An Optimized Dual Extraction Method for the Simultaneous and Accurate Analysis of Polar Metabolites and Lipids Carried out on Single Biological Samples

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
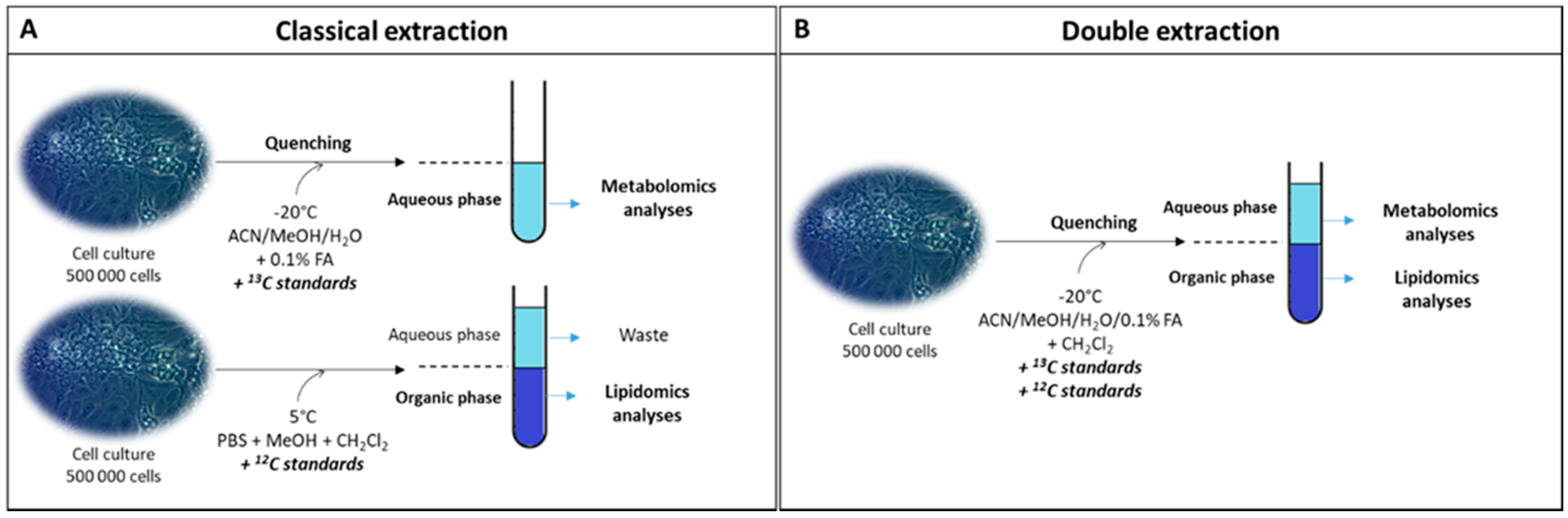
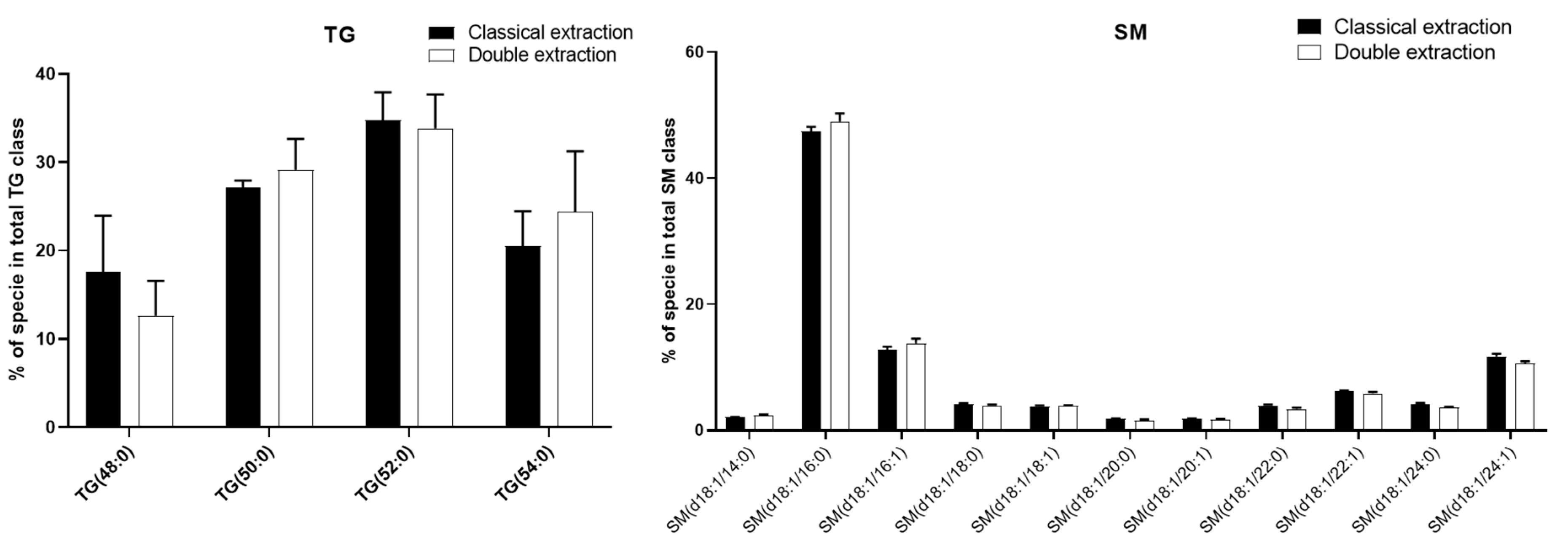
2.1. Qualitative and Quantitative Analysis of the Cell Lipidome and Metabolome Using a Newly Developed Dual Extraction Protocol
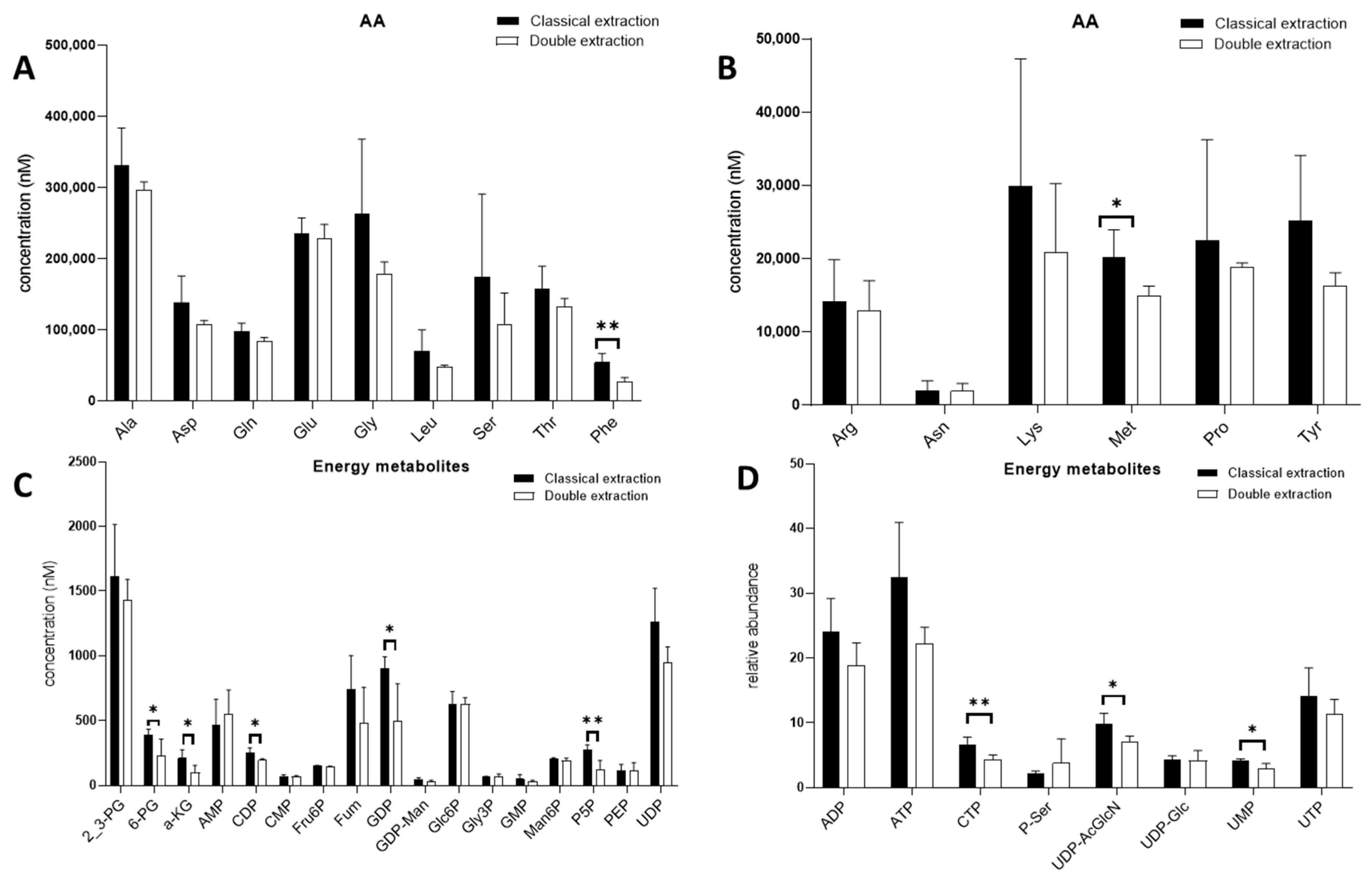
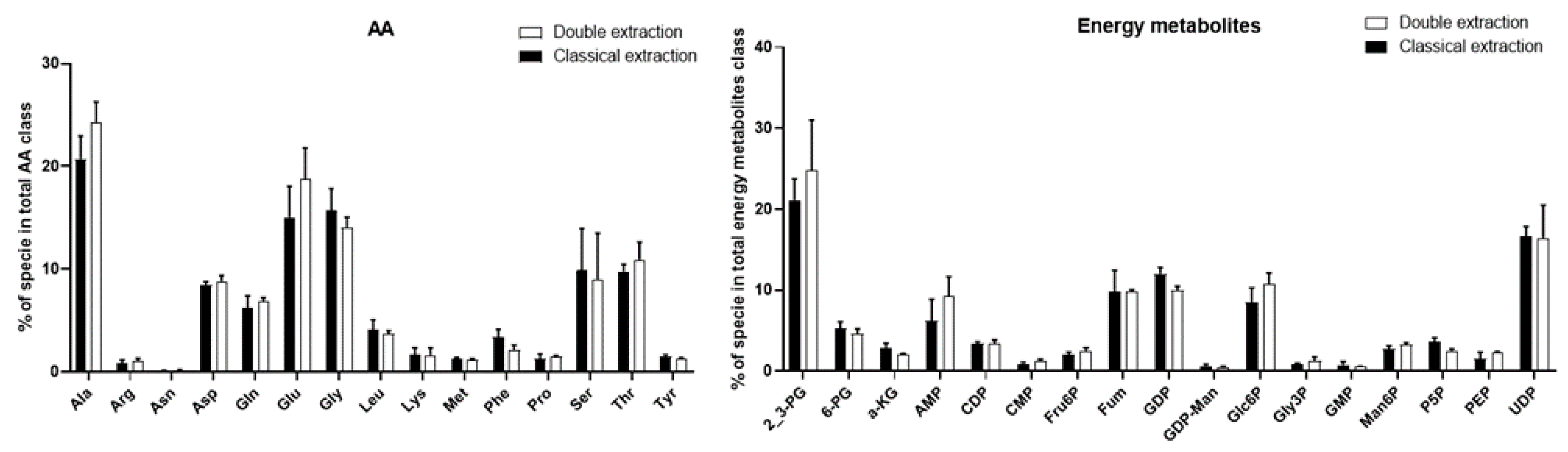
2.2. Polar Metabolites
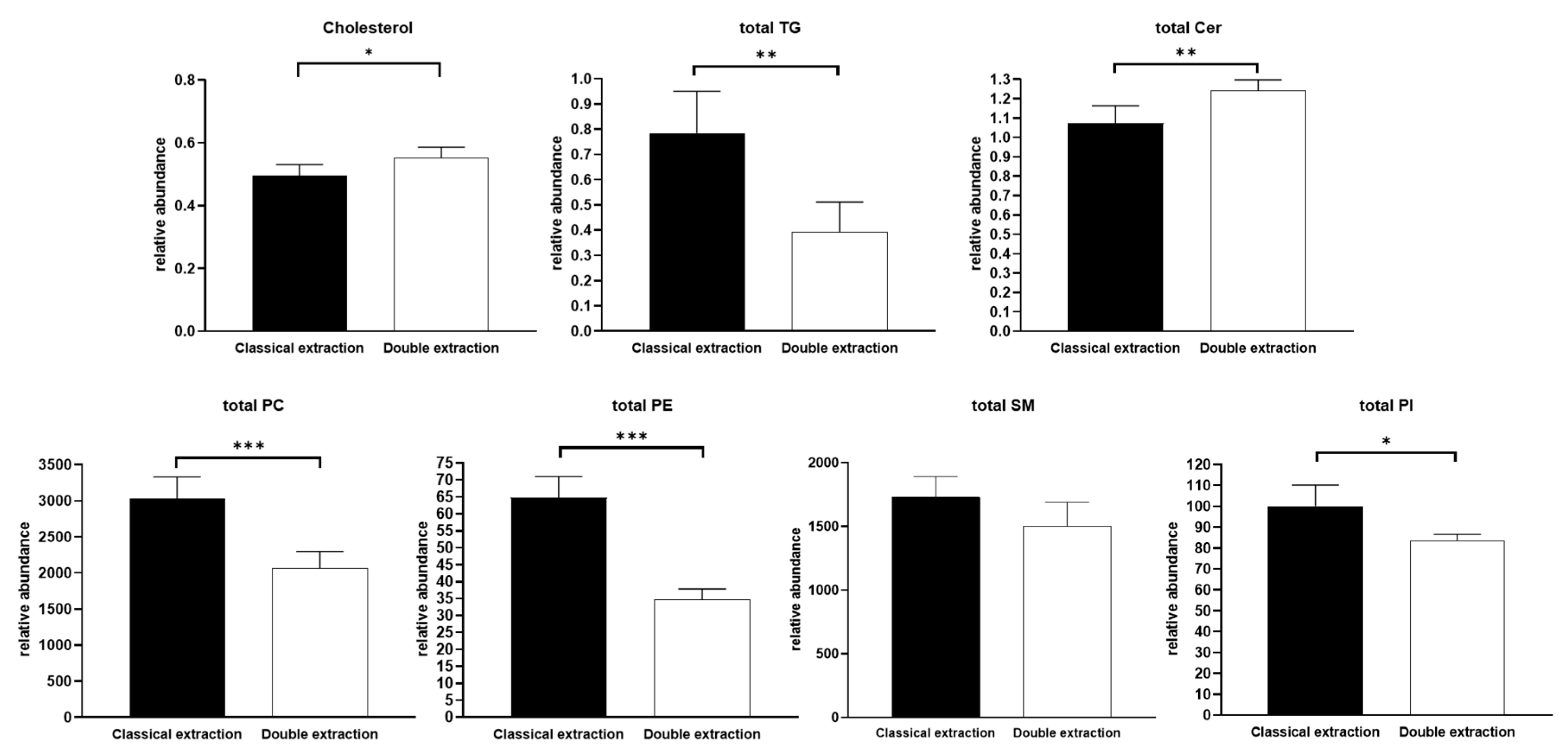
2.3. Apolar Metabolites
2.4. Methods Reproducibility
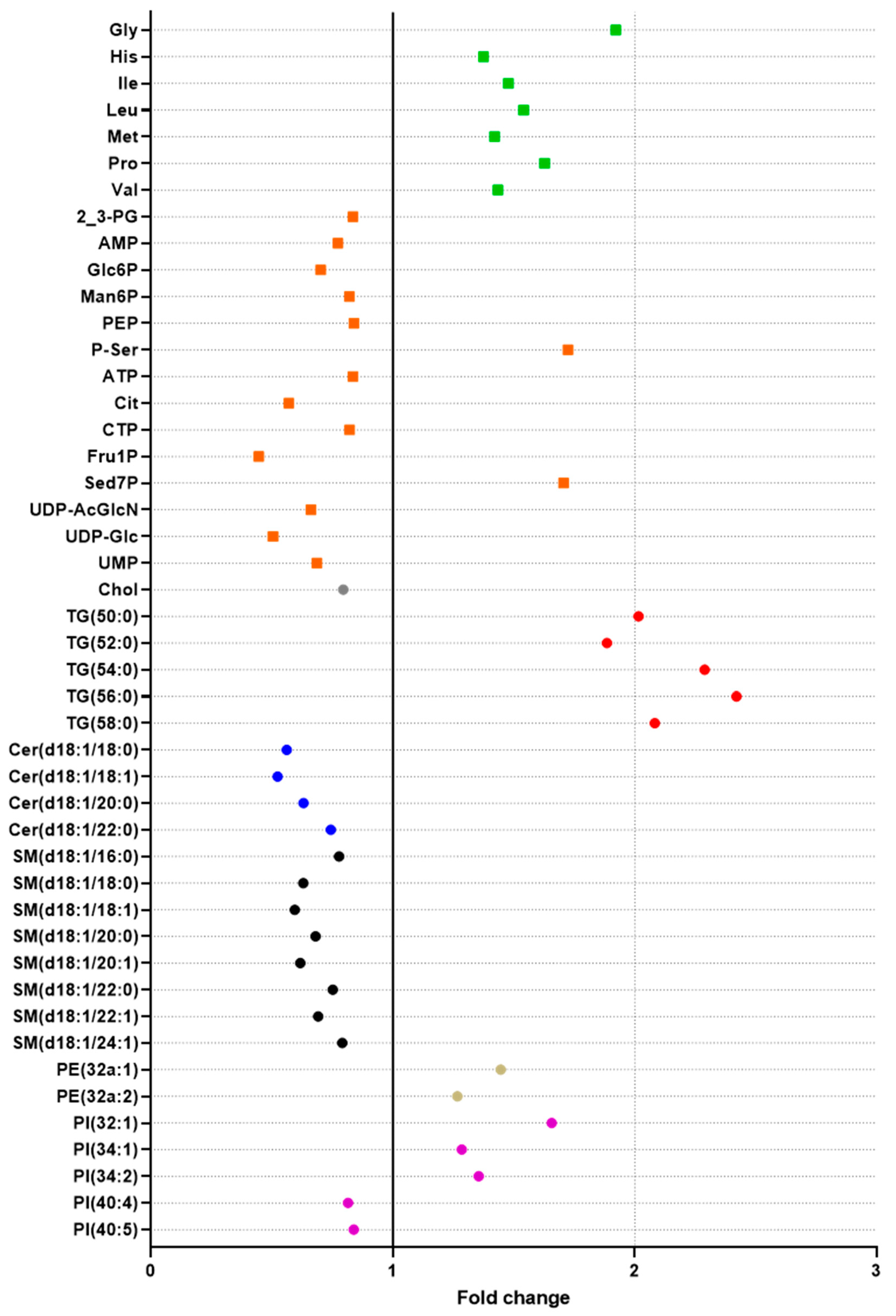
2.5. Exploration of the Metabolic Effects of Fenofibrate Using the Double Extraction Protocol
2.5.1. Effects on Lipids
2.5.2. Effects on Polar Metabolites
3. Materials and Methods
3.1. Reagents
3.2. Cell Culture and Treatment
3.3. Sampling
3.4. Classical Extraction Method
3.5. Double Extraction Method
3.6. Neutral Lipids Analysis
3.7. Phospholipids and Sphingolipids Analysis
3.8. Energy Metabolites Analysis
3.9. Amino Acids Analysis
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Veenstra, T.D. Metabolomics: The final frontier? Genome Med. 2012, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Damiani, C.; Gaglio, D.; Sacco, E.; Alberghina, L.; Vanoni, M. Systems metabolomics: From metabolomic snapshots to design principles. Curr. Opin. Biotechnol. 2020, 63, 190–199. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Gowda, G.A.N.; Zhang, S.; Gu, H.; Asiago, V.; Shanaiah, N.; Raftery, D. Metabolomics-based methods for early disease diagnostics. Expert Rev. Mol. Diagn. 2008, 8, 617–633. [Google Scholar] [CrossRef]
- Ramirez, T.; Daneshian, M.; Kamp, H.; Bois, F.Y.; Clench, M.R.; Coen, M.; Donley, B.; Fischer, S.M.; Ekman, E.R.; Fabian, E.; et al. Metabolomics in toxicology and preclinical research. ALTEX 2013, 30, 209–225. [Google Scholar] [CrossRef]
- Cabaton, N.J.; Poupin, N.; Canlet, C.; Tremblay-Franco, M.; Audebert, M.; Cravedi, J.-P.; Riu, A.; Jourdan, F.; Zalko, D. An untargeted metabolomics approach to investigate the metabolic modulations of HepG2 cells exposed to low doses of bisphenol A and 17β-estradiol. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Poupin, N.; Tremblay-Franco, M.; Amiel, A.; Canlet, C.; Rémond, D.; Debrauwer, L.; Dardevet, M.; Thiele, I.; Aurich, M.K.; Jourdan, F.; et al. Arterio-venous metabolomics exploration reveals major changes across liver and intestine in the obese Yucatan minipig. Sci. Rep. 2019, 9, 12527. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bohra, A.; Pandey, A.K.; Pandey, M.K.; Kumar, A. metabolomics for plant improvement: Status and prospects. Front. Plant Sci. 2017, 8, 1032. [Google Scholar] [CrossRef]
- Dunn, W.B.; Bailey, N.J.C.; Johnson, H.E. Measuring the metabolome: Current analytical technologies. Analyst 2005, 130, 606–625. [Google Scholar] [CrossRef]
- Vuckovic, D. Improving metabolome coverage and data quality: Advancing metabolomics and lipidomics for biomarker discovery. Chem. Commun. 2018, 54, 6728–6749. [Google Scholar] [CrossRef]
- Wawrzyniak, R.; Kosnowska, A.; Macioszek, S.; Bartoszewski, R.; Markuszewski, M.J. New plasma preparation approach to enrich metabolome coverage in untargeted metabolomics: Plasma protein bound hydrophobic metabolite release with proteinase K. Sci. Rep. 2018, 8, 9541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hong, Y.; Jiang, L.-L.; Yi, X.; Chen, Y.; Liu, L.; Chen, Z.; Wu, Y.; Cai, Z. Global metabolomic and lipidomic analysis reveal the synergistic effect of bufalin in combination with cinobufagin against HepG2 Cells. J. Proteome Res. 2020, 19, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Lebkuchen, A.; Carvalho, V.M.; Venturini, G.; Salgueiro, J.S.; Freitas, L.S.; Dellavance, A.; Martins, F.C.; Lorenzi-Filho, G.; Cardozo, K.H.M.; Drager, L.F. Metabolomic and lipidomic profile in men with obstructive sleep apnoea: Implications for diagnosis and biomarkers of cardiovascular risk. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Song, Z.; Chen, Z.; Guo, Z.; Jin, H.; Ding, C.; Hong, Y.; Cai, Z. Metabolic and lipidomic characterization of malignant pleural effusion in human lung cancer. J. Pharm. Biomed. Anal. 2020, 180, 113069. [Google Scholar] [CrossRef] [PubMed]
- Giesbertz, P.; Padberg, I.; Rein, D.; Ecker, J.; Höfle, A.S.; Spanier, B.; Daniel, H. Metabolite profiling in plasma and tissues of ob/ob and db/db mice identifies novel markers of obesity and type 2 diabetes. Diabetologia 2015, 58, 2133–2143. [Google Scholar] [CrossRef]
- Acharjee, A.; Ament, Z.; West, J.A.; Stanley, E.; Griffin, J.L. Integration of metabolomics, lipidomics and clinical data using a machine learning method. BMC Bioinform. 2016, 17, 440–449. [Google Scholar] [CrossRef]
- Nakayasu, E.S.; Nicora, C.D.; Sims, A.C.; Burnum-Johnson, K.E.; Kim, Y.-M.; Kyle, J.E.; Matzke, M.M.; Shukla, A.K.; Chu, R.K.; Schepmoes, A.A.; et al. MPLEx: A robust and universal protocol for single-sample integrative proteomic, metabolomic, and lipidomic analyses. mSystems 2016, 1, e00043-16. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Liu, X.; Li, A.; Hou, Z.; Wang, Y.; Zhang, Y. A novel approach to the simultaneous extraction and non-targeted analysis of the small molecules metabolome and lipidome using 96-well solid phase extraction plates with column-switching technology. J. Chromatogr. A 2015, 1409, 277–281. [Google Scholar] [CrossRef]
- Godzien, J.; Ciborowski, M.; Whiley, L.; Legido-Quigley, C.; Rupérez, F.J.; Barbas, C. In-vial dual extraction liquid chromatography coupled to mass spectrometry applied to streptozotocin-treated diabetic rats. Tips and pitfalls of the method. J. Chromatogr. A 2013, 1304, 52–60. [Google Scholar] [CrossRef]
- Whiley, L.; Godzien, J.; Rupérez, F.J.; Legido-Quigley, C.; Barbas, C. In-vial dual extraction for direct Lc-ms analysis of plasma for comprehensive and highly reproducible metabolic fingerprinting. Anal. Chem. 2012, 84, 5992–5999. [Google Scholar] [CrossRef]
- Chen, S.; Hoene, M.; Li, J.; Li, Y.; Zhao, X.; Häring, H.-U.; Schleicher, E.D.; Weigert, C.; Xu, G.; Lehmann, R. Simultaneous extraction of metabolome and lipidome with methyl tert-butyl ether from a single small tissue sample for ultra-high performance liquid chromatography/mass spectrometry. J. Chromatogr. A 2013, 1298, 9–16. [Google Scholar] [CrossRef]
- Rombouts, C.; De Spiegeleer, M.; Van Meulebroek, L.; De Vos, W.H.; Vanhaecke, L. Validated comprehensive metabolomics and lipidomics analysis of colon tissue and cell lines. Anal. Chim. Acta 2019, 1066, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.D.; Souza, A.L.; Gerszten, R.E.; Clish, C.B. Targeted metabolomics. Curr. Protoc. Mol. Boil. 2012, 98, 30.2.1–30.2.24. [Google Scholar] [CrossRef] [PubMed]
- Coman, C.; Solari, F.A.; Hentschel, A.; Sickmann, A.; Zahedi, R.P.; Ahrends, R. Simultaneous metabolite, protein, lipid extraction (SIMPLEX): A combinatorial multimolecular omics approach for systems Biology. Mol. Cell. Proteom. 2016, 15, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Rampler, E.; Egger, D.; Schoeny, H.; Rusz, M.; Pacheco, M.P.; Marino, G.; Kasper, C.; Nägele, T.; Koellensperger, G. The power of LC-MS based multiomics: Exploring adipogenic differentiation of human mesenchymal stem/stromal cells. Molecules 2019, 24, 3615. [Google Scholar] [CrossRef]
- Serkova, N.J.; Standiford, T.J.; Stringer, K.A. The emerging field of quantitative blood metabolomics for biomarker discovery in critical illnesses. Am. J. Respir. Crit. Care Med. 2011, 184, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Heuillet, M.; Millard, P.; Cissé, M.Y.; Linares, L.K.; Letisse, F.; Manie, S.N.; Le Cam, L.; Portais, J.-C.; Bellvert, F. Simultaneous measurement of metabolite concentration and isotope incorporation by mass spectrometry. Anal. Chem. 2020, 92, 5890–5896. [Google Scholar] [CrossRef]
- Wu, L.; Mashego, M.R.; Van Dam, J.C.; Proell, A.M.; Vinke, J.L.; Ras, C.; Van Winden, W.A.; Van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal. Biochem. 2005, 336, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.D.; Yuan, J.; Kimball, E.H.; Rabinowitz, J.D. Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat. Protoc. 2008, 3, 1299–1311. [Google Scholar] [CrossRef]
- Millard, P.; Massou, S.; Wittmann, C.; Portais, J.-C.; Létisse, F. Sampling of intracellular metabolites for stationary and non-stationary 13C metabolic flux analysis in Escherichia coli. Anal. Biochem. 2014, 465, 38–49. [Google Scholar] [CrossRef]
- Ribbenstedt, A.; Ziarrusta, H.; Benskin, J.P. Development, characterization and comparisons of targeted and non-targeted metabolomics methods. PLoS ONE 2018, 13, e0207082. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Villaret-Cazadamont, J.; Claus, S.P.; Canlet, C.; Guillou, H.; Cabaton, N.J.; Ellero-Simatos, S. Important considerations for sample collection in metabolomics studies with a special focus on applications to liver functions. Metabolites 2020, 10, 104. [Google Scholar] [CrossRef]
- Pinu, F.; Villas-Boas, S.G.; Aggio, R. Analysis of intracellular metabolites from microorganisms: Quenching and extraction protocols. Metabolites 2017, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Martano, G.; Delmotte, N.; Kiefer, P.; Christen, P.; Kentner, D.; Bumann, D.; Vorholt, J.A. Fast sampling method for mammalian cell metabolic analyses using liquid chromatography–mass spectrometry. Nat. Protoc. 2014, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- A Sellick, C.; Hansen, R.; Stephens, G.; Goodacre, R.; Dickson, A.J. Metabolite extraction from suspension-cultured mammalian cells for global metabolite profiling. Nat. Protoc. 2011, 6, 1241–1249. [Google Scholar] [CrossRef]
- León, Z.; García-Cañaveras, J.C.; Donato, M.; Lahoz, A. Mammalian cell metabolomics: Experimental design and sample preparation. Electrophoresis 2013, 34, 2762–2775. [Google Scholar] [CrossRef]
- Villas-Boas, S.G.; Højer-Pedersen, J.; Åkesson, M.; Smedsgaard, J.; Nielsen, J. Global metabolite analysis of yeast: Evaluation of sample preparation methods. Yeast 2005, 22, 1155–1169. [Google Scholar] [CrossRef]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Nonlinear partial differential equations and applications: Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef]
- Legler, J.; Zalko, D.; Jourdan, F.; Jacobs, M.; Fromenty, B.; Balaguer, P.; Bourguet, W.; Kos, V.M.; Nadal, A.; Beausoleil, C.; et al. The GOLIATH project: Towards an internationally harmonised approach for testing metabolism disrupting compounds. Int. J. Mol. Sci. 2020, 21, 3480. [Google Scholar] [CrossRef]
- Merrick, B.A.; Paules, R.S.; Tice, R. Intersection of toxicogenomics and high throughput screening in the Tox21 program: An NIEHS perspective. Int. J. Biotechnol. 2015, 14, 7–27. [Google Scholar] [CrossRef]
- Houck, M.M.; Siegel, J.A. (Eds.) Chapter 6—Separation methods. In Fundamentals of Forensic Science, 3rd ed.; Academic Press: San Diego, CA, USA, 2015; pp. 121–151. ISBN 978-0-12-800037-3. [Google Scholar]
- Poole, C.F.; Poole, S.K. Extraction of organic compounds with room temperature ionic liquids. J. Chromatogr. A 2010, 1217, 2268–2286. [Google Scholar] [CrossRef] [PubMed]
- Humbert, L.; Hoizey, G.; Lhermitte, M. Chapter 7—Drugs involved in drug-facilitated crimes (DFC): Analytical aspects: 1—Blood and urine. In Toxicological Aspects of Drug-Facilitated Crimes; Kintz, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 159–180. ISBN 978-0-12-416748-3. [Google Scholar]
- Rabinowitz, J.D.; Kimball, E. Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal. Chem. 2007, 79, 6167–6173. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.V.; McDunn, J.E.; Gunst, P.R.; Smith, E.; Milburn, M.V.; Troyer, D.; A Lawton, K. Cancer detection and biopsy classification using concurrent histopathological and metabolomic analysis of core biopsies. Genome Med. 2012, 4, 33. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2011, 22, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.; Guo, A.C.; Chaudhary, K.K.; Liu, P.; Yallou, F.S.; Dong, E.; Aziat, F.; Wishart, D.S. Multi-platform characterization of the human cerebrospinal fluid metabolome: A comprehensive and quantitative update. Genome Med. 2012, 4, 38. [Google Scholar] [CrossRef]
- Ma, S.; Liu, S.; Wang, Q.; Chen, L.; Yang, P.; Sun, H. Fenofibrate-induced hepatotoxicity: A case with a special feature that is different from those in the LiverTox database. J. Clin. Pharm. Ther. 2019, 45, 204–207. [Google Scholar] [CrossRef]
- Soria, A.; Bocos, C.; Herrera, E. Opposite metabolic response to fenofibrate treatment in pregnant and virgin rats. J. Lipid Res. 2002, 43, 74–81. [Google Scholar]
- Lu, Y.; Boekschoten, M.V.; Wopereis, S.; Muller, M.; Kersten, S. Comparative transcriptomic and metabolomic analysis of fenofibrate and fish oil treatments in mice. Physiol. Genom. 2011, 43, 1307–1318. [Google Scholar] [CrossRef]
- Yang, Q.; Nagano, T.; Shah, Y.; Cheung, C.; Ito, S.; Gonzalez, F.J. The PPARα-Humanized Mouse: A model to investigate species differences in liver toxicity mediated by Pparα. Toxicol. Sci. 2007, 101, 132–139. [Google Scholar] [CrossRef]
- Croyal, M.; Kaabia, Z.; León, L.; Ramin-Mangata, S.; Baty, T.; Fall, F.; Billon-Crossouard, S.; Aguesse, A.; Hollstein, T.; Sullivan, D.; et al. Fenofibrate decreases plasma ceramide in type 2 diabetes patients: A novel marker of CVD? Diabetes Metab. 2018, 44, 143–149. [Google Scholar] [CrossRef]
- Shepherd, J. Mechanism of action of fibrates. Postgrad. Med J. 1993, 69, S34–S41. [Google Scholar] [PubMed]
- Yan, F.; Wang, Q.; Xu, C.; Cao, M.; Zhou, X.; Wang, T.; Yu, C.; Jing, F.; Chen, W.; Gao, L.; et al. Peroxisome proliferator-activated receptor α activation induces hepatic steatosis, suggesting an adverse effect. PLoS ONE 2014, 9, e99245. [Google Scholar] [CrossRef] [PubMed]
- Rogue, A.; Antherieu, S.; Vluggens, A.; Umbdenstock, T.; Claude, N.; De La Moureyre-Spire, C.; Weaver, R.J.; Guillouzo, A. PPAR agonists reduce steatosis in oleic acid-overloaded HepaRG cells. Toxicol. Appl. Pharmacol. 2014, 276, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Harano, Y.; Yasui, K.; Toyama, T.; Nakajima, T.; Mitsuyoshi, H.; Mimani, M.; Hirasawa, T.; Itoh, Y.; Okanoue, T. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver shionogi mice with hereditary fatty liver. Liver Int. 2006, 26, 613–620. [Google Scholar] [CrossRef]
- Tsoko, M.; Beauseigneur, F.; Gresti, J.; Demarquoy, J.; Clouet, P. Hypolipidaemic effects of fenofibrate are not altered by mildronate-mediated normalization of carnitine concentration in rat liver. Biochimie 1998, 80, 943–948. [Google Scholar] [CrossRef]
- Franco, M.E.; Fernandez-Luna, M.T.; Ramirez, A.J.; Lavado, R. Metabolomic-based assessment reveals dysregulation of lipid profiles in human liver cells exposed to environmental obesogens. Toxicol. Appl. Pharmacol. 2020, 398, 115009. [Google Scholar] [CrossRef]
- Oosterveer, M.H.; Grefhorst, A.; Van Dijk, T.H.; Havinga, R.; Staels, B.; Kuipers, F.; Groen, A.K.; Reijngoud, D.-J. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. J. Biol. Chem. 2009, 284, 34036–34044. [Google Scholar] [CrossRef]
- Ohta, T.; Masutomi, N.; Tsutsui, N.; Sakairi, T.; Mitchell, M.W.; Milburn, M.V.; Ryals, J.A.; Beebe, K.D.; Guo, L. Untargeted metabolomic profiling as an evaluative tool of fenofibrate-induced toxicology in fischer 344 male rats. Toxicol. Pathol. 2009, 37, 521–535. [Google Scholar] [CrossRef]
- Chachad, S.S.; Gole, M.; Malhotra, G.; Naidu, R. Comparison of pharmacokinetics of two fenofibrate tablet formulations in healthy human subjects. Clin. Ther. 2014, 36, 967–973. [Google Scholar] [CrossRef]
- Heindel, J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2016, 68, 3–33. [Google Scholar] [CrossRef]
- Heuillet, M.; Bellvert, F.; Cahoreau, E.; Létisse, F.; Millard, P.; Portais, J.-C. Methodology for the validation of isotopic analyses by mass spectrometry in stable-isotope labeling experiments. Anal. Chem. 2018, 90, 1852–1860. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villaret-Cazadamont, J.; Poupin, N.; Tournadre, A.; Batut, A.; Gales, L.; Zalko, D.; Cabaton, N.J.; Bellvert, F.; Bertrand-Michel, J. An Optimized Dual Extraction Method for the Simultaneous and Accurate Analysis of Polar Metabolites and Lipids Carried out on Single Biological Samples. Metabolites 2020, 10, 338. https://doi.org/10.3390/metabo10090338
Villaret-Cazadamont J, Poupin N, Tournadre A, Batut A, Gales L, Zalko D, Cabaton NJ, Bellvert F, Bertrand-Michel J. An Optimized Dual Extraction Method for the Simultaneous and Accurate Analysis of Polar Metabolites and Lipids Carried out on Single Biological Samples. Metabolites. 2020; 10(9):338. https://doi.org/10.3390/metabo10090338
Chicago/Turabian StyleVillaret-Cazadamont, Joran, Nathalie Poupin, Anthony Tournadre, Aurélie Batut, Lara Gales, Daniel Zalko, Nicolas J. Cabaton, Floriant Bellvert, and Justine Bertrand-Michel. 2020. "An Optimized Dual Extraction Method for the Simultaneous and Accurate Analysis of Polar Metabolites and Lipids Carried out on Single Biological Samples" Metabolites 10, no. 9: 338. https://doi.org/10.3390/metabo10090338
APA StyleVillaret-Cazadamont, J., Poupin, N., Tournadre, A., Batut, A., Gales, L., Zalko, D., Cabaton, N. J., Bellvert, F., & Bertrand-Michel, J. (2020). An Optimized Dual Extraction Method for the Simultaneous and Accurate Analysis of Polar Metabolites and Lipids Carried out on Single Biological Samples. Metabolites, 10(9), 338. https://doi.org/10.3390/metabo10090338