, the presence of a para-halogen substituent (R= 4-Br, 4-Cl) led to nearly racemic products 47 in moderate chemical yield whereas good conversion of the substrates (87–91%) coupled with low optical purity (21–41% ee) of the corresponding monoamides were observed with unsubstituted (R= H) and meta-substituted (R= 3-Br, 3-Me) phenyl derivatives. Ortho-substituents (R= 2-Br, 2-Cl, 2-Me) beneficially affected the reaction giving the expected products in excellent chemical (90–95%) and enantiomeric (>99.5%) yields, suggesting a role of the steric hindrance in aiding the discrimination of the two enantiotopic amide groups. However, this behavior was not general since the 2-OMe derivative gave sluggish reaction without selectivity (30% yield, 5% ee).
On the contrary, in the series of 3-arylmethylglutaramides 48 high substrate conversion (84–92%) and complete sterocontrol was observed in most cases, independently from the nature and position of the substituent on the phenyl ring (R = H, 2-Br, 3-Br, 4-Br, 4-Cl, 4-F, 4-Me), leading to enantiopure R-monoamides 49 (>99.5% ee) with the exception of substrates with R= 4-CF3 and 4-OMe that gave products with slightly lower optical purity (77% and 91% ee, respectively). However, the 2-Br substituted glutaramide required longer reaction time (65 h) compared to the other substrates in the series, that were fully converted within 8–28 h. These results suggested that the C-2 benzyl groups in derivatives 48, less bulky and more flexible in comparison with the phenyl ones present in 46, allows a more efficient accommodation of the substrates in the active site of amidase, resulting in increased chiral discrimination ability of the enzyme and lowered dependence on the remote substituents. Both series of monoamides 47 and 49 displayed R-configuration, indicating a stereopreference of the microbial amidase for the pro-R enantiotopic amide group (Figure 22).
4. Biocatalyzed Desymmetrization of Diketones
Ketones are an important class of prochiral substrates that can be converted into chiral compounds by the stereoselective attack of a given reagent on one side or the other one of the plane containing the carbonyl group and an efficient face differentiation ultimately results in the formation of one enantiomer predominantly (see Figure 1B). However, in this section we will limit the discussion to symmetric diketones bearing also a prochiral carbon centre, for which both enantiotopic and enantiofacial discriminations are involved in the formation of a single optically active product.
Indeed, for a generic diketone 50 two alcohols, 51 or 52, with opposite configuration at the initially prochiral centre of the substrate can be formed depending on the enantiotoposelective stereopreference of the catalyst and each one of them could be obtained as a single isomer or as a diastereomeric mixture of 51a/51b or 52a/52b according to the catalyst ability to differentiate the two faces of the involved ketone group. Although an effective enantiotopic differentiation is sufficient to obtain optically active products after separation of their diastereoisomeric mixtures with conventional procedures, the formation of products as single isomers is often observed so that the desymmetrization of ketones is an important tool in synthetic chemistry for the generation of two stereogenic carbons from relatively simply and achiral molecules (Figure 23).
The biocatalytic reduction of ketones with purified enzymes, as reductases or alcohol dehydrogenases, has been widely exploited for the production optically active alcohols [78]. These enzymes require a cofactor, NADH or NADPH, from which a hydride is transferred to the carbonyl group of the substrate, and a hydrogen source to recycle the cofactor from its oxidized form NAD+ or NADP+. As an alternative, the use of microbial whole cells can be advantageous since it overcomes the need of enzyme purification and can supply the complementary enzymes for the cofactor regeneration. However, some limits in substrate and product diffusion through the cell membrane may decrease the reaction rate and the co-existence of different cellular enzymes can influence the selectivity [58].
Among the microorganisms that display reducing ability the yeast Saccharomyces cerevisiae, which has been used for millennia in the fermentation of food and beverages, has gained huge popularity in chemical synthesis for its ready availability, acceptance of a variety of carbonyl-containing substrates and tolerance to different reaction conditions [79,80].
In the stereocontrolled total synthesis of paspaline, a natural diterpene alkaloid with potent activity as potassium channel antagonist, the desymmetrization of diketone 53 through its enantiotoposelective monoreduction, coupled with excellent face discrimination, allowed to introduce the proper stereochemistry at the stereogenic carbons of ketoalcohol 54 and, simultaneously, the suitable functional group for assembling the pyran ring [81] (Figure 24).
For the selective reduction of ethylsecodione 55, Romano and coworkers screened different microorganisms and they found that S. cerevisiae CEN.PK113-7D and Pichia minuta CBS 1708 catalyzed the formation of (13R,17S)-56 with ee >90% whereas Pichia glucozyma CBS 5766 and Rhodotorula glutinis NRRL 1587 displayed opposite enantiotoposelectivity giving (13S,17S)-56 in 94% and 90% ee respectively. In all these cases the conversion of substrate was in the 78–85% range but the reaction rate was remarkable high for P. glucozyma CBS 5766 (2 h) compared to the other microorganisms (24–48 h). By optimizing the experimental conditions, the reduction in the presence of S. cerevisiae reached outstanding levels of enantioselectivity (ee >98%), comparable with those obtained with a purified recombinant ketoreductase from P. glucozyma (KRED1-PGlu), also giving high chemical yield of the target alcohol. With both established stereocenters, the alcohol (13R,17S)-56 is a key intermediate in the synthesis of the second-generation oral contraceptives, as desogestrel, levonorgestrel or gestodene, through the construction of the steroidal A/B bicyclic core [82].
Ketone 55 was selectively reduced also in continuous-flow conditions by using a mixed bed system composed by ketoreductase KRED1-Pglu and glucose dehydrogenase from Bacillus megaterium (BmGDH) for cofactor regeneration, each one separately immobilized on agarose, as biocatalyst. In the alkaline conditions (pH ≥10.0) required for the reaction with aldehyde-activated agarose, some loss of activity was observed for BmGDH enzyme, even in the presence of 20% glycerol as stabilizing additive, whereas KRED1-Pglu fully retained its initial activity. Notably, the enzyme stability towards DMSO, a co-solvent often required to ensure the solubilization of the substrates, markedly increased after immobilization. The packed bed reactor, filled with immobilized KRED1-Pglu and BmGDH in 1:25 ratio, was fed with a buffer/DMSO solution containing 55, NADP+ and glucose to give (13R,17S)-56 in 65% yield after a residence time of 3h and no significant changes of the composition of the outflow stream were observed for 15 days [83] (Figure 25).
5. Desymmetrization of Challenging Substrates
In this section three examples of desymmetrization of substrates with peculiar structural features are presented, one for each of the groups of symmetric molecules considered in this review, as a further evidence of the versatility and synthetic potential of biocatalyzed reactions.
Meso-compound 57 and prochiral biphenyl 60 can be viewed as a challenging substrate for lipases due to the phenolic nature of hydroxyl groups, which display different reactivity compared with the alcohol ones and are not directly bonded to stereogenic carbons. Although lipase-catalyzed reactions have been applied for the preparation of regioselectively acylated polyphenols [84,85], there are few reports on enantioselective processes based on the functionalization of the phenol group(s) [86,87].
In the development of an efficient synthesis of yimitasvir (DAG 181), a novel inhibitor of nonstructural protein 5A (NS5A) with promising potential in the therapy of hepatitis C infection [88], the use of a chiral starting material was crucial to overcome the limits in the yield related with the separation at the late stage of the synthesis of the diastereomeric products obtained from functionalization of 57.
After a preliminary screening of lipases and the selection of the optimal diester as substrate, Feng and coworkers [89] tested the hydrolysis of dipropionate derivative 57a in the presence of lipase from P. fluorescens (AK Amano) and a careful tuning of the reaction was carried out by monitoring the effects of an organic co-solvent to improve substrate solubilization, the pH of the buffered solution, the temperature and the amount of base additive for effective adjustment of pH throughout the reaction. In the best conditions, in ethyl acetate: buffer pH 7.0 1:1 at 15 °C and in the presence of 0.9–1.1 eqv. of NaHCO3, monopropionate 58 was obtained in excellent chemical and optical yields, which were maintained also in a 30 L scale-up of the reaction at the industrially acceptable concentration of 57 (110 g/L) and 0.2% enzyme loading (Figure 26).
The 1,1′-biaryl structural motif is widely diffused in natural and biologically active molecules [90,91], many of which are axially chiral in consequence of a high energy barrier to rotation around the biaryl bond that prevents the interconversion between the different conformers. Depending on the size and the number of substituents, the rotation around the 1,1′-bond of a generic biphenyl 59 could be hampered and two enantiomers (called atropisomers) with opposite mutual disposition of the substituents with respect the axis joining the two aryl rings and axial chirality M or P exist as distinct and configurationally stable compounds (Figure 27).
A variety of synthetic methodologies have been developed for the atroposelective coupling of different aryl substrates [92,93,94], but some interest has been also directed to the kinetic resolution of the racemates [95,96] or desymmetrization of σ-symmetric biphenyls [97,98,99]. In σ-symmetric biphenyls, two identical substituents in the 2- and 5-positions of the same phenyl ring are related by a symmetry plane containing the other phenyl ring and are enantiotopic, so that the molecule becomes axially chiral following their stereoselective differentiation (Figure 28).
Desymmetrization of compounds 60a–f has been recently reported by Akai and coworkers [100] as the first example of enantiotoposelective lipase-catalyzed esterification of σ-symmetric biphenyl diols. Different lipases were able to catalyze the reaction in organic solvent with vinyl acetate as acyl donor and in the optimal conditions, with immobilized B. cepacia lipase (Amano PS-IM) in toluene, (S)-monoacetates 61a–f were obtained in excellent enantiopurity, resulting from both the stereoselectivity of the enzyme and the contribute of kinetic amplification phenomenon through the formation of the optically inactive diacetates 62a–f (4–18%). Complete conversion of the substrates was observed within 26 h and the presence of Na2CO3 was shown essential in accelerating the reaction [101]. The same lipase was also active in promoting the complementary hydrolysis reaction of diacetates 62a–f leading to (R)-monoacetates 61a–f in enantiopure form as exclusive products (Figure 29).
The enantioselective transformation of molecules bearing a “remote stereocentre”, defined as a stereogenic centre separated by at least three atoms or an aromatic group from the group to be transformed, is a challenging task in organic synthesis and an effective stereocontrol is more difficult to achieve in acyclic systems due to the lack of conformational constraints. Since the possibility of solving remote centres is a valuable tool to introduce chirality in any part of a synthetic route, it has attracted interest especially for its relevance in the synthesis of complex biologically active molecules [102,103].
Biocatalysis is a valuable methodology for kinetic resolution or desymmetrization of remote stereocentres [104,105] because the stereochemical outcome of the reactions is governed through specific conformations of the enzyme–substrate complex as a whole and even “remote” stereocentres can give rise to sufficiently differentiated interactions between the substrate and the catalytic site of the enzyme.
Diketone 63, bearing two enantiotopic reactive carbonyl groups three bonds far away from the pro-stereogenic carbon, is an interesting example of molecule with remote stereocentres whose desymmetrization has been recently investigated by Skoupi et al. [106] in the course of their study of a synthetic strategy to acyclic meso-diols. A series of reductases from S. cerevisiae, Homo sapiens and Lactobacillus kefir were cloned, expressed and purified, then tested in the reduction of 63 in the presence of a recombinant glucose-dehydrogenase from Bacillus subtilis for cofactor recycling.
Six out of the nine screened reductases from S. cerevisiae were selective for quantitative monoreduction of 63 giving (5S,8S)-64 with remarkable diastereoselectivity (anti:syn > 95:1) and stereopreference for the (S)-configured alcohol. On the contrary, reductase from L. kefir showed low enantiotoposelectivity giving two diastereoisomeric alcohols (5S,8R)-64 and (5R, 8R)-64 in 60:40 ratio, possibly due to negligible discriminating interactions with the nitro group, but the opposite stereopreference of the enzyme toward the formation of (R)-configured alcohol was exploited for the sequential reduction of 63 to give meso-anti,anti-(2R,5s,8R)-diol 65 (Figure 30).
6. Conclusions
The biocatalytic approach to the desymmetrization of meso- or prochiral compounds is nowadays an important tool in the synthesis of chiral molecules, offering in many cases a more valuable and sustainable alternative to chemical catalysis. Thanks to their activity both in aqueous medium or in organic solvent and the complementary reactions that they catalyze, lipases are still the most employed enzymes for stereoselective modifications of a large variety of molecular scaffolds. The portfolio of biocatalytic reactions is continuously expanding and even more selective enzymes are available through recombinant techniques. On the other hand, the use of whole microbial cells is attracting growing interest for the possibility to exploit multiple enzymatic reactions with a single catalyst, so reducing demand of cost, chemicals and time. Although productivity of enzymatic reactions has been often considered too low for large-scale applications, improvement in this field are expected through the use of immobilization techniques coupled to in-flow reactors.
Author Contributions
Conceptualization, Data Curation, Writing Review & Editing, A.P. and C.S. Writing Original Draft Preparation, A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Dedicated to the memory of our colleague Giovanni Nicolosi.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crossley, R. The relevance of chirality to the study of biological activity. Tetrahedron 1992, 48, 8155–8178. [Google Scholar] [CrossRef]
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. Development of New Stereoisomeric Drugs; U.S. Food & Drug: Washington, DC, USA, 1992. Available online: https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm122883.html (accessed on 7 April 2017).
- Mannschreck, A.; Kiesswetter, R.; von Angerer, E. Unequal activities of enantiomers via biological receptors: Examples of chiral drug, pesticide, and fragrance molecules. J. Chem. Educ. 2007, 84, 2012–2018. [Google Scholar] [CrossRef]
- Prusov, E.V. Stereoselective Synthesis of Drugs and Natural Products; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 1–1836. [Google Scholar] [CrossRef]
- Willis, M.C. Enantioselective desymmetrization. J. Chem. Soc. Perkin Trans. 1 1999, 1765–1784. [Google Scholar] [CrossRef]
- Merad, J.; Candy, M.; Pons, J.-M.; Bressy, C. Catalytic enantioselective desymmetrization of Meso compounds in total synthesis of natural products: Towards an economy of chiral reagents. Synth. Stuttg. 2017, 49, 1938–1954. [Google Scholar] [CrossRef] [Green Version]
- IUPAC. Basic terminology of stereochemistry. Pure Appl. Chem. 1996, 68, 2193–2222. [Google Scholar] [CrossRef]
- Vigneron, J.P.; Dhaenens, M.; Horeau, A. Nouvelle methode pour porter au maximum la purete optique d’un produit partiellement dedouble sans l’aide d’aucune substance chirale. Tetrahedron 1973, 29, 1055–1059. [Google Scholar] [CrossRef]
- Garcìa-Urdiales, E.; Alfonso, I.; Gotor, V. Enantioselective enzymatic desymmetrizations in organic synthesis. Chem. Rev. 2005, 105, 313–354. [Google Scholar] [CrossRef]
- Borissov, A.; Davies, T.Q.; Ellis, S.R.; Fleming, T.A.; Richardson, M.S.W.; Dixon, D.J. Organocatalytic enantioselective desymmetrization. Chem. Soc. Rev. 2016, 45, 5474–5540. [Google Scholar] [CrossRef]
- Suzuki, T. Recent topics in the desymmetrization of meso-diols. Tetrahedron Lett. 2017, 58, 4731–4739. [Google Scholar] [CrossRef]
- Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Catalytic enantioselective desymmetrization reactions to all-carbon quaternary stereocenters. Chem. Rev. 2016, 116, 7330–7396. [Google Scholar] [CrossRef]
- Patel, R.N. Biocatalysis: Synthesis of key intermediates for development of pharmaceuticals. ACS Catal. 2011, 1056–1074. [Google Scholar] [CrossRef]
- Bode, S.E.; Wolberg, M.; Müller, M. Stereoselective synthesis of 1,3-diols. Synthesis 2006, 4, 557–588. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D. Broadening the scope of biocatalysis in sustainable organic synthesis. ChemSusChem 2019, 12, 2859–2881. [Google Scholar] [CrossRef]
- Zeymer, C.; Hilvert, D. Directed evolution of protein catalysts. Annu. Rev. Biochem. 2018, 87, 131–157. [Google Scholar] [CrossRef]
- Reetz, M.T. Lipases as practical biocatalysts. Curr. Opin. Chem. Biol. 2002, 6, 145–150. [Google Scholar] [CrossRef]
- Gotor-Fernández, V.; Brieva, R.; Gotor, V. Lipases: Useful biocatalysts for the preparation of pharmaceuticals. J. Mol. Catal. B Enzym. 2006, 40, 111–120. [Google Scholar] [CrossRef]
- Angajala, G.; Pavan, P.; Subashini, R. Lipases: An overview of its current challenges and prospectives in the revolution of biocatalysis. Biocatal. Agric. Biotechnol. 2016, 7, 257–270. [Google Scholar] [CrossRef]
- Carrea, G.; Riva, S. Medium Engineering. In Asymmetric Organic Synthesis with Enzymes; Gotor, V., Alfonso, I., García-Urdiales, E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; Chapter 1; pp. 3–18. [Google Scholar]
- Castillo, E.; Casas-Godoy, L.; Sandoval, G. Medium-engineering: A useful tool for modulating lipase activity and selectivity. Biocatalysis 2015, 178–188. [Google Scholar] [CrossRef]
- Quilles, J.C.J.; Brito, R.R.; Borges, J.P.; Aragonc, C.C.; Fernandez-Lorente, G.; Bocchini-Martins, D.A.; Gomes, E.; da Silvaa, R.; Boscoloa, M.; Guisan, J.M. Modulation of the activity and selectivity of the immobilized lipases by surfactants and solvents. Biochem. Eng. J. 2015, 93, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Christena, R.L.; Rajaram, Y.R.S. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zoumpanioti, M.; Stamatis, H.; Xenakis, A. Microemulsion-based organogels as matrices for lipase immobilization. Biotechnol. Adv. 2010, 28, 395–406. [Google Scholar] [CrossRef]
- Rodriguesa, R.C.; Virgen-Ortízb, J.J.; dos Santosc, J.C.S.; Berenguer-Murciad, Á.; Alcantarae, A.R.; Barbosaf, O.; Ortizg, C.; Fernandez-Lafuente, R. Immobilization of lipases on hydrophobic supports: Immobilization mechanism, advantages, problems, and solutions. Biotechnol. Adv. 2019, 37, 746–770. [Google Scholar] [CrossRef] [Green Version]
- Uppenberg, J.; Oehrner, N.; Norin, M.; Hult, K.; Kleywegt, G.J.; Patkar, S.; Waagen, V.; Anthonsen, T.; Jones, T.A. Crystallographic and molecular-modeling studies of lipase B from Candida antarctica reveal a stereospecificity pocket for secondary alcohols. Biochemistry 1995, 34, 16838–16851. [Google Scholar] [CrossRef]
- Lang, D.A.; Mannesse, M.L.M.; De Haas, G.H.; Verheij, H.M.; Dijkstra, B.W. Structural basis of the chiral selectivity of Pseudomonas cepacia lipase. Eur. J. Biochem. 1998, 254, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.I.; Lan, D.; Durrani, R.; Huan, W.; Zhao, Z.; Wang, Y. The lid domain in lipases: Structural and functional determinant of enzymatic properties. Front. Bioeng. Biotechnol. 2017, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Theil, F.; Schick, H.; Lapitskaya, M.A.; Pivnitsky, K.K. Enzymes in organic synthesis, 4. Investigation of the pancreatin-catalyzed acylation of cis-cyclopent-2-ene-1,4-diol with various trichloroethyl and vinyl alkanoates. Liebigs Annal. Chem. 1991, 195–200. [Google Scholar] [CrossRef]
- Ghorpade, S.R.; Kharul, R.K.; Joshi, R.R.; Kalkote, U.R.; Ravindranathan, T. Desymmetrization of meso-cyclopenten-cis-1,4-diol to 4-(R)-hydroxycyclopent-2-en-1-(S)-acetate by irreversible transesterification using Chirazyme®. Tetrahedron Asymmetry 1999, 891–899. [Google Scholar] [CrossRef]
- Sharifabad, M.E.; Hodgson, B.; Jellite, M.; Mercer, T.; Sen, T. Enzyme immobilised novel core-shell superparamagnetic nanocomposites for enantioselective formation of 4-(R)-hydroxycyclopent-2-en-1-(S)-acetate. Chem. Commun. 2014, 50, 11185–11187. [Google Scholar] [CrossRef]
- Hinze, J.; Süss, P.; Strohmaier, S.; Bornscheuer, U.T.; Wardenga, R.; von Langermann, J. Recombinant pig liver esterase-catalyzed synthesis of (1S,4R)-4-hydroxy-2-cyclopentenyl acetate combined with subsequent enantioselective crystallization. Org. Process Res. Dev. 2016, 20, 1258–1264. [Google Scholar] [CrossRef]
- Putta, S.; Reddy, A.M.; Shelu, G.R.; Reddy, B.V.S.; Kumaraguru, T. Preparation of (1R,4S)-4-hydroxycyclopent-2-en-1-yl acetate via Novozym-435® catalyzed desymmetrization of cis-3,5-diacetoxy-1-cyclopentene. Tetrahedron 2018, 74, 6673–6679. [Google Scholar] [CrossRef]
- Moni, L.; Banfi, L.; Cartagenova, D.; Cavalli, A.; Lambruschini, C.; Martino, E.; Orru, R.V.A.; Ruijter, E.; Saya, J.M.; Sgrignani, J.; et al. Zinc (II)-mediated diastereoselective Passerini reactions of biocatalytically desymmetrised renewable inputs. Org. Chem. 2020, 7, 380–398. [Google Scholar] [CrossRef]
- Estermann, H.; Prasad, K.; Shapiro, M.J.; Bolsterli, J.J.; Walkinshaw, M.D. Enzyme-catalyzed asymmetrization of bis(hydroxymethyl)-tetrahydrofurans. Tetrahedron Lett. 1990, 31, 445–448. [Google Scholar] [CrossRef]
- Naemura, K.; Fukuda, R.; Konishi, N.M.; Hirose, Y.; Tobe, Y. Enzyme-catalyzed asymmetric acylation and hydrolysis of cis-2,5-disubstituted tetrahydrofuran derivatives: Contribution to development of models for reactions catalyzed by porcine liver esterase and porcine pancreatic lipase. Tetrahedron Asymmetry 1993, 4, 911–918. [Google Scholar] [CrossRef]
- Moni, L.; Banfi, L.; Basso, A.; Carcone, L.; Rasparini, M.; Riva, R. Ugi and Passerini reactions of biocatalytically derived chiral aldehydes: Application to the synthesis of bicyclic pyrrolidines and of antiviral agent telaprevir. J. Org. Chem. 2015, 80, 3411–3428. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Sarkar, A. Enantioselective syntheses of (–)-alloyohimbane and (–)-yohimbane by an efficient enzymatic desymmetrization process. Eur. J. Org. Chem. 2016, 6001–6009. [Google Scholar] [CrossRef] [Green Version]
- Wirz, B.; Idinga, H.; Hilpert, H. Multiselective enzymatic reactions for the synthesis of protected homochiral cis- and trans-1,3,5-cyclohexanetriols. Tetrahedron Asymmetry 2000, 11, 4171–4178. [Google Scholar] [CrossRef]
- González-García, T.; Verstuyf, A.; Verlinden, L.; Fernández, S.; Ferrero, M. Enzymatic desymmetrization of 19-nor-vitamin D3 a-ring synthon precursor: Synthesis, structure elucidation, and biological activity of 1α,25-Dihydroxy-3-epi-19-nor-vitamin D3 and 1β,25-dihydroxy-19-nor-vitamin D3. Adv. Synth. Catal. 2018, 360, 2762–2772. [Google Scholar] [CrossRef]
- Ribeiro, M.F.P.; Pais, K.C.; de Jesus, B.S.M.; Fernandez-Lafuente, R.; Freire, D.M.G.; Manoel, E.A.; Simas, A.B.C. Lipase regioselective O-acetylations of a myo-inositol derivative: Efficient desymmetrization of 1,3-di-O-benzyl-myo-inositol. Eur. J. Org. Chem. 2018, 386–391. [Google Scholar] [CrossRef]
- Liang, J.; Cochran, J.E.; Dorsch, W.A.; Davies, I.; Clark, M.P. Development of a scalable synthesis of an azaindolyl-pyrimidine inhibitor of influenza virus replication. Org. Process Res. Dev. 2016, 20, 965–969. [Google Scholar] [CrossRef]
- Matsuda, T.; Yamanaka, R.; Nakamura, K. Recent progress in biocatalysis for asymmetric oxidation and reduction. Tetrahedron Asymmetry 2009, 20, 513–557. [Google Scholar] [CrossRef]
- Martínez, A.T.; Ruiz-Dueñas, F.J.; Camarero, S.; Serrano, A.; Linde, D.; Lund, H.; Vind, J.; Tovborg, M.; Herold-Majumdar, O.M.; Hofrichter, M.; et al. Oxidoreductases on their way to industrial biotransformations. Biotechnol. Adv. 2017, 35, 815–831. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Fernández-Fueyo, E.; Hollmann, F.; Paul, C.E.; Pesic, M.; Schmidt, S.; Wang, Y.; Younes, S.; Zhang, W. Biocatalytic oxidation reactions: A chemist’s perspective. Angew. Chem. Int. Ed. 2018, 57, 9238–9261. [Google Scholar] [CrossRef]
- Kroutil, W.; Mang, H.; Edegger, K.; Faber, K. Recent advances in the biocatalytic reduction of ketones and oxidation of sec-alcohols. Curr. Opin. Chem. Biol. 2004, 8, 120–126. [Google Scholar] [CrossRef]
- Musa, M.M.; Phillips, R.S. Recent advances in alcohol dehydrogenase-catalyzed asymmetric production of hydrophobic alcohols. Catal. Sci. Technol. 2011, 1, 1311–1323. [Google Scholar] [CrossRef]
- Nealon, C.M.; Musa, M.M.; Patel, J.M.; Phillips, R.S. Controlling substrate specificity and stereospecificity of alcohol dehydrogenases. ACS Catal. 2015, 5, 2100–2114. [Google Scholar] [CrossRef]
- Zheng, Y.G.; Yin, H.H.; Yu, D.F.; Chen, X.; Tang, X.-L.; Zhang, X.-J.; Xue, Y.-P.; Wang, Y.-J.; Liu, Z.-Q. Recent advances in biotechnological applications of alcohol dehydrogenases. Appl. Microbiol. Biotechnol. 2017, 101, 987–1001. [Google Scholar] [CrossRef]
- Jones, J.B. Horse Liver Alcohol Dehydrogenase: An Illustrative Example of the Potential of Enzymes in Organic Synthesis. In Enzyme Engineering; Chibata, I., Fukui, S., Wingard, L.B., Eds.; Springer: Boston, MA, USA, 1982; pp. 107–116. [Google Scholar] [CrossRef]
- De Smidt, O.; Du Preez, J.C.; Albertyn, J. The alcohol dehydrogenases of Saccharomyces cerevisiae: A comprehensive review. FEMS Yeast Res. 2008, 8, 967–978. [Google Scholar] [CrossRef] [Green Version]
- Van der Donk, W.A.; Zhao, H. Recent developments in pyridine nucleotide regeneration. Curr. Opin. Biotechnol. 2003, 14, 421–442. [Google Scholar] [CrossRef]
- Hollmann, F.; Arends, I.W.C.E.; Buehler, K. Biocatalytic redox reactions for organic synthesis: Nonconventional regeneration methods. ChemCatChem 2010, 2, 762–782. [Google Scholar] [CrossRef]
- Hildebrandt, P.; Musidlowska, A.; Bornscheuer, U.T.; Altenbuchner, J. Cloning, functional expression and biochemical characterization of a stereoselective alcohol dehydrogenase from Pseudomonas fluorescens DSM50106. Appl. Microbiol. Biotechnol. 2002, 59, 483–487. [Google Scholar] [CrossRef]
- Pennacchio, A.; Pucci, B.; Secundo, F.; La Cara, F.; Rossi, M.; Raia, C.A. Purification and characterization of a novel recombinant highly enantioselective short-chain NAD(H)-dependent alcohol dehydrogenase from Thermus thermophiles. Appl. Environ. Microbiol. 2008, 74, 3949–3958. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, K.; Schroer, K.; Lütz, S.; Liese, A. Biocatalytic ketone reduction-A powerful tool for the production of chiral alcohols-Part II: Whole-cell reductions. Appl. Microbiol. Biotechnol. 2007, 76, 249–255. [Google Scholar] [CrossRef]
- Carballeira, J.D.; Quezada, M.A.; Hoyos, P.; Simeó, Y.; Hernaiza, M.J.; Alcantara, A.R.; Sinisterra, J.V. Microbial cells as catalysts for stereoselective red-ox reactions. Biotechnol. Adv. 2009, 27, 686–714. [Google Scholar] [CrossRef]
- Cui, Z.; Zhao, Y.; Mao, Y.; Shi, T.; Lu, L.; Ma, H.; Wanga, Z.; Chen, T. In vitro biosynthesis of optically pure D-(−)-acetoin from meso-2,3-butanediol using 2,3-butanediol dehydrogenase and NADH oxidase. J. Chem. Technol. Biotechnol. 2019, 94, 2547–2554. [Google Scholar] [CrossRef]
- Holec, C.; Sandkuhl, D.; Rother, D.; Kroutil, W.; Pietruszka, J. Chemoenzymatic synthesis towards the active agent travoprost. ChemCatChem 2015, 7, 3125–3130. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Horn, M.; Martinez-Rojas, E.; Görisch, H.; Tressl, R.; Garbe, L.A. Oxidation of 1,4-alkanediols into γ-lactones via γ-lactols using Rhodococcus erythropolis as biocatalyst. J. Mol. Catal. B Enzym. 2007, 49, 24–27. [Google Scholar] [CrossRef]
- Martinez-Rojas, E.; Olejniczak, T.; Neumann, K.; Garbe, L.-A.; Boratyński, F. Simple preparation of Rhodococcus erythropolis DSM 44534 as biocatalyst to oxidize diols into the optically active lactones. Chirality 2016, 28, 623–627. [Google Scholar] [CrossRef]
- Sugai, T.; Yamazaki, T.; Yokoyama, M.; Ohta, H. Biocatalysis in organic synthesis: The use of nitrile- and amide-hydrolyzing microorganisms. Biosci. Biotechnol. Biochem. 1997, 61, 1419–1427. [Google Scholar] [CrossRef]
- Bhalla, T.C.; Kumar, V.; Kumar, V.; Thakur, N.S. Nitrile metabolizing enzymes in biocatalysis and biotransformation. Appl. Biochem. Biotechnol. 2018, 185, 925–946. [Google Scholar] [CrossRef]
- Wang, M.-X. Enantioselective biotransformations of nitriles in organic synthesis. Acc. Chem. Res. 2015, 48, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-J.; Chen, P.; Ao, Y.-F.; Wang, Q.-Q.; Wang, D.-X.; Wang, M.-X. Highly efficient biocatalytic desymmetrization of meso carbocyclic 1,3-dicarboxamides: A versatile route for enantiopure 1,3-disubstituted cyclohexanes and cyclopentanes. Org. Chem. Front. 2019, 6, 808–812. [Google Scholar] [CrossRef]
- Ao, Y.-F.; Wang, D.-X.; Zhao, L.; Wang, M.-X. Synthesis of quaternary-carbon-containing and functionalized enantiopure pentanecarboxylic acids from biocatalytic desymmetrization of meso-cyclopentane-1,3-dicarboxamides. Chem. Asian J. 2015, 10, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Khong, D.T.; Pamarthy, V.S.; Gallagher, T.; Judeh, Z.M.A. Chemoenzymatic synthesis of chiral 1-benzyl-5-(hydroxymethyl)-2-piperidone enabled by lipase AK-mediated desymmetrization of prochiral 1,3-diol and its diacetate. Eur. J. Org. Chem. 2016, 3084–3089. [Google Scholar] [CrossRef] [Green Version]
- Bhuniya, R.; Nanda, S. Asymmetric total synthesis of (–)-rasfonin. Tetrahedron 2013, 69, 1153–1165. [Google Scholar] [CrossRef]
- Tokairin, Y.; Konno, H. Preparation of (2R, 3R, 4R)-3-hydroxy-2,4,6-trimethylheptanoic acid via enzymatic desymmetrization. Tetrahedron 2017, 73, 39–45. [Google Scholar] [CrossRef]
- Kołodziejskaa, R.; Kwit, M.; Studzinska, R.; Jelecki, M. Enantio- and diastereoselective acylation of prochiral hydroxyl group of pyrimidine acyclonucleosides. J. Mol. Catal. B Enzym. 2016, 133, 98–106. [Google Scholar] [CrossRef]
- Kołodziejska, R.; Studzińska, R.; Pawluk, H. Lipase-catalyzed enantioselective transesterification of prochiral 1-((1,3-dihydroxypropan-2-yloxy)methyl)-5,6,7,8-tetrahydroquinazoline-2,4 (1H,3H)-dione in ionic liquids. Chirality 2018, 30, 206–214. [Google Scholar] [CrossRef]
- McLaughlin, M.; Kong, J.; Belyk, K.M.; Chen, B.; Gibson, A.W.; Keen, S.P.; Lieberman, D.R.; Milczek, E.M.; Moore, J.C.; Murray, D.; et al. Enantioselective synthesis of 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) via enzymatic desymmetrization. Org. Lett. 2017, 19, 926–929. [Google Scholar] [CrossRef]
- Romano, D.; Villa, R.; Molinari, F. Preparative biotransformations: Oxidation of alcohols. ChemCatChem 2012, 4, 739–749. [Google Scholar] [CrossRef]
- Brenna, E.; Cannavale, F.; Crotti, M.; De Vitis, V.; Gatti, F.G.; Migliazza, G.; Molinari, F.; Parmeggiani, F.; Romano, D.; Santangelo, S. Synthesis of enantiomerically enriched 2-hydroxymethylalkanoic acids by oxidative desymmetrisation of achiral 1,3-diols mediated by Acetobacter aceti. ChemCatChem 2016, 8, 3796–3803. [Google Scholar] [CrossRef] [Green Version]
- De Vitis, V.; Dall’Oglio, F.; Pinto, A.; De Micheli, C.; Molinari, F.; Conti, P.; Romano, D.; Tamborini, L. Chemoenzymatic synthesis in flow reactors: A rapid and convenient preparation of captopril. ChemistryOpen 2017, 6, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Ao, Y.-F.; Zhang, L.-B.; Wang, Q.-Q.; Wang, D.-X.; Wang, M.-X. Biocatalytic desymmetrization of prochiral 3-aryl and 3-arylmethyl glutaramides: Different remote substituent effect on catalytic efficiency and enantioselectivity. Adv. Synth. Catal. 2018, 360, 4594–4603. [Google Scholar] [CrossRef]
- Nakamura, K.; Yamanaka, R.; Matsuda, T.; Harada, T. Recent developments in asymmetric reduction of ketones with biocatalysts. Tetrahedron Asymmetry 2003, 14, 2659–2681. [Google Scholar] [CrossRef]
- Wolfson, A.; Dlugy, C.; Tavor, D. Baker’s yeast catalyzed asymmetric reduction of prochiral ketones in different reaction mediums. Org. Commun. 2013, 6, 1–11. [Google Scholar]
- De Souza Pereira, R. The use of baker’s yeast in the generation of asymmetric centers to produce chiral drugs and other compounds. Crit. Rev. Biotechnol. 1998, 18, 25–64. [Google Scholar] [CrossRef]
- Sharpe, R.J.; Johnson, J.S. A global and local desymmetrization approach to the synthesis of steroidal alkaloids: Stereocontrolled total synthesis of paspaline. J. Am. Chem. Soc. 2015, 137, 4968–4971. [Google Scholar] [CrossRef] [Green Version]
- Contente, M.L.; Molinari, F.; Serra, I.; Pinto, A.; Romano, D. Stereoselective enzymatic reduction of ethyl secodione: Preparation of a key intermediate for the total synthesis of steroids. Eur. J. Org. Chem. 2016, 1260–1263. [Google Scholar] [CrossRef]
- Dall’Oglio, F.; Contente, M.L.; Conti, P.; Molinari, F.; Monfredi, D.; Pinto, A.; Romano, D.; Ubiali, D.; Tamborini, L.; Serra, I. Flow-based stereoselective reduction of ketones using an immobilized ketoreductase/glucose dehydrogenase mixed bed system. Catal. Commun. 2017, 93, 29–32. [Google Scholar] [CrossRef]
- Chebil, L.; Humeau, C.; Falcimaigne, A.; Engasser, J.-M.; Ghoul, M. Enzymatic acylation of flavonoids. Process Biochem. 2006, 41, 2237–2251. [Google Scholar] [CrossRef]
- De Araújo, M.E.M.B.; Franco, Y.E.M.; Messias, M.C.F.; Longato, G.B.; Pamphile, J.A.; de Carvalho, P. Biocatalytic synthesis of flavonoid esters by lipases and their biological benefits. Planta Med. 2017, 7–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascoyne, D.G.; Finkbeiker, H.L.; Chan, K.P.; Gordon, J.L.; Stewart, K.R.; Kazlauskas, R.J. Molecular Basis for Enantioselectivity of Lipase from Chromobacterium viscosum toward the Diesters of 2,3-Dihydro-3-(4′-hydroxyphenyl)-1,1,3-trimethyl-1H-inden-5-ol. J. Org. Chem. 2001, 66, 3041–3048. [Google Scholar] [CrossRef]
- Lehmann, S.V.; Breinholt, J.; Bury, P.S.; Nielsen, T.E. Enzymatic resolution to (−)-ormeloxifene intermediates from their racemates using immobilized Candida rugosa lipase. Chirality 2000, 12, 568–573. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Xie, H.; Ren, Q.; Tan, Y.; Luo, H. Bridged Ring Compounds as Hepatitis C Virus (HCV) Inhibitors and Pharmaceutical Applications Thereof. U.S. Patent 0,193,44, 5 August 2013. [Google Scholar]
- Feng, Y.; Wang, Z.; Luo, Z.; Lai, J.; Xie, H.; Luo, Z.; Zhang, L.; Li, R.; Zhang, Y. Development of an efficient and scalable biocatalytic route to (1S,4R)-8-Hydroxy-1,2,3,4-tetrahydro-1,4-methano-naphthalen-5-yl propionate via enantioselective enzymatic desymmetrization of a prochiral diester. Org. Process Res. Dev. 2019, 23, 1243–1251. [Google Scholar] [CrossRef]
- Bringmann, G.; Günther, C.; Ochse, M.; Schupp, O.; Tasler, S. Biaryls in nature: A multi-facetted class of stereochemically, biosynthetically, and pharmacologically intriguing secondary metabolites. Fortschr. Chem. Org. Naturst. 2001, 82, 1–249. [Google Scholar] [CrossRef]
- Baudoin, O.; Guéritte, F. Natural Bridged Biaryls with Axial Chirality and Antimitotic Properties. In Studies in Natural Products Chemistry, 1st ed.; Atta-ur-Rahman, Ed.; Elsevier Science: Amsterdam, The Netherlands, 2003; Volume 29, part J; pp. 355–417. [Google Scholar] [CrossRef]
- Bringmannm, G.; Price, A.J.; Keller, M.P.A.; Gresser, M.J.; Garner, J.; Breuning, M. Atroposelective synthesis of axially chiral biaryl compounds. Angew. Chem. Int. Ed. 2005, 44, 5384–5427. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-B.; Tan, B. Construction of axially chiral compounds via asymmetric organocatalysis. Acc. Chem. Res. 2018, 51, 534–547. [Google Scholar] [CrossRef]
- Loxq, P.; Manoury, E.; Poli, R.; Deydier, E.; Labande, A. Synthesis of axially chiral biaryl compounds by asymmetric catalytic reactions with transition metals. Coord. Chem. Rev. 2016, 308, 131–190. [Google Scholar] [CrossRef]
- Ma, G.; Sibi, M.P. Catalytic kinetic resolution of biaryl compounds. Chem. Eur. J. 2015, 21, 11644–11657. [Google Scholar] [CrossRef]
- Moustafa, G.A.I.; Oki, Y.; Akai, S. Lipase-catalyzed dynamic kinetic resolution of C1-and C2-symmetric racemic axially chiral 2,2′-dihydroxy-1,1′-biaryls. Angew. Chem. Int. Ed. 2018, 57, 10278–10282. [Google Scholar] [CrossRef]
- Harada, T.; Tuyet, T.M.T.; Oku, A. Asymmetric desymmetrization of 2,2‘,6,6‘-tetrahydroxybiphenyl through annulation with enantiomerically pure bis (mesylate). Org. Lett. 2000, 2, 1319–1322. [Google Scholar] [CrossRef]
- Staniland, S.; Yuan, B.; Giménez-Agulló, N.; Marcelli, T.; Willies, S.C.; Grainger, D.M.; Turner, N.J.; Clayden, J. Enzymatic desymmetrising redox reactions for the asymmetric synthesis of biaryl atropisomers. Chem. Eur. J. 2014, 20, 13084–13088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, T.; Konegawa, T.; Nakamura, T.; Suzuki, K. Facile and highly enantioselective synthesis of axially chiral biaryls by enzymatic desymmetrization. Synlett 2002, 122–124. [Google Scholar] [CrossRef]
- Kasama, K.; Aoyama, H.; Akai, S. Enantiodivergent synthesis of axially chiral biphenyls from σ-symmetric 1,1’-biphenyl-2,6-diol derivatives by single lipase-catalyzed acylative and hydrolytic desymmetrization. Eur. J. Org. Chem. 2020, 654–661. [Google Scholar] [CrossRef]
- Moustafa, G.A.I.; Kasama, K.; Higashio, K.; Akai, S. Base-promoted lipase-catalyzed kinetic resolution of atropisomeric 1,1′-biaryl-2,2′-diols. RSC Adv. 2019, 9, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, A.G. Recent advances in acyclic stereocontrol. Tetrahedron 2011, 67, 9639–9667. [Google Scholar] [CrossRef]
- Chauhan, P.; Kaya, U.; Enders, D. Advances in organocatalytic 1,6-addition reactions: Enantioselective construction of remote stereogenic centers. Adv. Synth. Catal. 2017, 359, 888–912. [Google Scholar] [CrossRef]
- Alfaro Blasco, M.; Gröger, H. Enzymatic resolution of racemates with a ‘remote’ stereogenic center as an efficient tool in drug, flavor and vitamin synthesis. Bioorg. Med. Chem. 2014, 22, 5539–5546. [Google Scholar] [CrossRef]
- Cunha, R.L.O.R.; Ferreira, E.A.; Oliveira, C.S.; Omori, Á.T. Biocatalysis for desymmetrization and resolution of stereocenters beyond the reactive center: How far is far enough? Biotechnol. Adv. 2015, 33, 614–623. [Google Scholar] [CrossRef]
- Skoupi, M.; Vaxelaire, C.; Strohmann, C.; Christmann, M.; Niemeyer, C.M. Enantiogroup-differentiating biocatalytic reductions of prochiral Cs-symmetrical dicarbonyl compounds to meso compounds. Chem. Eur. J. 2015, 21, 8701–8705. [Google Scholar] [CrossRef]
Figure 1.
(a) General representation of prochiral molecules; (b) General representation of prochiral molecules with a trigonal group; (c) General representation of meso-molecules; (d) General representation of meso-molecules containing additional stereogenic centre in the symmetry plane.
Figure 1.
(a) General representation of prochiral molecules; (b) General representation of prochiral molecules with a trigonal group; (c) General representation of meso-molecules; (d) General representation of meso-molecules containing additional stereogenic centre in the symmetry plane.
Scheme 1.
Schematic representation of kinetic amplification in the desymmetrization of meso-compounds.
Scheme 1.
Schematic representation of kinetic amplification in the desymmetrization of meso-compounds.
Scheme 2.
Schematic representation of lipase-catalyzed reactions.
Scheme 3.
Preparation of both enantiomers of a target compound by desymmetrization of meso-substrates with lipase-catalyzed complementary reactions.
Scheme 3.
Preparation of both enantiomers of a target compound by desymmetrization of meso-substrates with lipase-catalyzed complementary reactions.
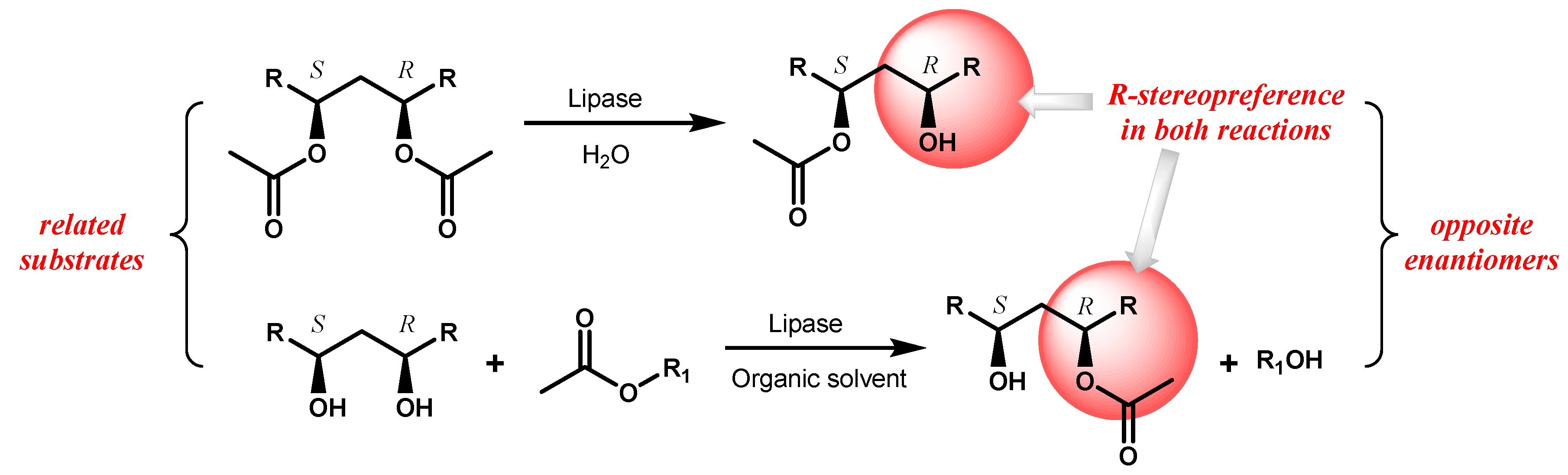
Figure 2.
Synthesis of both enantiomers of monoacetate 1 by lipase-catalyzed reactions.
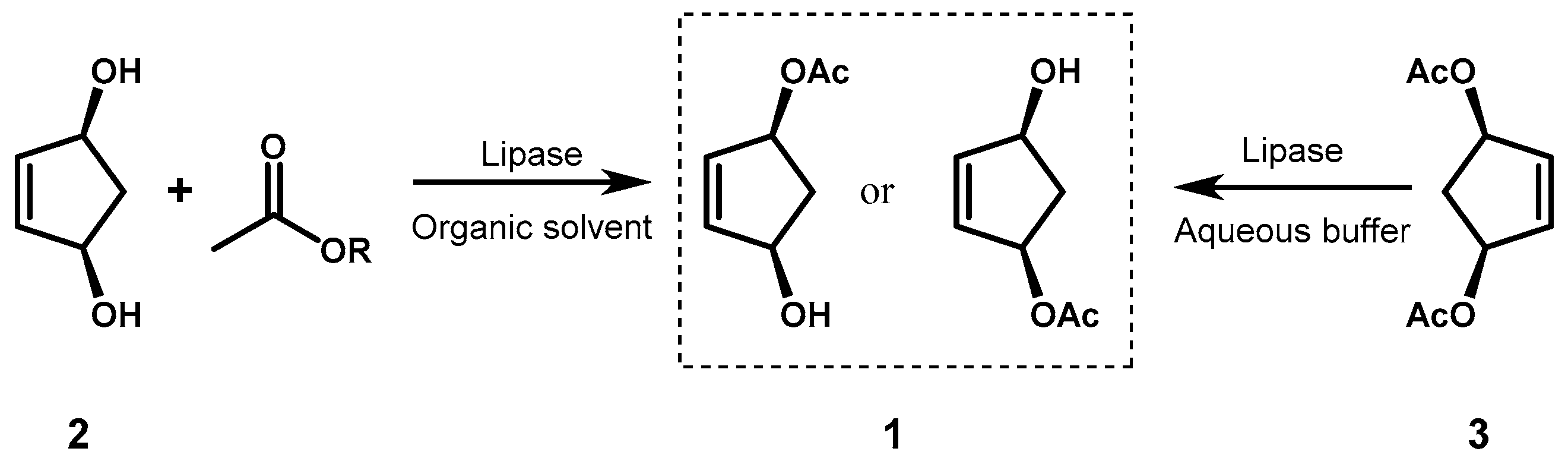
Figure 3.
Synthesis of both enantiomers of monoacetate 1 by lipase-catalyzed (a) hydrolysis and (b) alcoholysis of 3 with enzymes with opposite stereopreference.
Figure 3.
Synthesis of both enantiomers of monoacetate 1 by lipase-catalyzed (a) hydrolysis and (b) alcoholysis of 3 with enzymes with opposite stereopreference.

Figure 4.
Desymmetrization of diol 5 by lipase-catalyzed esterification.
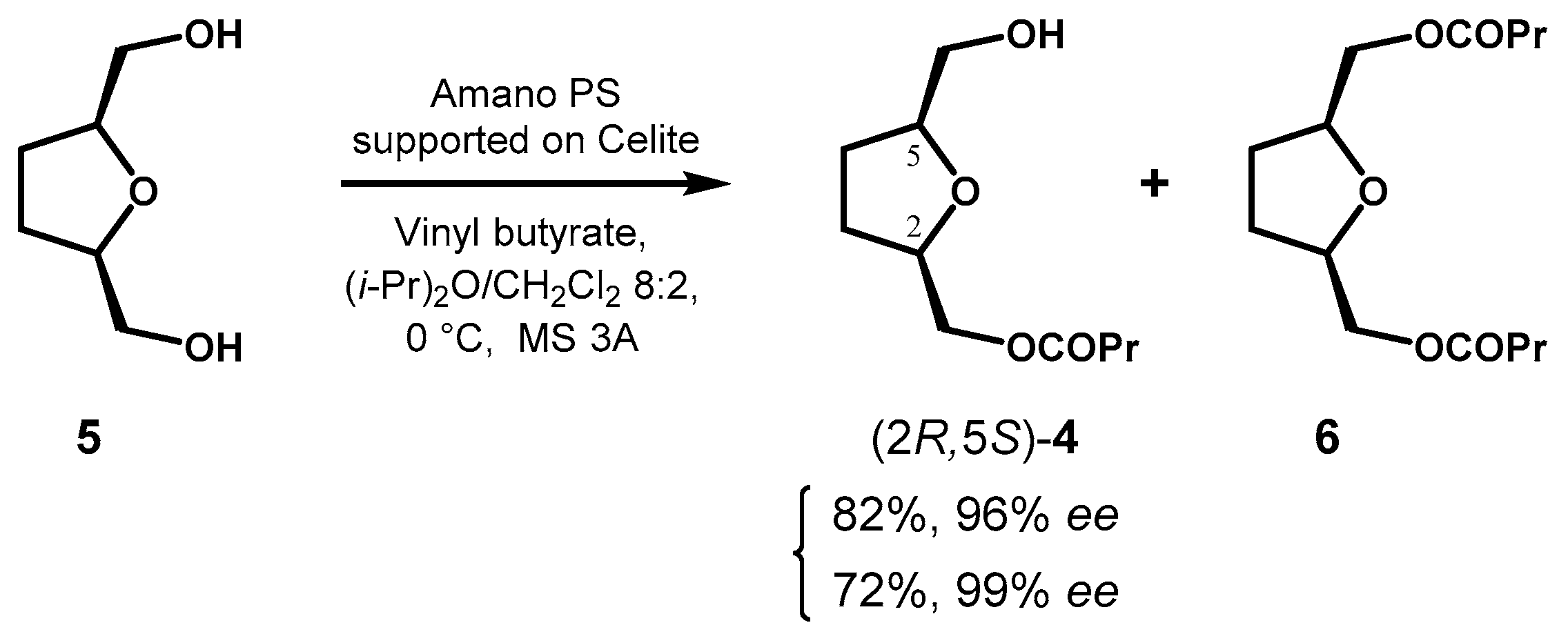
Figure 5.
Complementary desymmetrization reactions for the access to both enantiomers of mono- acetate 8.
Figure 5.
Complementary desymmetrization reactions for the access to both enantiomers of mono- acetate 8.
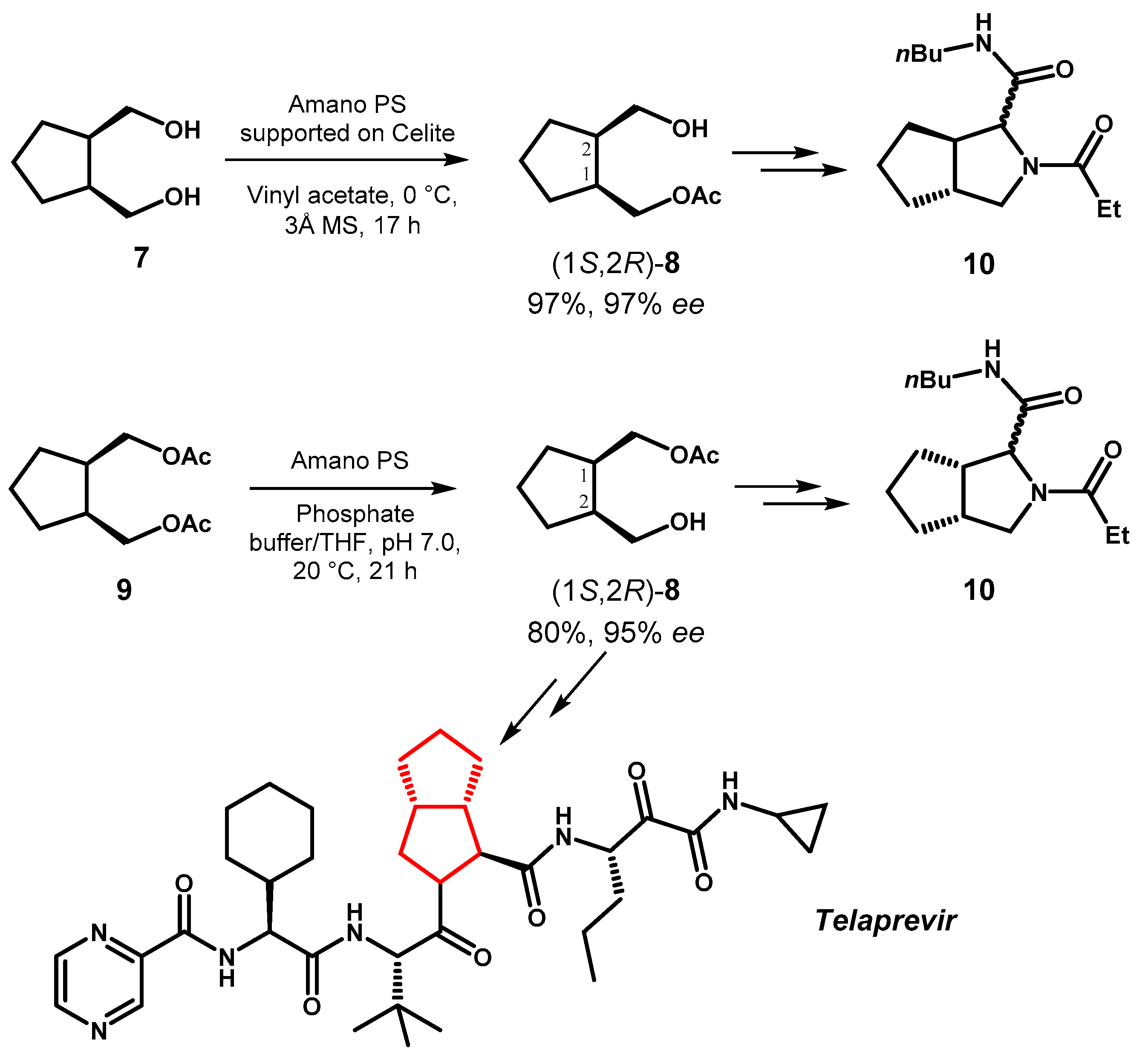
Figure 6.
Synthesis of yohimbine alkaloids and darunavir analogue through hydrolytic desymmetrization of 11.
Figure 6.
Synthesis of yohimbine alkaloids and darunavir analogue through hydrolytic desymmetrization of 11.

Figure 7.
Synthesis analogues of vitamin D3 through enantiotoposelective esterification of diol 13.
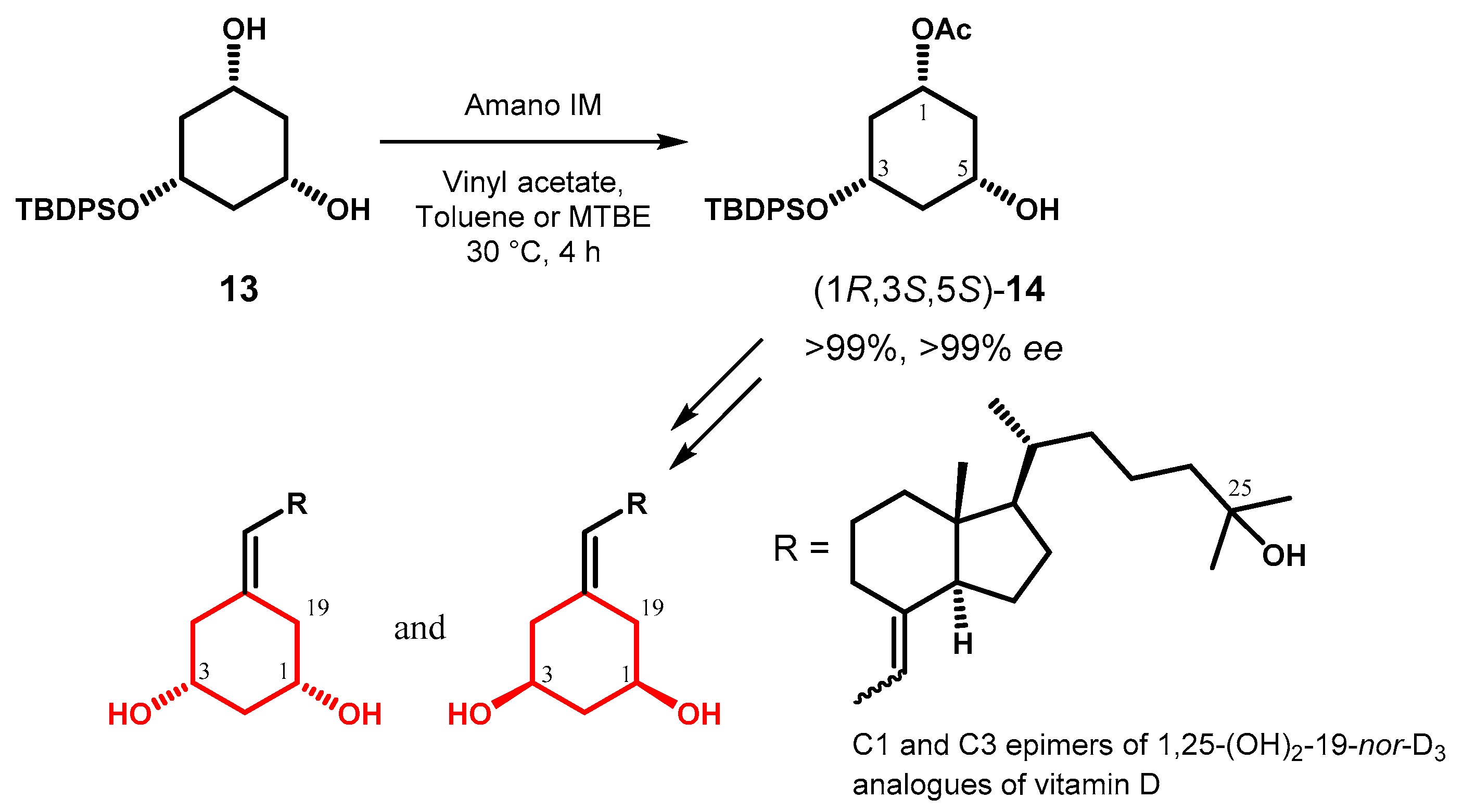
Figure 8.
Desymmetrization of myo-inositol 15 via enantiotoposelective esterification.

Figure 9.
Desymmetrization of diester 17 via enantiotoposelective hydrolysis.

Figure 10.
Desymmetrization of diol 20 via enantiotoposelective oxidation.
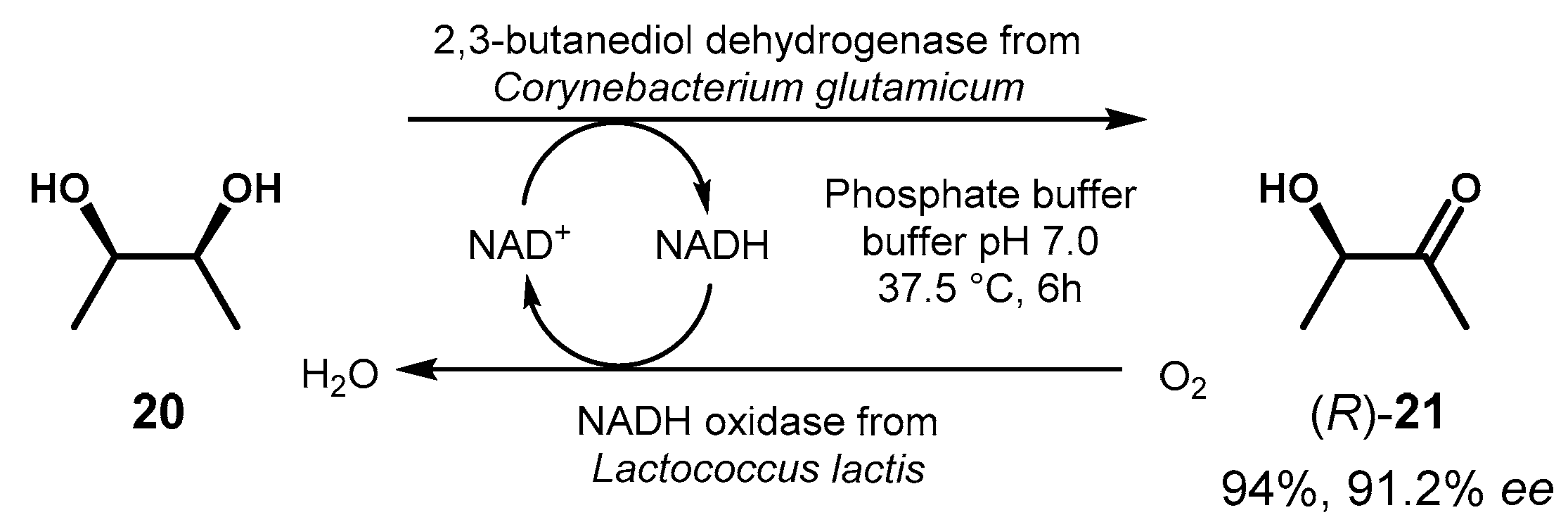
Figure 11.
Enantiotoposelective oxidation of 2 as key step in the synthesis of travoprost.

Figure 12.
Synthesis of chiral γ-lactones via oxidative desymmetrization of diols 23a–c.
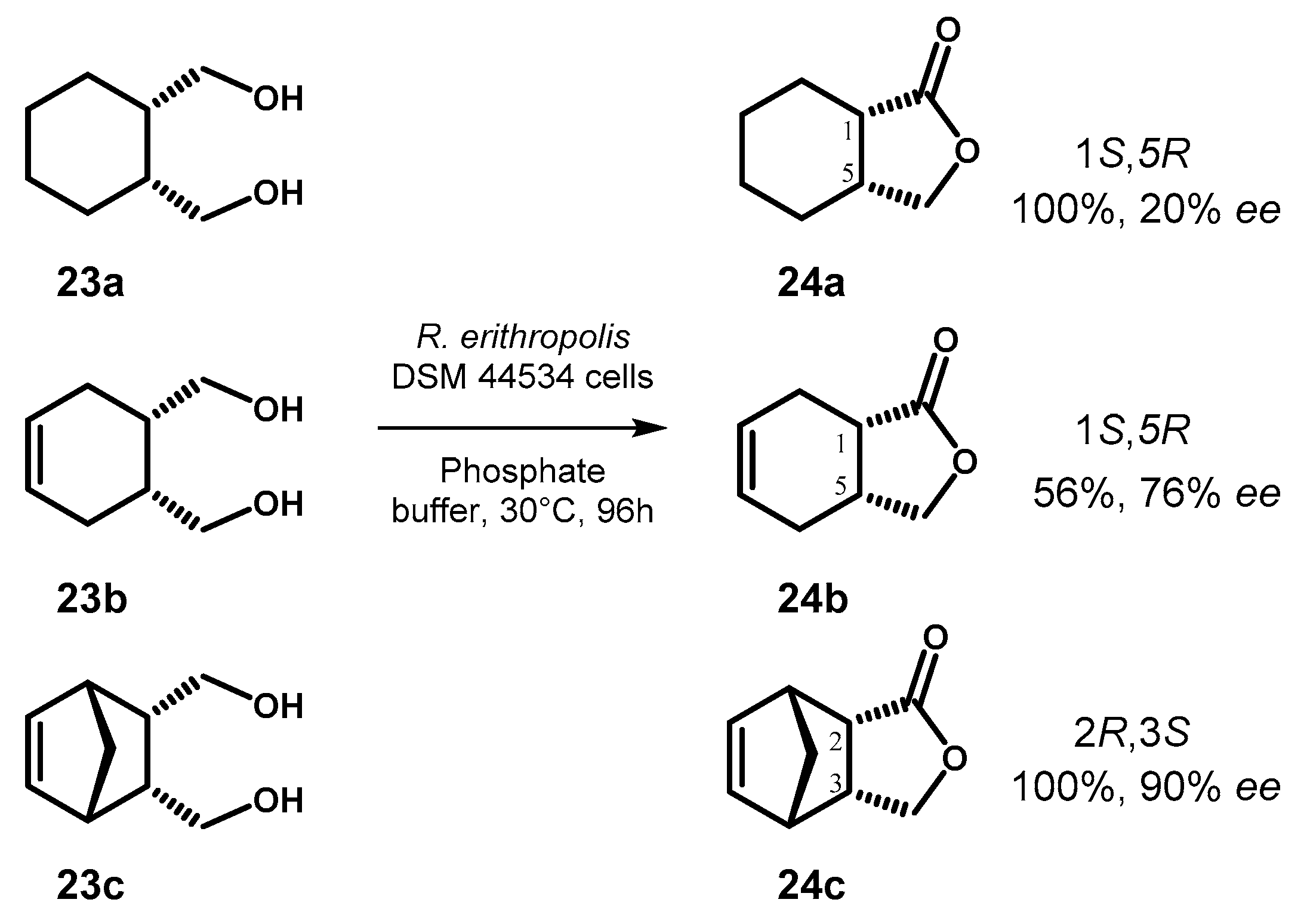
Figure 13.
Amidase-catalyzed desymmetrization of cyclic cis-dicarboxamides 25a–c.
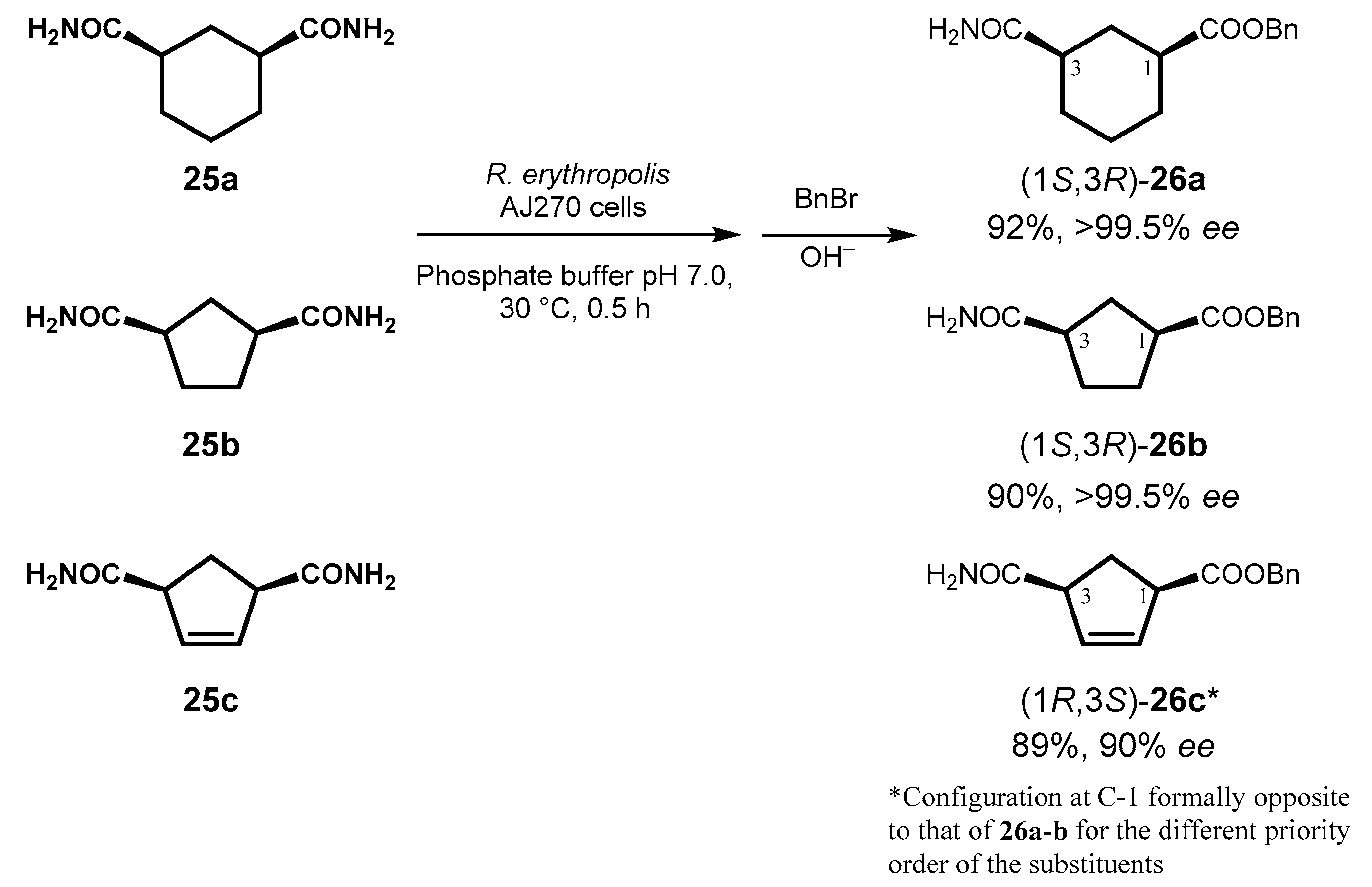
Figure 14.
Amidase-catalyzed desymmetrization of cyclic cis-dicarboxamides 27a–c and 29.
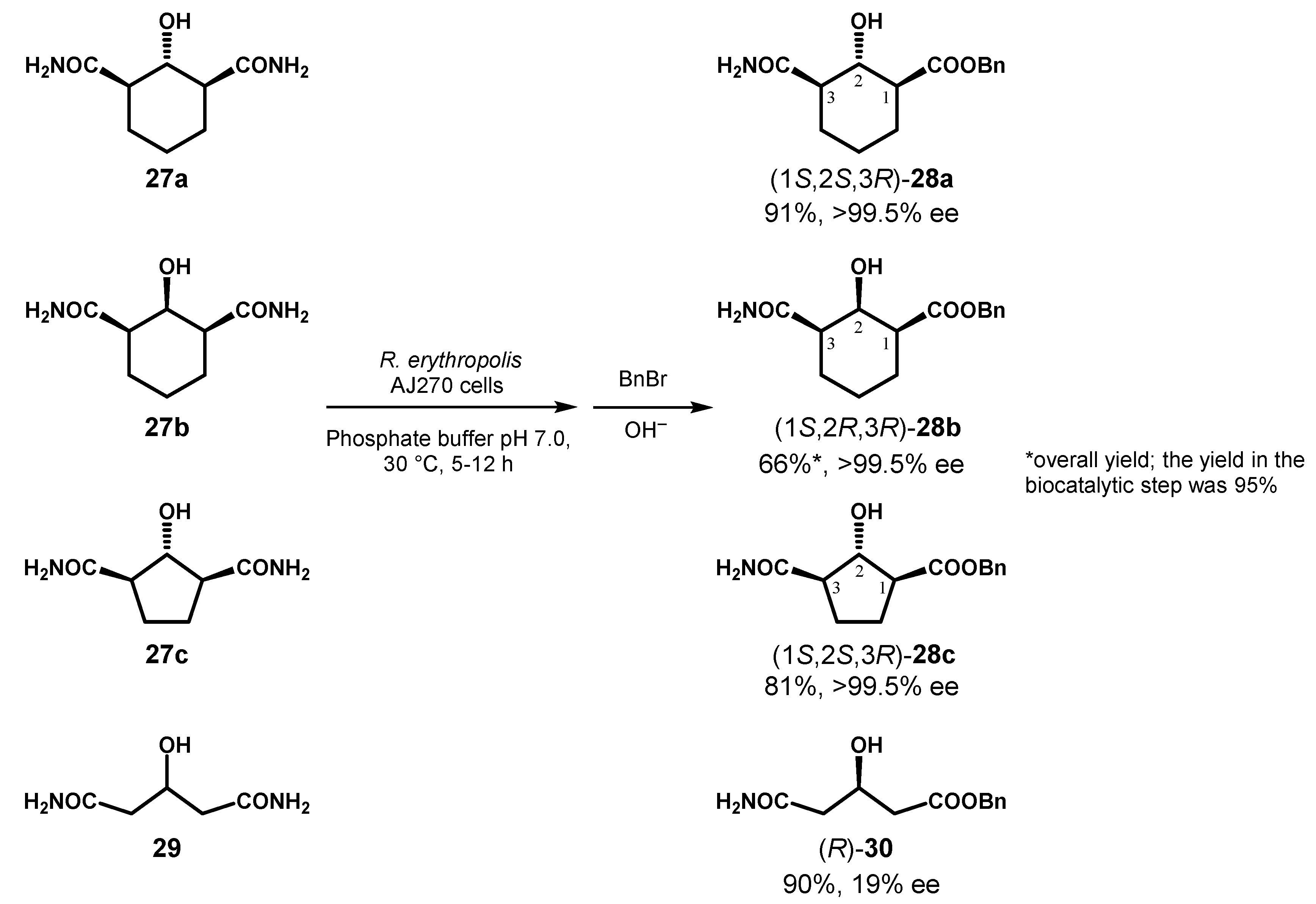
Figure 15.
Amidase-catalyzed desymmetrization of cyclic cis-dicarboxamides 31a–e and 33a–b containing a C-2 all-carbon quaternary stereogenic centre.
Figure 15.
Amidase-catalyzed desymmetrization of cyclic cis-dicarboxamides 31a–e and 33a–b containing a C-2 all-carbon quaternary stereogenic centre.

Figure 16.
Lipase-catalyzed desymmetrization of propane-1,3-diol 35.
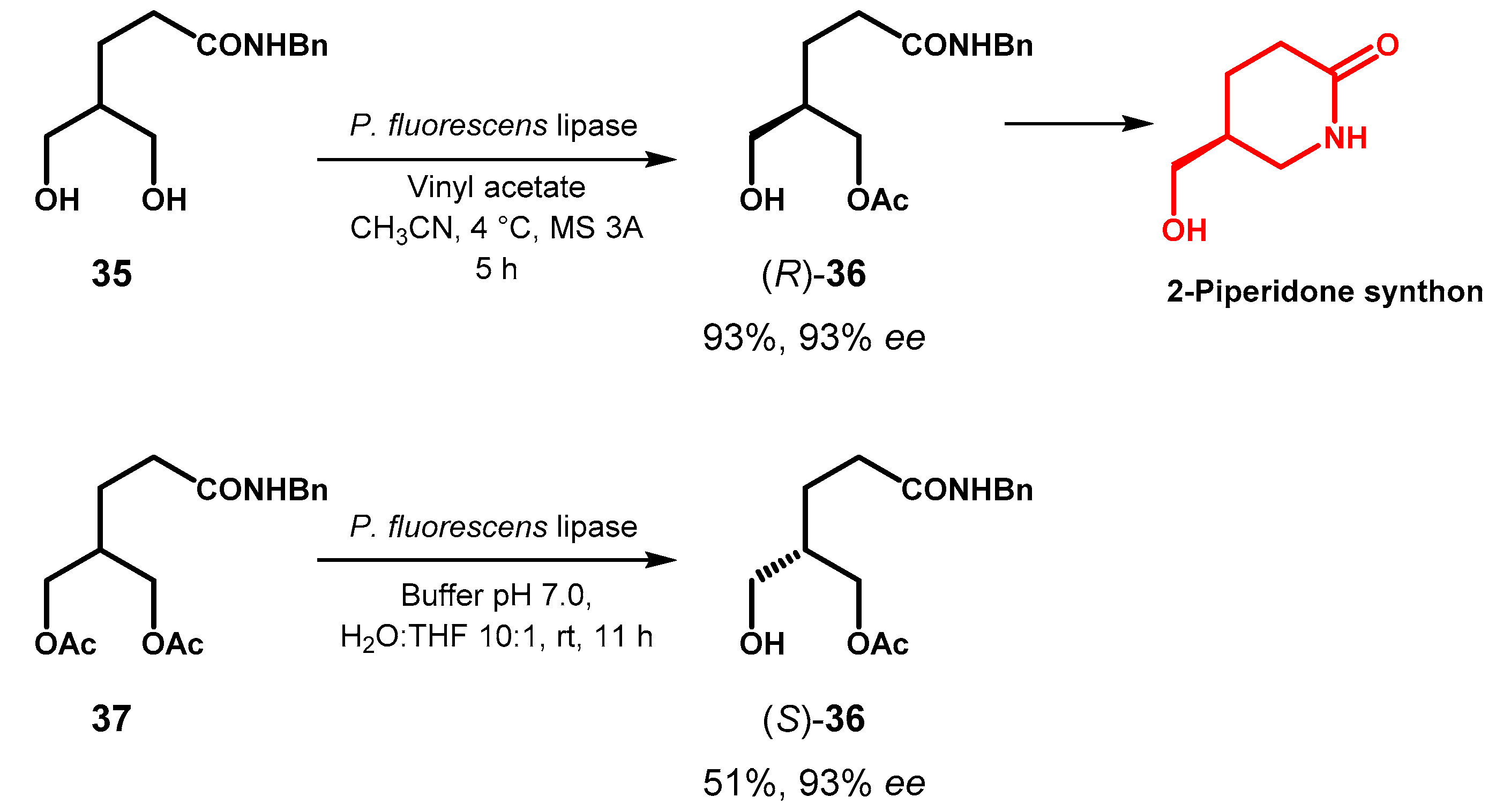
Figure 17.
Lipase-catalyzed desymmetrization of propane-1,3-diacetate 38.
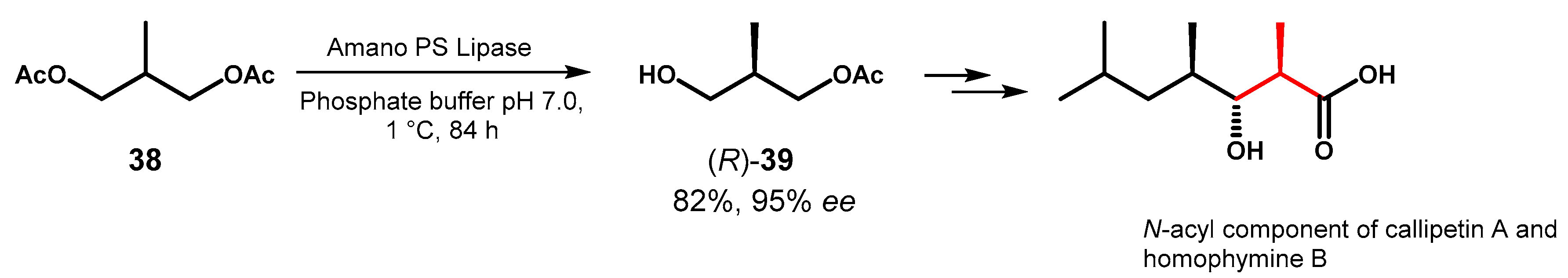
Figure 18.
Lipase-catalyzed desymmetrization of nucleoside-substituted glycerol 40.
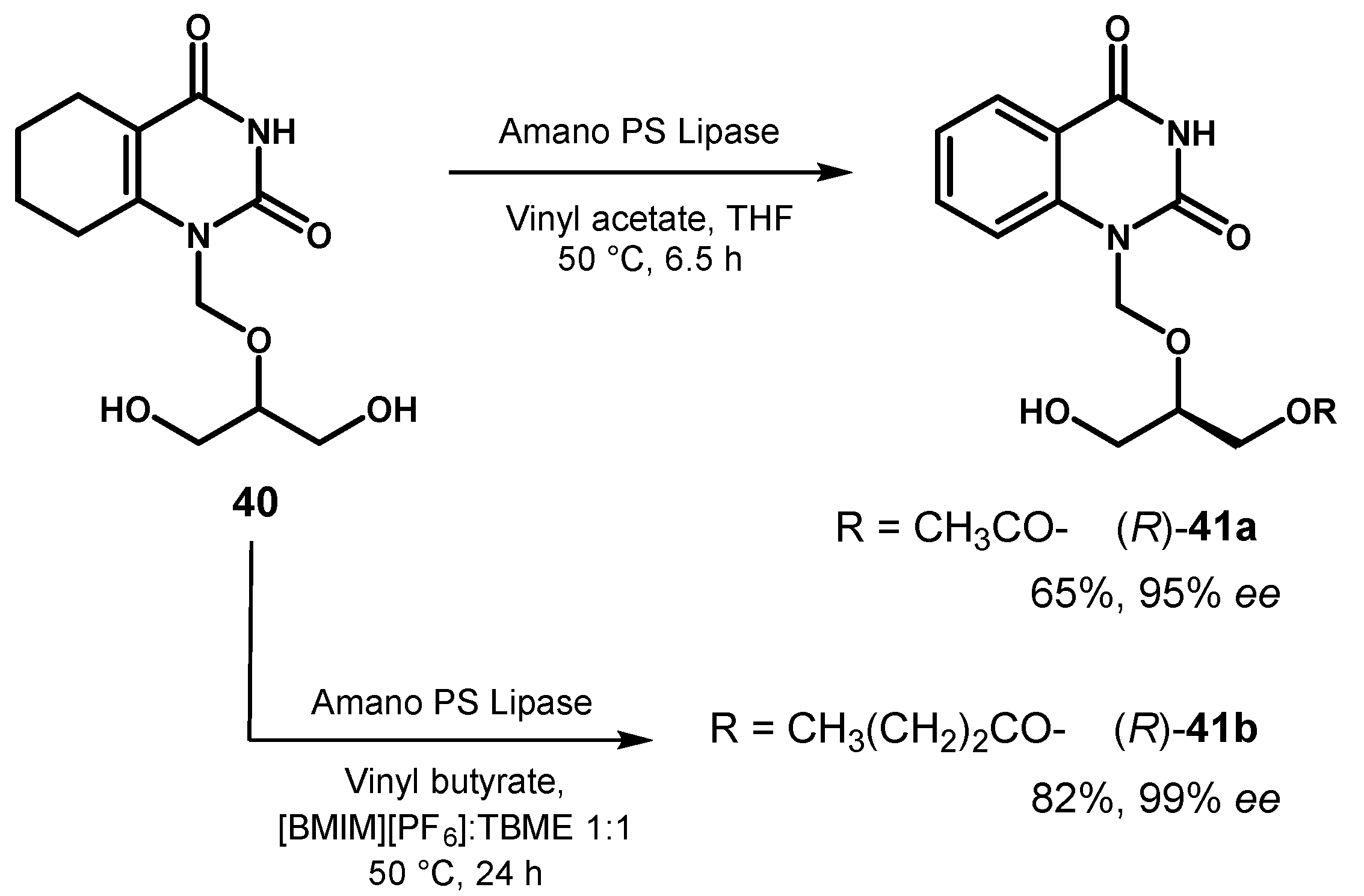
Figure 19.
Synthesis of EfdA via desymmetrization of the prochiral glycerol derivative 42.
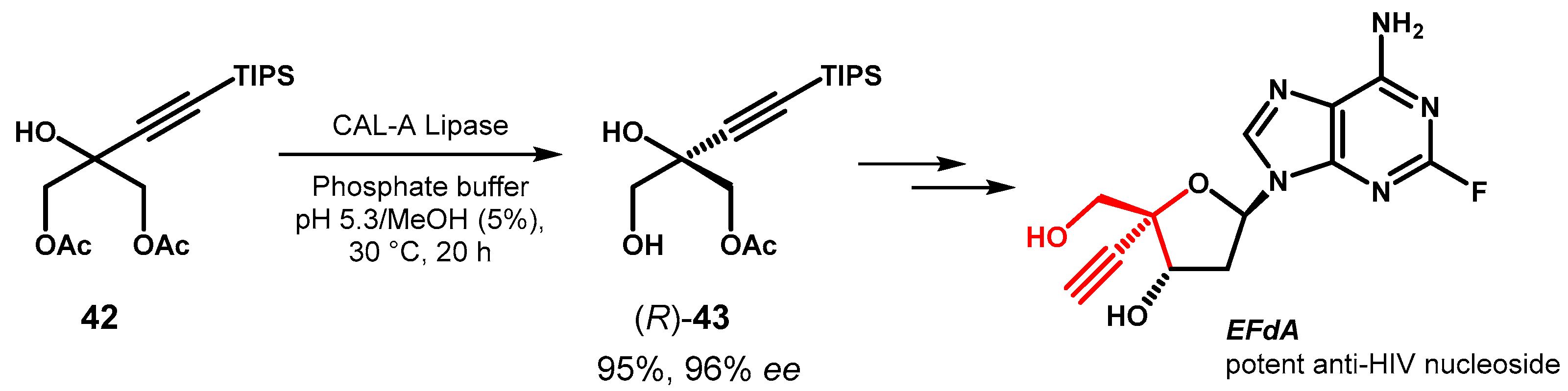
Figure 20.
Oxidative desymmetrization of 2-alkyl propane-1,3-diols 44a–c catalyzed by microbial whole cells of Acetobacter aceti.
Figure 20.
Oxidative desymmetrization of 2-alkyl propane-1,3-diols 44a–c catalyzed by microbial whole cells of Acetobacter aceti.
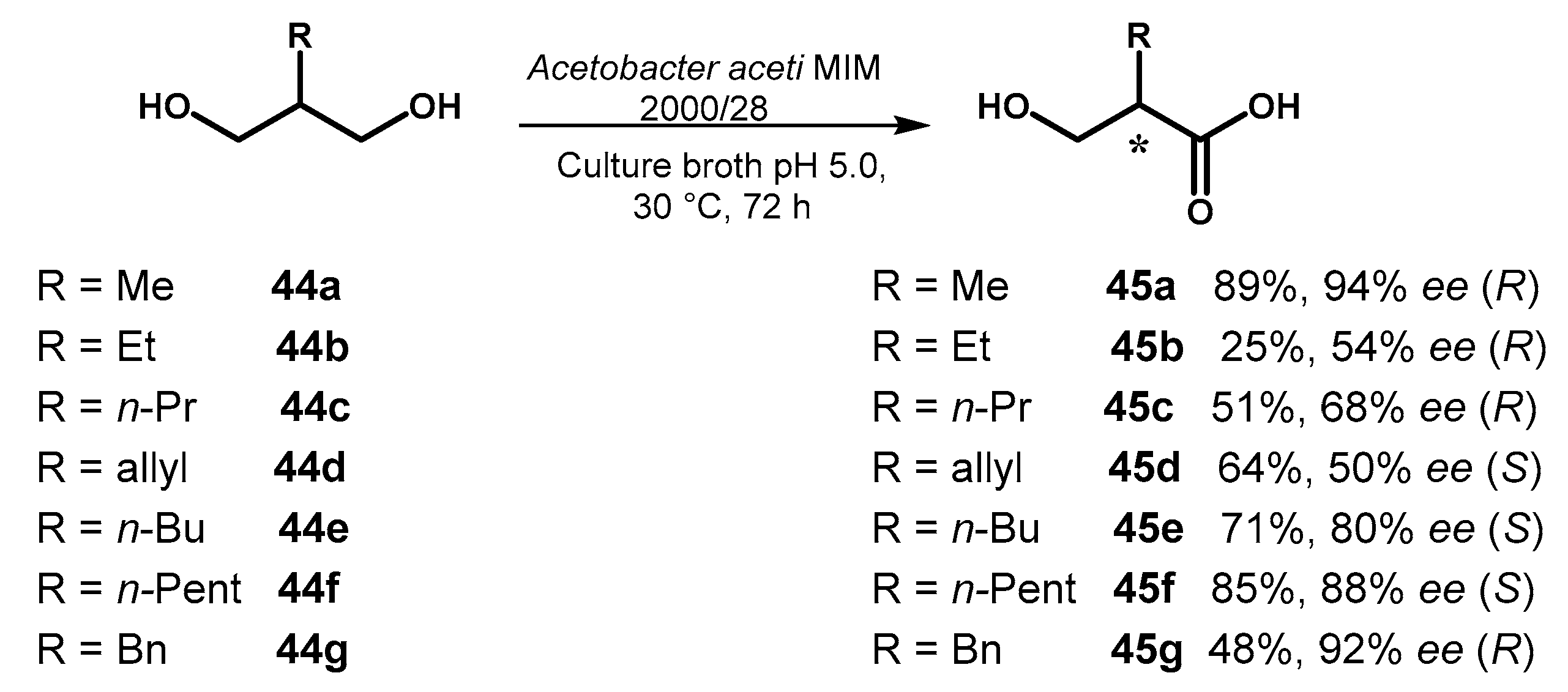
Figure 21.
In-flow oxidative desymmetrization of 44a for the synthesis of captopril.
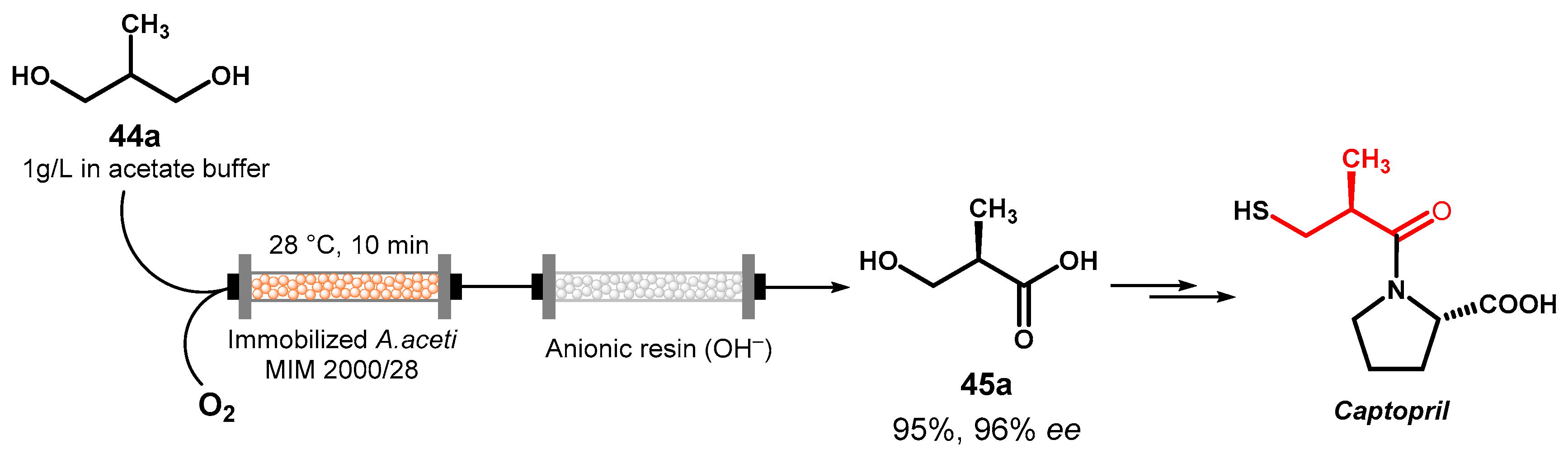
Figure 22.
Amidase-catalyzed desymmetrization of 2-phenyl bisamides 46 and 2-benzyl bisamides 48.
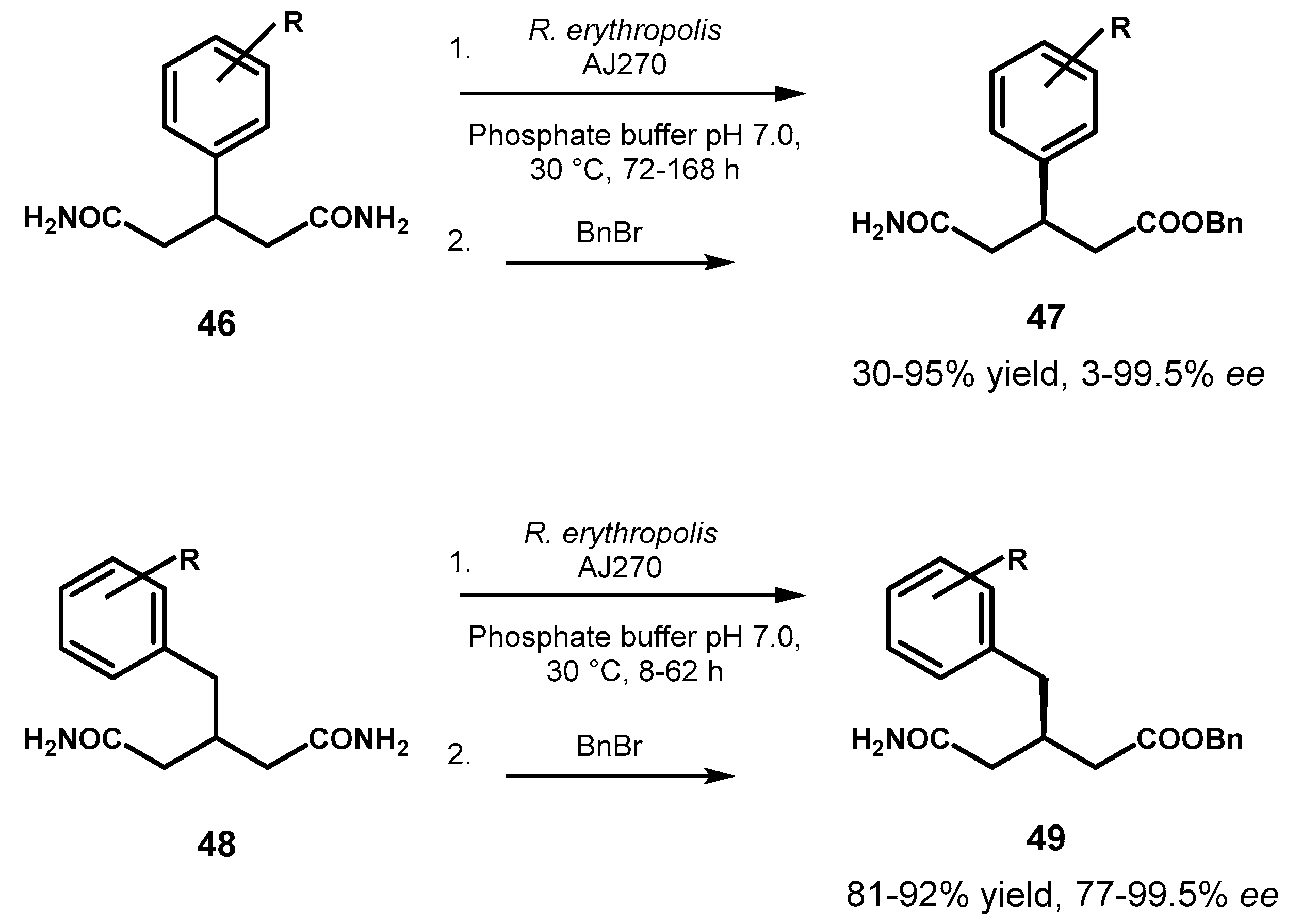
Figure 23.
Possible products in the desymmetrization of a generic diketone 50.
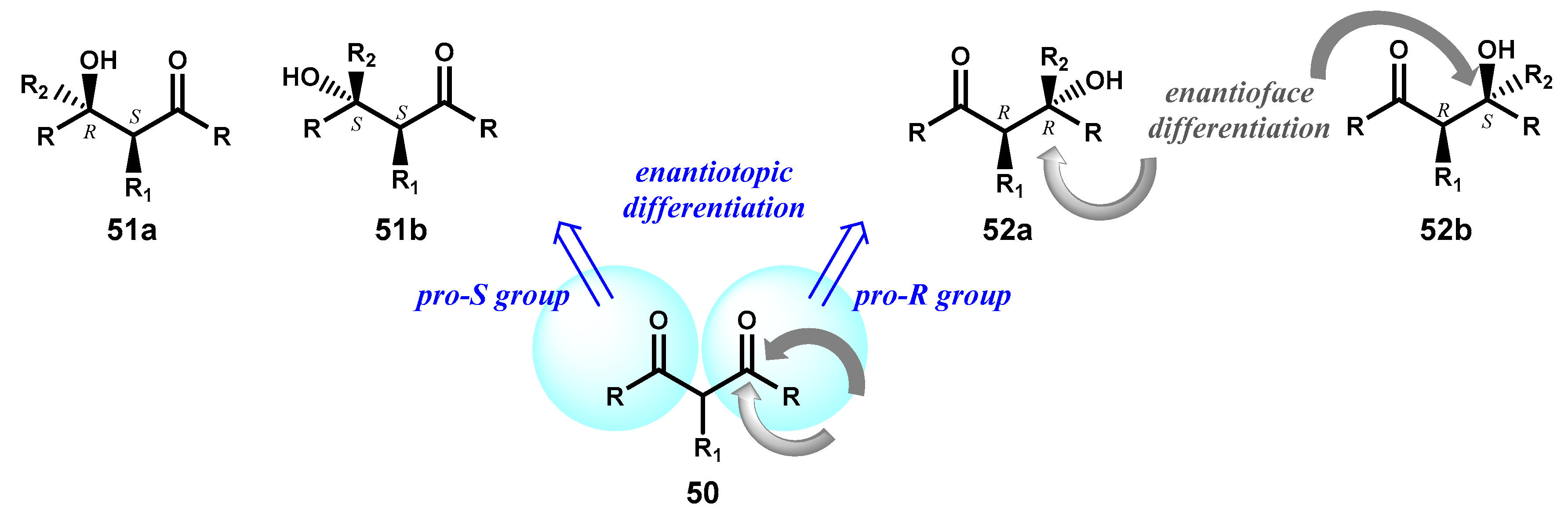
Figure 24.
Yeast-catalyzed desymmetrization of diketone 53 as key step in the synthesis of paspaline.
Figure 24.
Yeast-catalyzed desymmetrization of diketone 53 as key step in the synthesis of paspaline.
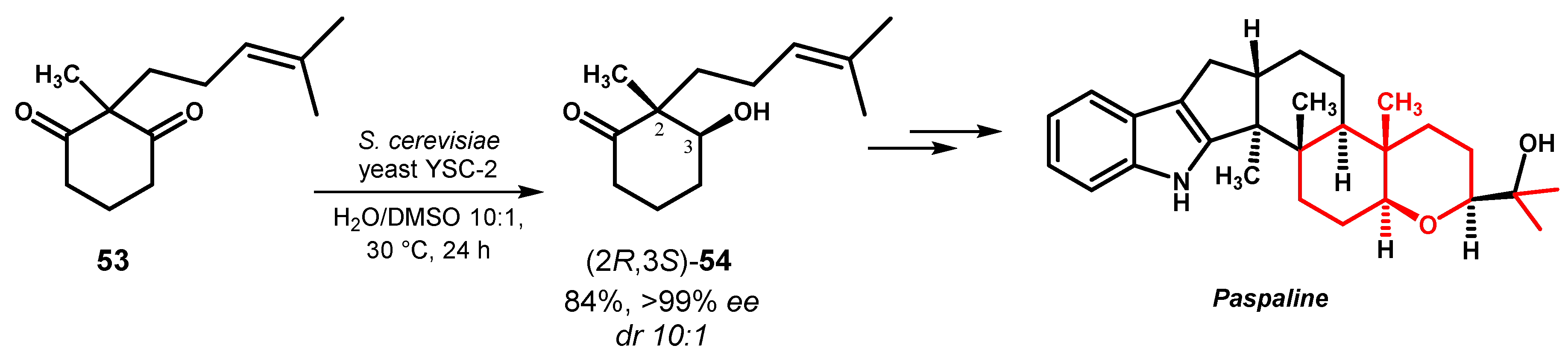
Figure 25.
Desymmetrization of diketone 55 in batch or in-flow conditions.
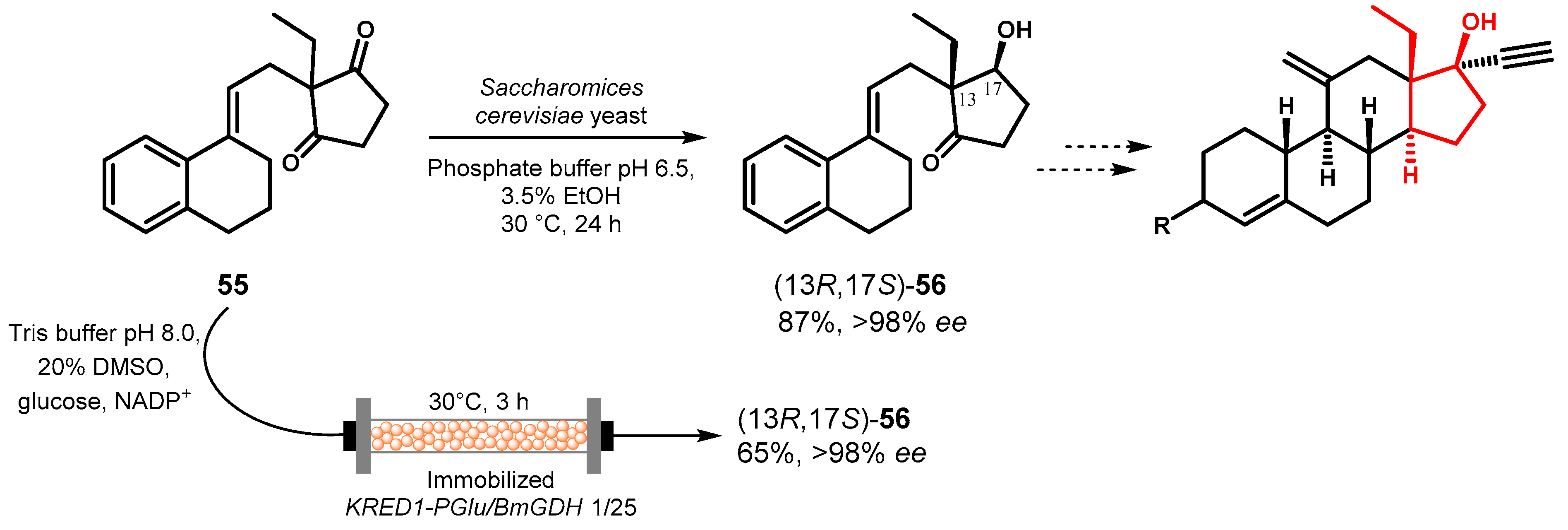
Figure 26.
Desymmetrization of bisphenol propionate 57a by lipase-catalyzed hydrolysis.
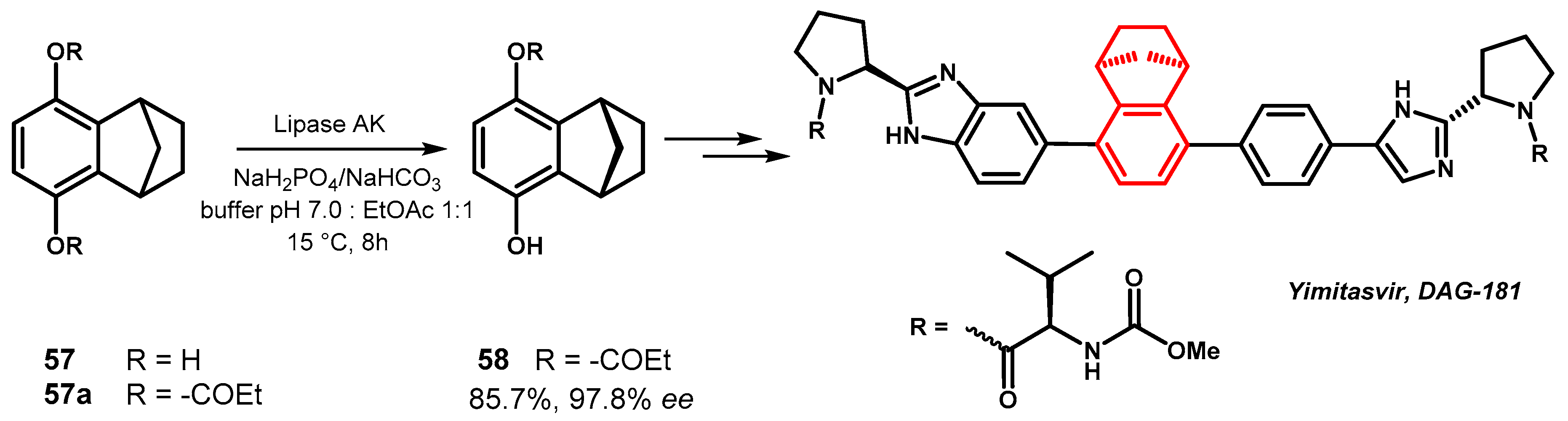
Figure 27.
Enantiomeric structures for a generic conformationally stable biphenyl compound.
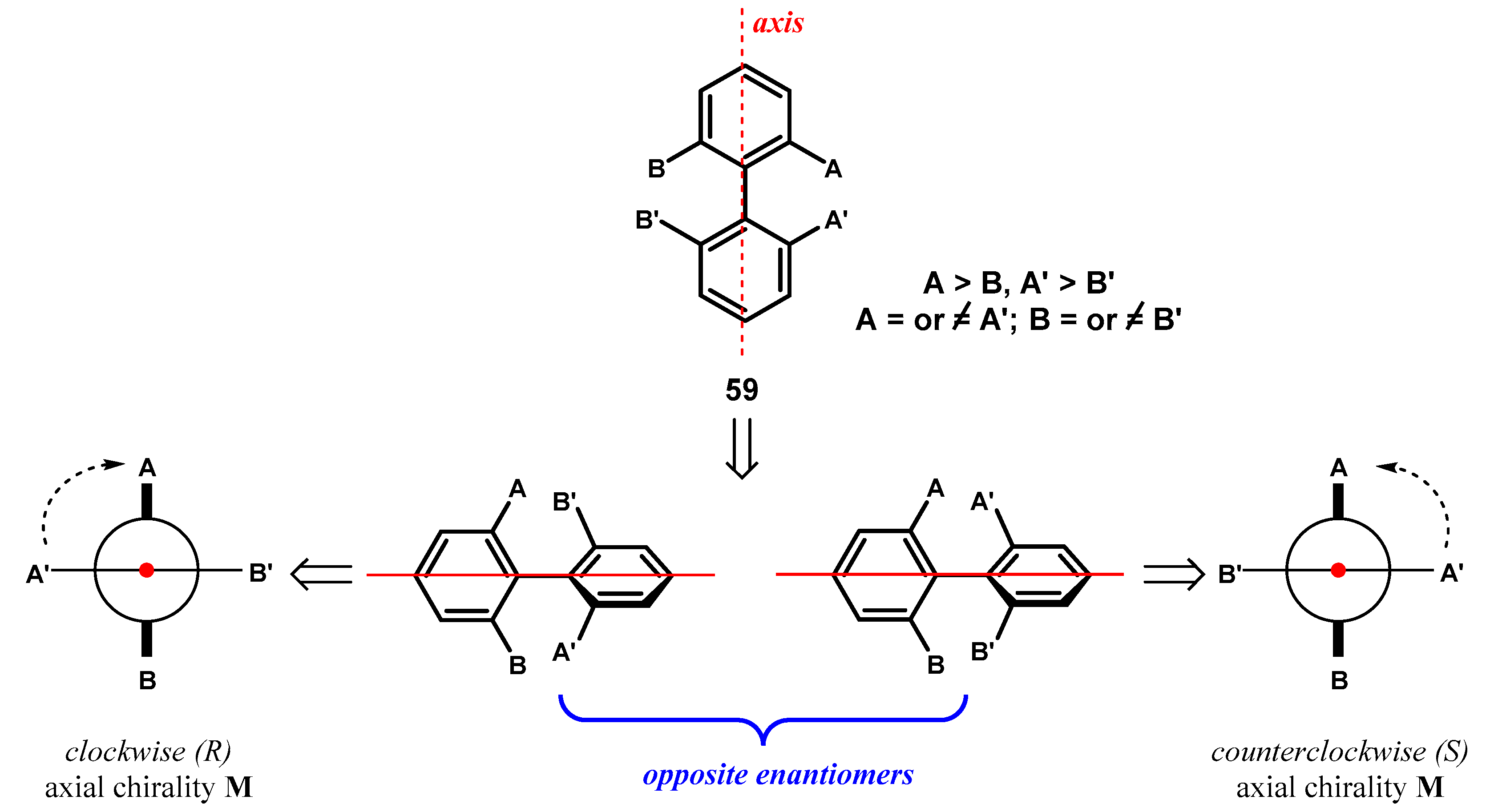
Figure 28.
Prochirality in σ-symmetric biphenyls.
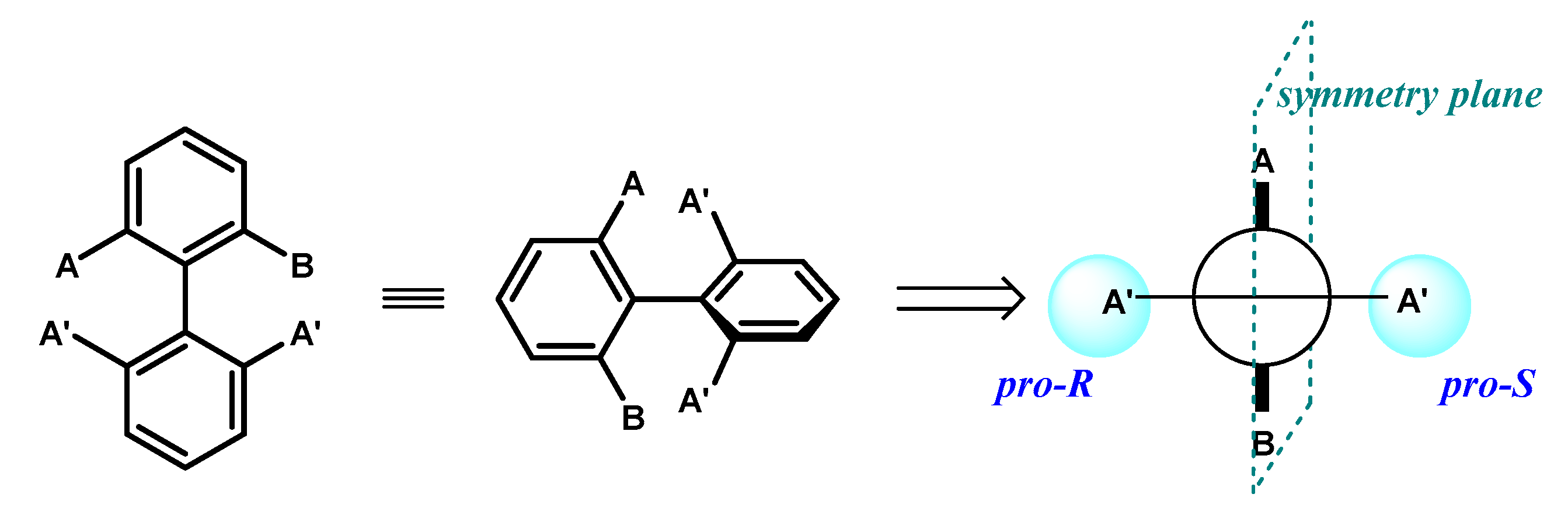
Figure 29.
Enantiodivergent synthesis of optically active biphenyls 61a–f.
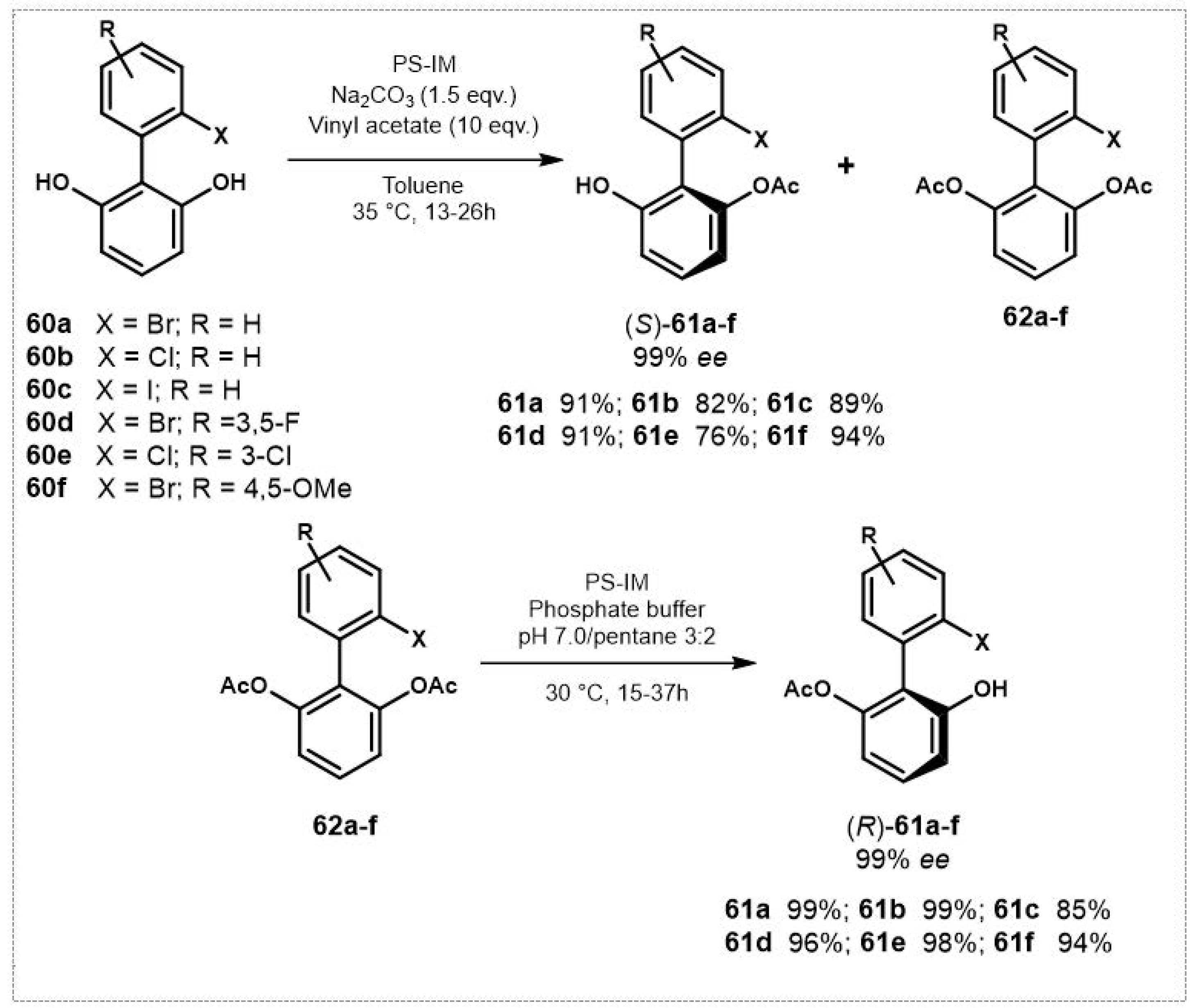
Figure 30.
Desymmetrization of diketone 63 and sequential enzyme reduction to meso-anti,anti-diol 65.
Figure 30.
Desymmetrization of diketone 63 and sequential enzyme reduction to meso-anti,anti-diol 65.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Patti, A.; Sanfilippo, C. Breaking Molecular Symmetry through Biocatalytic Reactions to Gain Access to Valuable Chiral Synthons. Symmetry 2020, 12, 1454. https://doi.org/10.3390/sym12091454
AMA Style
Patti A, Sanfilippo C. Breaking Molecular Symmetry through Biocatalytic Reactions to Gain Access to Valuable Chiral Synthons. Symmetry. 2020; 12(9):1454. https://doi.org/10.3390/sym12091454
Chicago/Turabian StylePatti, Angela, and Claudia Sanfilippo. 2020. "Breaking Molecular Symmetry through Biocatalytic Reactions to Gain Access to Valuable Chiral Synthons" Symmetry 12, no. 9: 1454. https://doi.org/10.3390/sym12091454
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.