Rewiring of Lipid Metabolism in Adipose Tissue Macrophages in Obesity: Impact on Insulin Resistance and Type 2 Diabetes


Abstract
1. Introduction
2. ATM Heterogeneity in Obesity: Activation and Function
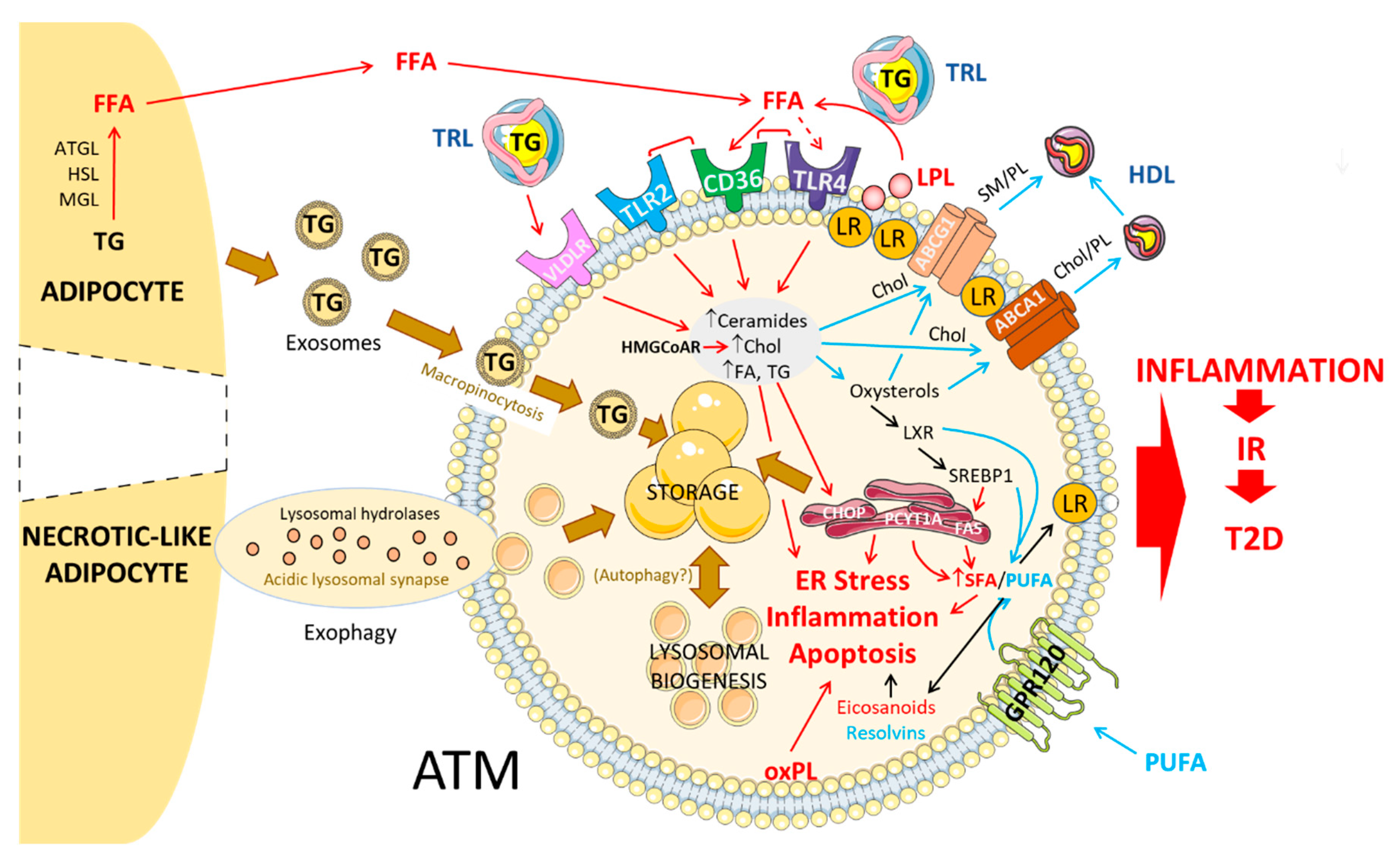
3. Lipid Metabolism in Healthy and Obese AT
3.1. Circulating Lipids: Mechanisms of Uptake into ATMs
3.1.1. FFA
3.1.2. VLDL/VLDLR
3.2. Adipocyte-Released Lipids: Mechanism of Uptake into ATMs
3.2.1. Lipolysis-Dependent Lipid Release
3.2.2. Exosomes
3.2.3. Exophagy
3.3. Fate of Lipids in ATM
3.3.1. Storage in the Form of LD
3.3.2. Lysosome-Mediated Lipid Handling
3.3.3. Autophagy
3.3.4. Lipid Efflux/Export
3.4. Regulation of Intracellular Lipid Metabolism in ATM
4. Membrane Dynamics and Inflammatory Signaling in ATM
4.1. Altered Chol ATM Content and Membrane Signaling
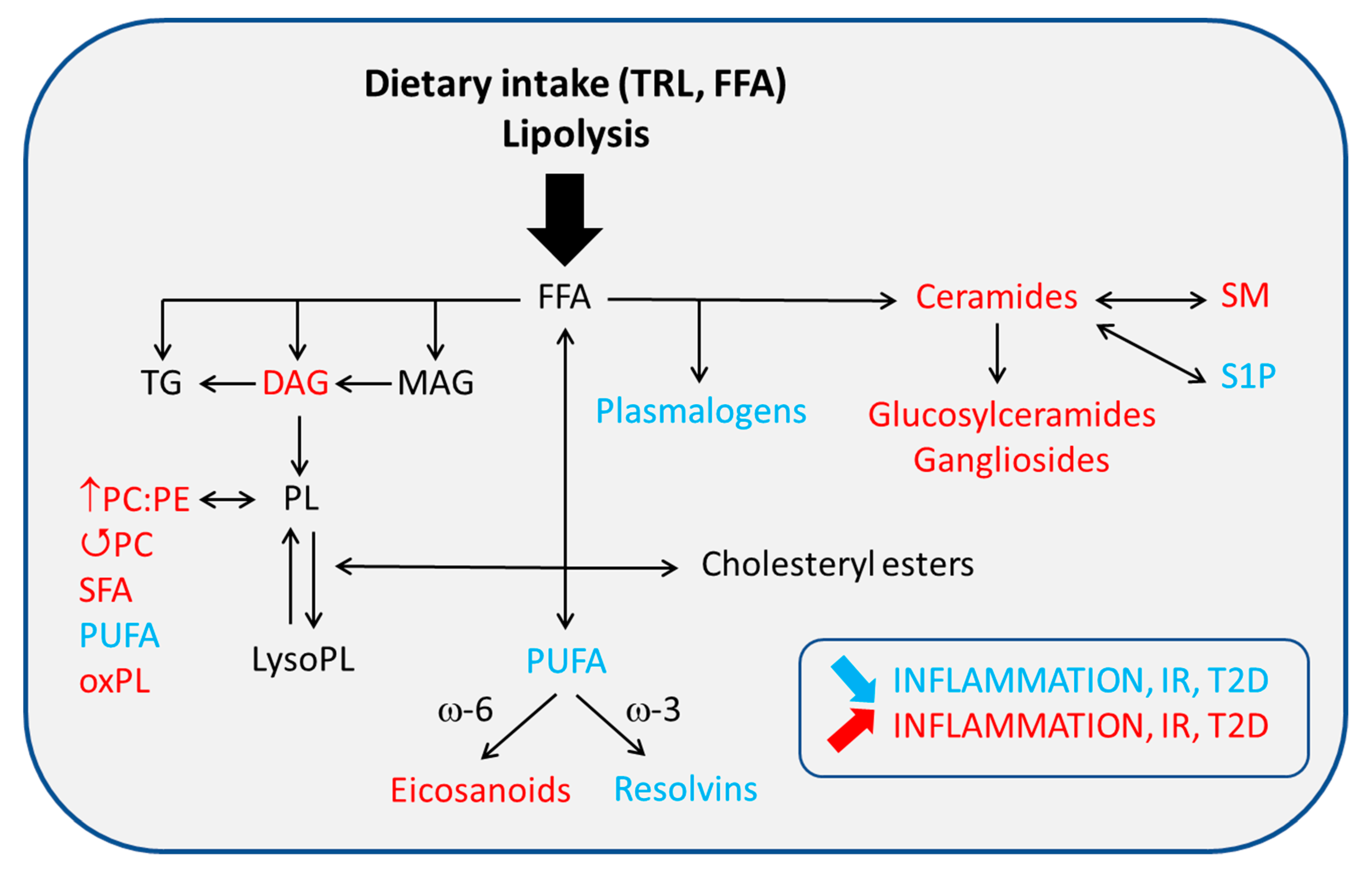
4.2. Altered PL/FA ATM Content and Membrane Signaling
4.2.1. Glycerophospholipids (GPL)
4.2.2. Sphingolipids (SL)
4.3. FA Nature and ATM-Mediated Inflammation
4.3.1. SFA
4.3.2. UFA
4.3.3. Eicosanoids and Other Bioactive Lipid Mediators
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| ABCA1/G1 | ATP-binding cassette A1/G1 |
| AT | Adipose tissue |
| ATM | Adipose tissue macrophages |
| Chol | Cholesterol |
| CLS | Crown-like structures |
| DHA | Docosahexaenoic acid |
| DIO | Diet-induced obesity |
| DNL | De novo lipogenesis |
| EPA | Eicosapentaenoic acid |
| FAS | Fatty acid synthase |
| FFA | Free Fatty Acid |
| HDL | High-density lipoprotein |
| HFD | High fat diet |
| IR | Insulin resistance |
| LD | Lipid droplet |
| LDL | Low-density lipoprotein |
| LPL | Lipoprotein lipase |
| LXR | Liver X Receptor |
| MCP-1/CCL2 | Monocyte chemoattractant protein-1/Chemokine C-C motif ligand 2 |
| Mme | Metabolically activated |
| MUFA | Monounsaturated fatty acid |
| PA | Palmitic acid |
| PE | Phosphatidylethanolamine |
| PG | Prostaglandin |
| PL | Phospholipid |
| PldG | Phosphatidylglycerol |
| PUFA | Polyunsaturated Fatty Acid |
| SFA | Saturated Fatty Acid |
| SL | Sphingolipid |
| SREBP | Sterol Regulatory Element-Binding Protein |
| T2D | Type 2 Diabetes |
| TG | Triglyceride |
| TLR | Toll-like receptor |
| VLDL | Very low-density lipoprotein |
References
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet Lond. Engl. 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Roglic, G.; Varghese, C.; Riley, L.; Harvey, A. (Eds.) Global Report on Diabetes; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Nimptsch, K.; Konigorski, S.; Pischon, T. Diagnosis of obesity and use of obesity biomarkers in science and clinical medicine. Metabolism 2019, 92, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, H.; Pecht, T.; Shaco-Levy, R.; Harman-Boehm, I.; Kirshtein, B.; Kuperman, Y.; Chen, A.; Blüher, M.; Shai, I.; Rudich, A. Adipose Tissue Foam Cells Are Present in Human Obesity. J. Clin. Endocrinol. Metab. 2013, 98, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H.; Lundgren, P.; Bleriot, C.; Liu, Z.; Deczkowska, A.; et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 2019, 178, 686–698. [Google Scholar] [CrossRef]
- Muir, L.A.; Kiridena, S.; Griffin, C.; DelProposto, J.B.; Geletka, L.; Martinez-Santibañez, G.; Zamarron, B.F.; Lucas, H.; Singer, K.; O’Rourke, R.W.; et al. Frontline Science: Rapid adipose tissue expansion triggers unique proliferation and lipid accumulation profiles in adipose tissue macrophages. J. Leukoc. Biol. 2018, 103, 615–628. [Google Scholar] [CrossRef]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic Dysfunction Drives a Mechanistically Distinct Proinflammatory Phenotype in Adipose Tissue Macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef]
- Coats, B.R.; Schoenfelt, K.Q.; Barbosa-Lorenzi, V.C.; Peris, E.; Cui, C.; Hoffman, A.; Zhou, G.; Fernandez, S.; Zhai, L.; Hall, B.A.; et al. Metabolically Activated Adipose Tissue Macrophages Perform Detrimental and Beneficial Functions during Diet-Induced Obesity. Cell Rep. 2017, 20, 3149–3161. [Google Scholar] [CrossRef]
- O’Reilly, M.E.; Kajani, S.; Ralston, J.C.; Lenighan, Y.M.; Roche, H.M.; McGillicuddy, F.C. Nutritionally Derived Metabolic Cues Typical of the Obese Microenvironment Increase Cholesterol Efflux Capacity of Adipose Tissue Macrophages. Mol. Nutr. Food Res. 2019, 63, 1800713. [Google Scholar] [CrossRef]
- Carroll, R.G.; Zasłona, Z.; Galván-Peña, S.; Koppe, E.L.; Sévin, D.C.; Angiari, S.; Triantafilou, M.; Triantafilou, K.; Modis, L.K.; O’Neill, L.A. An unexpected link between fatty acid synthase and cholesterol synthesis in proinflammatory macrophage activation. J. Biol. Chem. 2018, 293, 5509–5521. [Google Scholar] [CrossRef]
- Petkevicius, K.; Virtue, S.; Bidault, G.; Jenkins, B.; Çubuk, C.; Morgantini, C.; Aouadi, M.; Dopazo, J.; Serlie, M.J.; Koulman, A.; et al. Accelerated phosphatidylcholine turnover in macrophages promotes adipose tissue inflammation in obesity. eLife 2019, 8. [Google Scholar] [CrossRef]
- Köberlin, M.S.; Snijder, B.; Heinz, L.X.; Baumann, C.L.; Fauster, A.; Vladimer, G.I.; Gavin, A.-C.; Superti-Furga, G. A Conserved Circular Network of Coregulated Lipids Modulates Innate Immune Responses. Cell 2015, 162, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Li, P.-P.; Thapar, D.; Chapman, J.; Olefsky, J.M.; Neels, J.G. Ablation of CD11c-Positive Cells Normalizes Insulin Sensitivity in Obese Insulin Resistant Animals. Cell Metab. 2008, 8, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Jiao, P.; Nie, Y.; Kim, T.; Jun, D.; van Rooijen, N.; Yang, Z.; Xu, H. Clodronate Liposomes Improve Metabolic Profile and Reduce Visceral Adipose Macrophage Content in Diet-Induced Obese Mice. PLoS ONE 2011, 6, e24358. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Gao, M.; Qu, S.; Liu, D. Intraperitoneal Injection of Clodronate Liposomes Eliminates Visceral Adipose Macrophages and Blocks High-fat Diet-induced Weight Gain and Development of Insulin Resistance. AAPS J. 2013, 15, 1001–1011. [Google Scholar] [CrossRef]
- Aouadi, M.; Tencerova, M.; Vangala, P.; Yawe, J.C.; Nicoloro, S.M.; Amano, S.U.; Cohen, J.L.; Czech, M.P. Gene silencing in adipose tissue macrophages regulates whole-body metabolism in obese mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8278–8283. [Google Scholar] [CrossRef]
- Aouadi, M.; Vangala, P.; Yawe, J.C.; Tencerova, M.; Nicoloro, S.M.; Cohen, J.L.; Shen, Y.; Czech, M.P. Lipid storage by adipose tissue macrophages regulates systemic glucose tolerance. Am. J. Physiol.-Endocrinol. Metab. 2014, 307, E374–E383. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Nawaz, A.; Aminuddin, A.; Kado, T.; Takikawa, A.; Yamamoto, S.; Tsuneyama, K.; Igarashi, Y.; Ikutani, M.; Nishida, Y.; Nagai, Y.; et al. CD206+ M2-like macrophages regulate systemic glucose metabolism by inhibiting proliferation of adipocyte progenitors. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Orliaguet, L.; Dalmas, E.; Drareni, K.; Venteclef, N.; Alzaid, F. Mechanisms of Macrophage Polarization in Insulin Signaling and Sensitivity. Front. Endocrinol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- DiStefano, M.T.; Roth Flach, R.J.; Senol-Cosar, O.; Danai, L.V.; Virbasius, J.V.; Nicoloro, S.M.; Straubhaar, J.; Dagdeviren, S.; Wabitsch, M.; Gupta, O.T.; et al. Adipocyte-specific Hypoxia-inducible gene 2 promotes fat deposition and diet-induced insulin resistance. Mol. Metab. 2016, 5, 1149–1161. [Google Scholar] [CrossRef]
- Boutens, L.; Hooiveld, G.J.; Dhingra, S.; Cramer, R.A.; Netea, M.G.; Stienstra, R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia 2018, 61, 942–953. [Google Scholar] [CrossRef]
- Sharma, M.; Boytard, L.; Hadi, T.; Koelwyn, G.; Simon, R.; Ouimet, M.; Seifert, L.; Spiro, W.; Yan, B.; Hutchison, S.; et al. Enhanced glycolysis and HIF-1α activation in adipose tissue macrophages sustains local and systemic interleukin-1β production in obesity. Sci. Rep. 2020, 10, 5555. [Google Scholar] [CrossRef]
- Prieur, X.; Mok, C.Y.L.; Velagapudi, V.R.; Núñez, V.; Fuentes, L.; Montaner, D.; Ishikawa, K.; Camacho, A.; Barbarroja, N.; O’Rahilly, S.; et al. Differential Lipid Partitioning Between Adipocytes and Tissue Macrophages Modulates Macrophage Lipotoxicity and M2/M1 Polarization in Obese Mice. Diabetes 2011, 60, 797–809. [Google Scholar] [CrossRef]
- Miller, A.M.; Asquith, D.L.; Hueber, A.J.; Anderson, L.A.; Holmes, W.M.; McKenzie, A.N.; Xu, D.; Sattar, N.; McInnes, I.B.; Liew, F.Y. Interleukin-33 Induces Protective Effects in Adipose Tissue Inflammation During Obesity in Mice. Circ. Res. 2010, 107, 650–658. [Google Scholar] [CrossRef]
- Silva, H.M.; Báfica, A.; Rodrigues-Luiz, G.F.; Chi, J.; Santos, P.; d’Emery, A.; Reis, B.S.; van Hoytema Konijnenburg, D.P.; Crane, A.; Arifa, R.D.N.; et al. Vasculature-associated fat macrophages readily adapt to inflammatory and metabolic challenges. J. Exp. Med. 2019, 216, 786–806. [Google Scholar] [CrossRef]
- Hill, D.A.; Lim, H.-W.; Kim, Y.H.; Ho, W.Y.; Foong, Y.H.; Nelson, V.L.; Nguyen, H.C.B.; Chegireddy, K.; Kim, J.; Habertheuer, A.; et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc. Natl. Acad. Sci. USA 2018, 115, E5096–E5105. [Google Scholar] [CrossRef] [PubMed]
- Serbulea, V.; Upchurch, C.M.; Schappe, M.S.; Voigt, P.; DeWeese, D.E.; Desai, B.N.; Meher, A.K.; Leitinger, N. Macrophage phenotype and bioenergetics are controlled by oxidized phospholipids identified in lean and obese adipose tissue. Proc. Natl. Acad. Sci. USA 2018, 115, E6254–E6263. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Bechor, S.; Nachmias, D.; Elia, N.; Haim, Y.; Vatarescu, M.; Leikin-Frenkel, A.; Gericke, M.; Tarnovscki, T.; Las, G.; Rudich, A. Adipose tissue conditioned media support macrophage lipid-droplet biogenesis by interfering with autophagic flux. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 1001–1012. [Google Scholar] [CrossRef]
- Xu, X.; Grijalva, A.; Skowronski, A.; van Eijk, M.; Serlie, M.J.; Ferrante, A.W. Obesity Activates a Program of Lysosomal-Dependent Lipid Metabolism in Adipose Tissue Macrophages Independently of Classic Activation. Cell Metab. 2013, 18, 816–830. [Google Scholar] [CrossRef]
- Olivier, M.; Tanck, M.W.; Out, R.; Villard, E.F.; Lammers, B.; Bouchareychas, L.; Frisdal, E.; Superville, A.; Van Berkel, T.; Kastelein, J.J.; et al. Human ATP–Binding Cassette G1 Controls Macrophage Lipoprotein Lipase Bioavailability and Promotes Foam Cell Formation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2223–2231. [Google Scholar] [CrossRef]
- Poledne, R.; Kralova Lesna, I.; Kralova, A.; Fronek, J.; Cejkova, S. The relationship between non-HDL cholesterol and macrophage phenotypes in human adipose tissue. J. Lipid Res. 2016, 57, 1899–1905. [Google Scholar] [CrossRef]
- He, J.; Xu, X.; Francisco, A.; Ferrante, A.; Krakoff, J. Markers of adipose tissue macrophage content are negatively associated with serum HDL-C concentrations. Atherosclerosis 2011, 215, 243–246. [Google Scholar] [CrossRef][Green Version]
- Crawford, D.C.; Nord, A.S.; Badzioch, M.D.; Ranchalis, J.; McKinstry, L.A.; Ahearn, M.; Bertucci, C.; Shephard, C.; Wong, M.; Rieder, M.J.; et al. A common VLDLR polymorphism interacts with APOE genotype in the prediction of carotid artery disease risk. J. Lipid Res. 2008, 49, 588–596. [Google Scholar] [CrossRef]
- Shin, K.C.; Hwang, I.; Choe, S.S.; Park, J.; Ji, Y.; Kim, J.I.; Lee, G.Y.; Choi, S.H.; Ching, J.; Kovalik, J.-P.; et al. Macrophage VLDLR mediates obesity-induced insulin resistance with adipose tissue inflammation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Schoiswohl, G.; Stefanovic-Racic, M.; Menke, M.N.; Wills, R.C.; Surlow, B.A.; Basantani, M.K.; Sitnick, M.T.; Cai, L.; Yazbeck, C.F.; Stolz, D.B.; et al. Impact of Reduced ATGL-Mediated Adipocyte Lipolysis on Obesity-Associated Insulin Resistance and Inflammation in Male Mice. Endocrinology 2015, 156, 3610–3624. [Google Scholar] [CrossRef] [PubMed]
- Albert, J.S.; Yerges-Armstrong, L.M.; Horenstein, R.B.; Pollin, T.I.; Sreenivasan, U.T.; Chai, S.; Blaner, W.S.; Snitker, S.; O’Connell, J.R.; Gong, D.-W.; et al. Null Mutation in Hormone-Sensitive Lipase Gene and Risk of Type 2 Diabetes. N. Engl. J. Med. 2014, 370, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, S.E.; Grijalva, A.; Xu, X.; Ables, E.; Nomani, A.; Ferrante, A.W. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science 2019, 363, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mei, H.; Chang, X.; Chen, F.; Zhu, Y.; Han, X. Adipocyte-derived microvesicles from obese mice induce M1 macrophage phenotype through secreted miR-155. J. Mol. Cell Biol. 2016, 8, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Wang, X.; Liu, X.; Du, H.; Sun, C.; Shao, X.; Tian, J.; Gu, X.; Wang, H.; Tian, J.; et al. Adipose-Derived Exosomes Exert Proatherogenic Effects by Regulating Macrophage Foam Cell Formation and Polarization. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Deng, Z.; Poliakov, A.; Hardy, R.W.; Clements, R.; Liu, C.; Liu, Y.; Wang, J.; Xiang, X.; Zhang, S.; Zhuang, X.; et al. Adipose Tissue Exosome-Like Vesicles Mediate Activation of Macrophage-Induced Insulin Resistance. Diabetes 2009, 58, 2498–2505. [Google Scholar] [CrossRef]
- Haka, A.S.; Barbosa-Lorenzi, V.C.; Lee, H.J.; Falcone, D.J.; Hudis, C.A.; Dannenberg, A.J.; Maxfield, F.R. Exocytosis of macrophage lysosomes leads to digestion of apoptotic adipocytes and foam cell formation. J. Lipid Res. 2016, 57, 980–992. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Barbosa-Lorenzi, V.C.; Singh, R.K. Digestive exophagy: Phagocyte digestion of objects too large for phagocytosis. Traffic 2020, 21, 6–12. [Google Scholar] [CrossRef]
- Fujimoto, T.; Parton, R.G. Not Just Fat: The Structure and Function of the Lipid Droplet. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- van Dierendonck, X.A.M.H.; de la Rosa Rodriguez, M.A.; Georgiadi, A.; Mattijssen, F.; Dijk, W.; van Weeghel, M.; Singh, R.; Borst, J.W.; Stienstra, R.; Kersten, S. HILPDA Uncouples Lipid Droplet Accumulation in Adipose Tissue Macrophages from Inflammation and Metabolic Dysregulation. Cell Rep. 2020, 30, 1811–1822. [Google Scholar] [CrossRef]
- Li, F.; Zhang, H. Lysosomal Acid Lipase in Lipid Metabolism and Beyond. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Grijalva, A.; Xu, X.; Ferrante, A.W. Autophagy Is Dispensable for Macrophage-Mediated Lipid Homeostasis in Adipose Tissue. Diabetes 2016, 65, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Schlager, S.; Vujic, N.; Korbelius, M.; Duta-Mare, M.; Dorow, J.; Leopold, C.; Rainer, S.; Wegscheider, M.; Reicher, H.; Ceglarek, U.; et al. Lysosomal lipid hydrolysis provides substrates for lipid mediator synthesis in murine macrophages. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.-C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, Y.; Liang, T.; Lu, X.; Zhang, C.; Liu, X.; Jiang, X.; Martin, R.C.; Cheng, M.; Cai, L. ER Stress and Autophagy Dysfunction Contribute to Fatty Liver in Diabetic Mice. Int. J. Biol. Sci. 2015, 11, 559–568. [Google Scholar] [CrossRef]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy Regulates Cholesterol Efflux from Macrophage Foam Cells via Lysosomal Acid Lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef]
- Hadadi-Bechor, S.; Haim, Y.; Pecht, T.; Gat, R.; Tarnovscki, T.; Gericke, M.; Rudich, A. Autophagy differentially regulates macrophage lipid handling depending on the lipid substrate (oleic acid vs. acetylated-LDL) and inflammatory activation state. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 158527. [Google Scholar] [CrossRef]
- Subramanian, S.; Han, C.Y.; Chiba, T.; McMillen, T.S.; Wang, S.A.; Haw, A.; Kirk, E.A.; O’Brien, K.D.; Chait, A. Dietary Cholesterol Worsens Adipose Tissue Macrophage Accumulation and Atherosclerosis in Obese LDL Receptor–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 685–691. [Google Scholar] [CrossRef]
- Umemoto, T.; Han, C.Y.; Mitra, P.; Averill, M.M.; Tang, C.; Goodspeed, L.; Omer, M.; Subramanian, S.; Wang, S.; Den Hartigh, L.J.; et al. Apolipoprotein AI and High-Density Lipoprotein Have Anti-Inflammatory Effects on Adipocytes via Cholesterol Transporters: ATP-Binding Cassette A-1, ATP-Binding Cassette G-1, and Scavenger Receptor B-1. Circ. Res. 2013, 112, 1345–1354. [Google Scholar] [CrossRef]
- Laurila, P.-P.; Surakka, I.; Sarin, A.-P.; Yetukuri, L.; Hyötyläinen, T.; Söderlund, S.; Naukkarinen, J.; Tang, J.; Kettunen, J.; Mirel, D.B.; et al. Genomic, Transcriptomic, and Lipidomic Profiling Highlights the Role of Inflammation in Individuals With Low High-density Lipoprotein Cholesterol. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Liu, Y.; Yang, W.; Storey, C.; McMillen, T.S.; Houston, B.A.; Heinecke, J.W.; LeBoeuf, R.C. Hematopoietic ABCA1 deletion promotes monocytosis and worsens diet-induced insulin resistance in mice. J. Lipid Res. 2016, 57, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chung, S.; Bi, X.; Chuang, C.-C.; Brown, A.L.; Liu, M.; Seo, J.; Cuffe, H.; Gebre, A.K.; Boudyguina, E.; et al. Myeloid cell-specific ABCA1 deletion does not worsen insulin resistance in HF diet-induced or genetically obese mouse models. J. Lipid Res. 2013, 54, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Edgel, K.A.; McMillen, T.S.; Wei, H.; Pamir, N.; Houston, B.A.; Caldwell, M.T.; Mai, P.-O.T.; Oram, J.F.; Tang, C.; LeBoeuf, R.C. Obesity and weight loss result in increased adipose tissue ABCG1 expression in db/db mice. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2012, 1821, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Tarling, E.J.; McMillen, T.S.; Tang, C.; LeBoeuf, R.C. ABCG1 regulates mouse adipose tissue macrophage cholesterol levels and ratio of M1 to M2 cells in obesity and caloric restriction. J. Lipid Res. 2015, 56, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Takei, A.; Nagashima, S.; Takei, S.; Yamamuro, D.; Murakami, A.; Wakabayashi, T.; Isoda, M.; Yamazaki, H.; Ebihara, C.; Takahashi, M.; et al. Myeloid HMG-CoA Reductase Determines Adipose Tissue Inflammation, Insulin Resistance, and Hepatic Steatosis in Diet-Induced Obese Mice. Diabetes 2020, 69, 158–164. [Google Scholar] [CrossRef]
- Archer, A.; Stolarczyk, É.; Doria, M.L.; Helguero, L.; Domingues, R.; Howard, J.K.; Mode, A.; Korach-André, M.; Gustafsson, J.-Å. LXR activation by GW3965 alters fat tissue distribution and adipose tissue inflammation in ob/ob female mice. J. Lipid Res. 2013, 54, 1300–1311. [Google Scholar] [CrossRef]
- Lee, J.-H.; Phelan, P.; Shin, M.; Oh, B.-C.; Han, X.; Im, S.-S.; Osborne, T.F. SREBP-1a–stimulated lipid synthesis is required for macrophage phagocytosis downstream of TLR4-directed mTORC1. Proc. Natl. Acad. Sci. USA 2018, 115, E12228–E12234. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698. [Google Scholar] [CrossRef]
- Wei, X.; Song, H.; Yin, L.; Rizzo, M.G.; Sidhu, R.; Covey, D.F.; Ory, D.S.; Semenkovich, C.F. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature 2016, 539, 294–298. [Google Scholar] [CrossRef]
- Ecker, J.; Liebisch, G.; Englmaier, M.; Grandl, M.; Robenek, H.; Schmitz, G. Induction of fatty acid synthesis is a key requirement for phagocytic differentiation of human monocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 7817–7822. [Google Scholar] [CrossRef] [PubMed]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem. Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Spann, N.J.; Link, V.M.; Muse, E.D.; Strid, T.; Edillor, C.; Kolar, M.J.; Matsuzaka, T.; Hayakawa, S.; Tao, J.; et al. SREBP1 Contributes to Resolution of Pro-inflammatory TLR4 Signaling by Reprogramming Fatty Acid Metabolism. Cell Metab. 2017, 25, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Varin, A.; Thomas, C.; Ishibashi, M.; Ménégaut, L.; Gautier, T.; Trousson, A.; Bergas, V.; de Barros, J.P.P.; Narce, M.; Lobaccaro, J.M.A.; et al. Liver X Receptor Activation Promotes Polyunsaturated Fatty Acid Synthesis in Macrophages: Relevance in the Context of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1357–1365. [Google Scholar] [CrossRef]
- Li, P.; Spann, N.J.; Kaikkonen, M.U.; Lu, M.; Oh, D.Y.; Fox, J.N.; Bandyopadhyay, G.; Talukdar, S.; Xu, J.; Lagakos, W.S.; et al. NCoR Repression of LXRs Restricts Macrophage Biosynthesis of Insulin-Sensitizing Omega 3 Fatty Acids. Cell 2013, 155, 200–214. [Google Scholar] [CrossRef]
- Wang, B.; Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Jalil, A.; Bourgeois, T.; Ménégaut, L.; Lagrost, L.; Thomas, C.; Masson, D. Revisiting the Role of LXRs in PUFA Metabolism and Phospholipid Homeostasis. Int. J. Mol. Sci. 2019, 20, 3787. [Google Scholar] [CrossRef]
- Hresko, R.C.; Kraft, T.E.; Quigley, A.; Carpenter, E.P.; Hruz, P.W. Mammalian Glucose Transporter Activity Is Dependent upon Anionic and Conical Phospholipids. J. Biol. Chem. 2016, 291, 17271–17282. [Google Scholar] [CrossRef]
- Stulnig, T.M.; Huber, J.; Leitinger, N.; Imre, E.-M.; Angelisová, P.; Nowotny, P.; Waldhäusl, W. Polyunsaturated Eicosapentaenoic Acid Displaces Proteins from Membrane Rafts by Altering Raft Lipid Composition. J. Biol. Chem. 2001, 276, 37335–37340. [Google Scholar] [CrossRef]
- Ruysschaert, J.-M.; Lonez, C. Role of lipid microdomains in TLR-mediated signalling. Biochim. Biophys. Acta BBA Biomembr. 2015, 1848, 1860–1867. [Google Scholar] [CrossRef]
- Pietiläinen, K.H.; Róg, T.; Seppänen-Laakso, T.; Virtue, S.; Gopalacharyulu, P.; Tang, J.; Rodriguez-Cuenca, S.; Maciejewski, A.; Naukkarinen, J.; Ruskeepää, A.-L.; et al. Association of Lipidome Remodeling in the Adipocyte Membrane with Acquired Obesity in Humans. PLoS Biol. 2011, 9, e1000623. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, B.H.; Brown, T.J.; Simon, I.; Spector, A.A. Effect of the Membrane Lipid Environment on the Properties of Insulin Receptors. Diabetes 1981, 30, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-W.; Park, W.-J.; Kuperman, Y.; Boura-Halfon, S.; Pewzner-Jung, Y.; Futerman, A.H. Ablation of very long acyl chain sphingolipids causes hepatic insulin resistance in mice due to altered detergent-resistant membranes. Hepatology 2013, 57, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Pilon, M. Revisiting the membrane-centric view of diabetes. Lipids Health Dis. 2016, 15, 167. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ishibashi, M.; Seimon, T.; Lee, M.; Sharma, S.M.; Fitzgerald, K.A.; Samokhin, A.O.; Wang, Y.; Sayers, S.; Aikawa, M.; et al. Free Cholesterol Accumulation in Macrophage Membranes Activates Toll-Like Receptors and p38 Mitogen-Activated Protein Kinase and Induces Cathepsin, K. Circ. Res. 2009, 104, 455–465. [Google Scholar] [CrossRef]
- Ito, A.; Hong, C.; Rong, X.; Zhu, X.; Tarling, E.J.; Hedde, P.N.; Gratton, E.; Parks, J.; Tontonoz, P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. eLife 2015, 4, e08009. [Google Scholar] [CrossRef]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Albert, C.J.; Hong, C.; Duerr, M.A.; Chamberlain, B.T.; Tarling, E.J.; Ito, A.; Gao, J.; Wang, B.; Edwards, P.A.; et al. LXRs Regulate ER Stress and Inflammation through Dynamic Modulation of Membrane Phospholipid Composition. Cell Metab. 2013, 18, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Kayser, B.D.; Lhomme, M.; Prifti, E.; Cunha, C.D.; Marquet, F.; Chain, F.; Naas, I.; Pelloux, V.; Dao, M.; Kontush, A.; et al. Phosphatidylglycerols are induced by gut dysbiosis and inflammation, and favorably modulate adipose tissue remodeling in obesity. FASEB J. 2019, 33, 4741–4754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Dong, Y.-Q.; Wang, P.; Zhang, X.; Yan, Y.; Sun, L.; Liu, B.; Zhang, D.; Zhang, H.; Liu, H.; et al. Adipocyte-derived Lysophosphatidylcholine Activates Adipocyte and Adipose Tissue Macrophage Nod-Like Receptor Protein 3 Inflammasomes Mediating Homocysteine-Induced Insulin Resistance. EBioMedicine 2018, 31, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Di Gioia, M.; Spreafico, R.; Springstead, J.R.; Mendelson, M.M.; Joehanes, R.; Levy, D.; Zanoni, I. Endogenous oxidized phospholipids reprogram cellular metabolism and boost hyperinflammation. Nat. Immunol. 2020, 21, 42–53. [Google Scholar] [CrossRef]
- Sun, X.; Seidman, J.S.; Zhao, P.; Troutman, T.D.; Spann, N.J.; Que, X.; Zhou, F.; Liao, Z.; Pasillas, M.; Yang, X.; et al. Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 189–206. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a Novel Macrophage Phenotype That Develops in Response to Atherogenic Phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef]
- Que, X.; Hung, M.-Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Määttä, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef]
- Halasiddappa, L.M.; Koefeler, H.; Futerman, A.H.; Hermetter, A. Oxidized Phospholipids Induce Ceramide Accumulation in RAW 264.7 Macrophages: Role of Ceramide Synthases. PLoS ONE 2013, 8, e70002. [Google Scholar] [CrossRef]
- Bochkov, V.; Gesslbauer, B.; Mauerhofer, C.; Philippova, M.; Erne, P.; Oskolkova, O.V. Pleiotropic effects of oxidized phospholipids. Free Radic. Biol. Med. 2017, 111, 6–24. [Google Scholar] [CrossRef]
- Choromańska, B.; Myśliwiec, P.; Razak Hady, H.; Dadan, J.; Myśliwiec, H.; Chabowski, A.; Mikłosz, A. Metabolic Syndrome is Associated with Ceramide Accumulation in Visceral Adipose Tissue of Women with Morbid Obesity. Obesity 2019, oby.22405. [Google Scholar] [CrossRef]
- Raichur, S.; Brunner, B.; Bielohuby, M.; Hansen, G.; Pfenninger, A.; Wang, B.; Bruning, J.C.; Larsen, P.J.; Tennagels, N. The role of C16:0 ceramide in the development of obesity and type 2 diabetes: CerS6 inhibition as a novel therapeutic approach. Mol. Metab. 2019, 21, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Chimin, P.; Andrade, M.L.; Belchior, T.; Paschoal, V.A.; Magdalon, J.; Yamashita, A.S.; Castro, É.; Castoldi, A.; Chaves-Filho, A.B.; Yoshinaga, M.Y.; et al. Adipocyte mTORC1 deficiency promotes adipose tissue inflammation and NLRP3 inflammasome activation via oxidative stress and de novo ceramide synthesis. J. Lipid Res. 2017, 58, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Camell, C.D.; Nguyen, K.Y.; Jurczak, M.J.; Christian, B.E.; Shulman, G.I.; Shadel, G.S.; Dixit, V.D. Macrophage-specific de Novo Synthesis of Ceramide Is Dispensable for Inflammasome-driven Inflammation and Insulin Resistance in Obesity. J. Biol. Chem. 2015, 290, 29402–29413. [Google Scholar] [CrossRef] [PubMed]
- Frisdal, E.; Le Lay, S.; Hooton, H.; Poupel, L.; Olivier, M.; Alili, R.; Plengpanich, W.; Villard, E.F.; Gilibert, S.; Lhomme, M.; et al. Adipocyte ATP-Binding Cassette G1 Promotes Triglyceride Storage, Fat Mass Growth, and Human Obesity. Diabetes 2015, 64, 840–855. [Google Scholar] [CrossRef]
- Song, G.; Zong, C.; Shao, M.; Yu, Y.; Liu, Q.; Wang, H.; Qiu, T.; Jiao, P.; Guo, Z.; Lee, P.; et al. Phospholipid transfer protein (PLTP) deficiency attenuates high fat diet induced obesity and insulin resistance. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 1305–1313. [Google Scholar] [CrossRef]
- Abbott, S.K.; Else, P.L.; Atkins, T.A.; Hulbert, A.J. Fatty acid composition of membrane bilayers: Importance of diet polyunsaturated fat balance. Biochim. Biophys. Acta BBA Biomembr. 2012, 1818, 1309–1317. [Google Scholar] [CrossRef]
- Poledne, R.; Malinska, H.; Kubatova, H.; Fronek, J.; Thieme, F.; Kauerova, S.; Kralova Lesna, I. Polarization of Macrophages in Human Adipose Tissue is Related to the Fatty Acid Spectrum in Membrane Phospholipids. Nutrients 2019, 12, 8. [Google Scholar] [CrossRef]
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 2009, 15, 1383–1391. [Google Scholar] [CrossRef]
- Gianfrancesco, M.A.; Dehairs, J.; L’homme, L.; Herinckx, G.; Esser, N.; Jansen, O.; Habraken, Y.; Lassence, C.; Swinnen, J.V.; Rider, M.H.; et al. Saturated fatty acids induce NLRP3 activation in human macrophages through K+ efflux resulting from phospholipid saturation and Na, K-ATPase disruption. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Talbot, N.A.; Wheeler-Jones, C.P.; Cleasby, M.E. Palmitoleic acid prevents palmitic acid-induced macrophage activation and consequent p38 MAPK-mediated skeletal muscle insulin resistance. Mol. Cell. Endocrinol. 2014, 393, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK Expression by Macrophages Promotes Obesity-Induced Insulin Resistance and Inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, E.D.; Lee, S.J.; Kim, J.Y.; Duran, A.; Linares, J.F.; Yajima, T.; Müller, T.D.; Tschöp, M.H.; Smith, S.R.; Diaz-Meco, M.T.; et al. A Macrophage NBR1-MEKK3 Complex Triggers JNK-Mediated Adipose Tissue Inflammation in Obesity. Cell Metab. 2014, 20, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid–induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Yamada, H.; Umemoto, T.; Kawano, M.; Kawakami, M.; Kakei, M.; Momomura, S.; Ishikawa, S.; Hara, K. High-density lipoprotein and apolipoprotein A-I inhibit palmitate-induced translocation of toll-like receptor 4 into lipid rafts and inflammatory cytokines in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2017, 484, 403–408. [Google Scholar] [CrossRef]
- Zhou, H.; Urso, C.J.; Jadeja, V. Saturated Fatty Acids in Obesity-Associated Inflammation. J. Inflamm. Res. 2020, 13, 1–14. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110. [Google Scholar] [CrossRef]
- Schilling, J.D.; Machkovech, H.M.; He, L.; Sidhu, R.; Fujiwara, H.; Weber, K.; Ory, D.S.; Schaffer, J.E. Palmitate and Lipopolysaccharide Trigger Synergistic Ceramide Production in Primary Macrophages. J. Biol. Chem. 2013, 288, 2923–2932. [Google Scholar] [CrossRef]
- Zhang, Y.; Rao, E.; Zeng, J.; Hao, J.; Sun, Y.; Liu, S.; Sauter, E.R.; Bernlohr, D.A.; Cleary, M.P.; Suttles, J.; et al. Adipose Fatty Acid Binding Protein Promotes Saturated Fatty Acid-induced Macrophage Cell Death through Enhancing Ceramide Production. J. Immunol. Baltim. Md 1950 2017, 198, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.W.; Kwon, M.-J.; Choi, A.M.K.; Kim, H.-P.; Nakahira, K.; Hwang, D.H. Fatty Acids Modulate Toll-like Receptor 4 Activation through Regulation of Receptor Dimerization and Recruitment into Lipid Rafts in a Reactive Oxygen Species-dependent Manner. J. Biol. Chem. 2009, 284, 27384–27392. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated Fatty Acids, but Not Unsaturated Fatty Acids, Induce the Expression of Cyclooxygenase-2 Mediated through Toll-like Receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [PubMed]
- Rogero, M.; Calder, P. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic Lipids and Lipoproteins Trigger CD36-TLR2-Dependent Apoptosis in Macrophages Undergoing Endoplasmic Reticulum Stress. Cell Metab. 2010, 12, 467–482. [Google Scholar] [CrossRef]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes: From molecules to man. Biochem. Soc. Trans. 2017, 45, 1105–1115. [Google Scholar] [CrossRef]
- Moon, Y.-A.; Hammer, R.E.; Horton, J.D. Deletion of ELOVL5 leads to fatty liver through activation of SREBP-1c in mice. J. Lipid Res. 2009, 50, 412–423. [Google Scholar] [CrossRef]
- Heemskerk, M.; Giera, M.; Bouazzaoui, F.; Lips, M.; Pijl, H.; van Dijk, K.; van Harmelen, V. Increased PUFA Content and 5-Lipoxygenase Pathway Expression Are Associated with Subcutaneous Adipose Tissue Inflammation in Obese Women with Type 2 Diabetes. Nutrients 2015, 7, 7676–7690. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Zhang, W.; Cai, Y.; Shan, P.; Wu, D.; Zhang, B.; Liu, H.; Khan, Z.A.; Liang, G. Arachidonic acid inhibits inflammatory responses by binding to myeloid differentiation factor-2 (MD2) and preventing MD2/toll-like receptor 4 signaling activation. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165683. [Google Scholar] [CrossRef]
- Guijas, C.; Pérez-Chacón, G.; Astudillo, A.M.; Rubio, J.M.; Gil-de-Gómez, L.; Balboa, M.A.; Balsinde, J. Simultaneous activation of p38 and JNK by arachidonic acid stimulates the cytosolic phospholipase A 2 -dependent synthesis of lipid droplets in human monocytes. J. Lipid Res. 2012, 53, 2343–2354. [Google Scholar] [CrossRef]
- Guijas, C.; Bermúdez, M.A.; Meana, C.; Astudillo, A.M.; Pereira, L.; Fernández-Caballero, L.; Balboa, M.A.; Balsinde, J. Neutral Lipids Are Not a Source of Arachidonic Acid for Lipid Mediator Signaling in Human Foamy Monocytes. Cells 2019, 8, 941. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Ma, Y.; Ji, Z.; Hao, Y.; Yan, X.; Zhong, Y.; Tang, X.; Ren, W. Arachidonic acid induces macrophage cell cycle arrest through the JNK signaling pathway. Lipids Health Dis. 2018, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.; Matabosch, X.; Llebaria, A.; Balboa, M.A.; Balsinde, J. Blockade of arachidonic acid incorporation into phospholipids induces apoptosis in U937 promonocytic cells. J. Lipid Res. 2006, 47, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Jacob, R.F.; Shrivastava, S.; Sherratt, S.C.R.; Chattopadhyay, A. Eicosapentaenoic acid reduces membrane fluidity, inhibits cholesterol domain formation, and normalizes bilayer width in atherosclerotic-like model membranes. Biochim. Biophys. Acta BBA Biomembr. 2016, 1858, 3131–3140. [Google Scholar] [CrossRef]
- Fournier, N.; Sayet, G.; Vedie, B.; Nowak, M.; Allaoui, F.; Solgadi, A.; Caudron, E.; Chaminade, P.; Benoist, J.-F.; Paul, J.-L. Eicosapentaenoic acid membrane incorporation impairs cholesterol efflux from cholesterol-loaded human macrophages by reducing the cholesteryl ester mobilization from lipid droplets. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 1079–1091. [Google Scholar] [CrossRef]
- Rombaldova, M.; Janovska, P.; Kopecky, J.; Kuda, O. Omega-3 fatty acids promote fatty acid utilization and production of pro-resolving lipid mediators in alternatively activated adipose tissue macrophages. Biochem. Biophys. Res. Commun. 2017, 490, 1080–1085. [Google Scholar] [CrossRef]
- LeMieux, M.J.; Kalupahana, N.S.; Scoggin, S.; Moustaid-Moussa, N. Eicosapentaenoic Acid Reduces Adipocyte Hypertrophy and Inflammation in Diet-Induced Obese Mice in an Adiposity-Independent Manner. J. Nutr. 2015, 145, 411–417. [Google Scholar] [CrossRef]
- Titos, E.; Rius, B.; González-Périz, A.; López-Vicario, C.; Morán-Salvador, E.; Martínez-Clemente, M.; Arroyo, V.; Clària, J. Resolvin D1 and Its Precursor Docosahexaenoic Acid Promote Resolution of Adipose Tissue Inflammation by Eliciting Macrophage Polarization toward an M2-Like Phenotype. J. Immunol. 2011, 187, 5408–5418. [Google Scholar] [CrossRef]
- Williams-Bey, Y.; Boularan, C.; Vural, A.; Huang, N.-N.; Hwang, I.-Y.; Shan-Shi, C.; Kehrl, J.H. Omega-3 Free Fatty Acids Suppress Macrophage Inflammasome Activation by Inhibiting NF-κB Activation and Enhancing Autophagy. PLoS ONE 2014, 9, e97957. [Google Scholar] [CrossRef]
- Jin, X.; Dimitriadis, E.K.; Liu, Y.; Combs, C.A.; Chang, J.; Varsano, N.; Stempinski, E.; Flores, R.; Jackson, S.N.; Muller, L.; et al. Macrophages Shed Excess Cholesterol in Unique Extracellular Structures Containing Cholesterol Microdomains. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1504–1518. [Google Scholar] [CrossRef]
- De Boer, A.A.; Monk, J.M.; Liddle, D.M.; Power, K.A.; Ma, D.W.L.; Robinson, L.E. Fish Oil-Derived Long-Chain n-3 Polyunsaturated Fatty Acids Reduce Expression of M1-Associated Macrophage Markers in an ex vivo Adipose Tissue Culture Model, in Part through Adiponectin. Front. Nutr. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-inflammatory and Insulin-Sensitizing Effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Im, D.-S. Functions of omega-3 fatty acids and FFA4 (GPR120) in macrophages. Eur. J. Pharmacol. 2016, 785, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated Fatty Acid–Enriched High-Fat Diets Impede Adipose NLRP3 Inflammasome–Mediated IL-1β Secretion and Insulin Resistance Despite Obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef]
- Lima, E.A.; Silveira, L.S.; Masi, L.N.; Crisma, A.R.; Davanso, M.R.; Souza, G.I.G.; Santamarina, A.B.; Moreira, R.G.; Roque Martins, A.; de Sousa, L.G.O.; et al. Macadamia Oil Supplementation Attenuates Inflammation and Adipocyte Hypertrophy in Obese Mice. Mediators Inflamm. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Souza, C.O.; Teixeira, A.A.; Biondo, L.A.; Silveira, L.S.; Calder, P.C.; Rosa Neto, J.C. Palmitoleic acid reduces the inflammation in LPS-stimulated macrophages by inhibition of NFκB, independently of PPARs. Clin. Exp. Pharmacol. Physiol. 2017, 44, 566–575. [Google Scholar] [CrossRef]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a Lipokine, a Lipid Hormone Linking Adipose Tissue to Systemic Metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef]
- Shridas, P.; Bailey, W.M.; Gizard, F.; Oslund, R.C.; Gelb, M.H.; Bruemmer, D.; Webb, N.R. Group X secretory phospholipase A2 negatively regulates ABCA1 and ABCG1 expression and cholesterol efflux in macrophages. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2014–2021. [Google Scholar] [CrossRef]
- Wooten, J.S.; Wu, H.; Raya, J.; Perrard, X.D.; Gaubatz, J.; Hoogeveen, R.C. The Influence of an Obesogenic Diet on Oxysterol Metabolism in C57BL/6J Mice. Cholesterol 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Sato, H.; Taketomi, Y.; Ushida, A.; Isogai, Y.; Kojima, T.; Hirabayashi, T.; Miki, Y.; Yamamoto, K.; Nishito, Y.; Kobayashi, T.; et al. The Adipocyte-Inducible Secreted Phospholipases PLA2G5 and PLA2G2E Play Distinct Roles in Obesity. Cell Metab. 2014, 20, 119–132. [Google Scholar] [CrossRef]
- Rubio, J.M.; Rodríguez, J.P.; Gil-de-Gómez, L.; Guijas, C.; Balboa, M.A.; Balsinde, J. Group V secreted phospholipase A2 is upregulated by IL-4 in human macrophages and mediates phagocytosis via hydrolysis of ethanolamine phospholipids. J. Immunol. Baltim. Md 1950 2015, 194, 3327–3339. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, M.A.; Boyanovsky, B.B.; Jordan, C.T.; Wadsworth, M.P.; Taatjes, D.J.; de Beer, F.C.; Webb, N.R. Group V Secretory Phospholipase A 2 Promotes Atherosclerosis: Evidence From Genetically Altered Mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, D.; Hashidate, T.; Shimizu, T.; Shindou, H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J. Lipid Res. 2014, 55, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Hurt-Camejo, E. Phospholipase A2 enzymes and the risk of atherosclerosis. Eur. Heart J. 2012, 33, 2899–2909. [Google Scholar] [CrossRef] [PubMed]
- Stachowska, E.; Dolegowska, B.; Dziedziejko, V.; Rybicka, M.; Kaczmarczyk, M.; Bober, J.; Machalinski, B.; Chlubek, D. Prostaglandin E2 (PGE2) and thromboxane A2 (TXA2) synthesis is regulated by conjugated linoleic acids (CLA) in human macrophages. J. Physiol. Pharmacol. 2009, 60, 77–85. [Google Scholar]
- García-Alonso, V.; Titos, E.; Alcaraz-Quiles, J.; Rius, B.; Lopategi, A.; López-Vicario, C.; Jakobsson, P.-J.; Delgado, S.; Lozano, J.; Clària, J. Prostaglandin E2 Exerts Multiple Regulatory Actions on Human Obese Adipose Tissue Remodeling, Inflammation, Adaptive Thermogenesis and Lipolysis. PLoS ONE 2016, 11, e0153751. [Google Scholar] [CrossRef]
- Luan, B.; Yoon, Y.-S.; Le Lay, J.; Kaestner, K.H.; Hedrick, S.; Montminy, M. CREB pathway links PGE2 signaling with macrophage polarization. Proc. Natl. Acad. Sci. USA 2015, 112, 15642–15647. [Google Scholar] [CrossRef]
- Chan, P.; Hsiao, F.; Chang, H.; Wabitsch, M.; Hsieh, P.S. Importance of adipocyte cyclooxygenase-2 and prostaglandin E 2 -prostaglandin E receptor 3 signaling in the development of obesity-induced adipose tissue inflammation and insulin resistance. FASEB J. 2016, 30, 2282–2297. [Google Scholar] [CrossRef]
- Saraswathi, V.; Ramnanan, C.J.; Wilks, A.W.; Desouza, C.V.; Eller, A.A.; Murali, G.; Ramalingam, R.; Milne, G.L.; Coate, K.C.; Edgerton, D.S. Impact of hematopoietic cyclooxygenase-1 deficiency on obesity-linked adipose tissue inflammation and metabolic disorders in mice. Metabolism. 2013, 62, 1673–1685. [Google Scholar] [CrossRef]
- Hu, X.; Cifarelli, V.; Sun, S.; Kuda, O.; Abumrad, N.A.; Su, X. Major role of adipocyte prostaglandin E 2 in lipolysis-induced macrophage recruitment. J. Lipid Res. 2016, 57, 663–673. [Google Scholar] [CrossRef]
- Clària, J.; Dalli, J.; Yacoubian, S.; Gao, F.; Serhan, C.N. Resolvin D1 and Resolvin D2 Govern Local Inflammatory Tone in Obese Fat. J. Immunol. 2012, 189, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, J.; Tang, Y.; Kosuri, M.; Bhatnagar, A.; Spite, M. Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J. 2011, 25, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Titos, E.; Rius, B.; López-Vicario, C.; Alcaraz-Quiles, J.; García-Alonso, V.; Lopategi, A.; Dalli, J.; Lozano, J.J.; Arroyo, V.; Delgado, S.; et al. Signaling and Immunoresolving Actions of Resolvin D1 in Inflamed Human Visceral Adipose Tissue. J. Immunol. 2016, 197, 3360–3370. [Google Scholar] [CrossRef] [PubMed]
- González-Périz, A.; Horrillo, R.; Ferré, N.; Gronert, K.; Dong, B.; Morán-Salvador, E.; Titos, E.; Martínez-Clemente, M.; López-Parra, M.; Arroyo, V.; et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by ω-3 fatty acids: A role for resolvins and protectins. FASEB J. 2009, 23, 1946–1957. [Google Scholar] [CrossRef]
- Martínez-Fernández, L.; González-Muniesa, P.; Laiglesia, L.M.; Sáinz, N.; Prieto-Hontoria, P.L.; Escoté, X.; Odriozola, L.; Corrales, F.J.; Arbones-Mainar, J.M.; Martínez, J.A.; et al. Maresin 1 improves insulin sensitivity and attenuates adipose tissue inflammation in ob/ob and diet-induced obese mice. FASEB J. 2017, 31, 2135–2145. [Google Scholar] [CrossRef]
- Börgeson, E.; Johnson, A.M.F.; Lee, Y.S.; Till, A.; Syed, G.H.; Ali-Shah, S.T.; Guiry, P.J.; Dalli, J.; Colas, R.A.; Serhan, C.N.; et al. Lipoxin A4 Attenuates Obesity-Induced Adipose Inflammation and Associated Liver and Kidney Disease. Cell Metab. 2015, 22, 125–137. [Google Scholar] [CrossRef]
- Börgeson, E.; McGillicuddy, F.C.; Harford, K.A.; Corrigan, N.; Higgins, D.F.; Maderna, P.; Roche, H.M.; Godson, C. Lipoxin A 4 attenuates adipose inflammation. FASEB J. 2012, 26, 4287–4294. [Google Scholar] [CrossRef]
- Fujimori, K.; Aritake, K.; Oishi, Y.; Nagata, N.; Maehara, T.; Lazarus, M.; Urade, Y. L-PGDS-produced PGD2 in premature, but not in mature, adipocytes increases obesity and insulin resistance. Sci. Rep. 2019, 9, 1931. [Google Scholar] [CrossRef]
- Virtue, S.; Masoodi, M.; de Weijer, B.A.M.; van Eijk, M.; Mok, C.Y.L.; Eiden, M.; Dale, M.; Pirraco, A.; Serlie, M.J.; Griffin, J.L.; et al. Prostaglandin profiling reveals a role for haematopoietic prostaglandin D synthase in adipose tissue macrophage polarisation in mice and humans. Int. J. Obes. 2015, 39, 1151–1160. [Google Scholar] [CrossRef][Green Version]
- Li, P.; Oh, D.Y.; Bandyopadhyay, G.; Lagakos, W.S.; Talukdar, S.; Osborn, O.; Johnson, A.; Chung, H.; Mayoral, R.; Maris, M.; et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med. 2015, 21, 239–247. [Google Scholar] [CrossRef]
- Horrillo, R.; González-Périz, A.; Martínez-Clemente, M.; López-Parra, M.; Ferré, N.; Titos, E.; Morán-Salvador, E.; Deulofeu, R.; Arroyo, V.; Clària, J. 5-Lipoxygenase Activating Protein Signals Adipose Tissue Inflammation and Lipid Dysfunction in Experimental Obesity. J. Immunol. 2010, 184, 3978–3987. [Google Scholar] [CrossRef] [PubMed]
- Maya-Monteiro, C.M.; Almeida, P.E.; D’Ávila, H.; Martins, A.S.; Rezende, A.P.; Castro-Faria-Neto, H.; Bozza, P.T. Leptin Induces Macrophage Lipid Body Formation by a Phosphatidylinositol 3-Kinase- and Mammalian Target of Rapamycin-dependent Mechanism. J. Biol. Chem. 2008, 283, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-J.; Pena Calderin, E.; Hill, B.G.; Bhatnagar, A.; Hellmann, J. Exercise Promotes Resolution of Acute Inflammation by Catecholamine-Mediated Stimulation of Resolvin D1 Biosynthesis. J. Immunol. 2019, 203, 3013–3022. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ruedl, C. Obesity retunes turnover kinetics of tissue-resident macrophages in fat. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef]
- Sima, C.; Montero, E.; Nguyen, D.; Freire, M.; Norris, P.; Serhan, C.N.; Van Dyke, T.E. ERV1 Overexpression in Myeloid Cells Protects against High Fat Diet Induced Obesity and Glucose Intolerance. Sci. Rep. 2017, 7, 12848. [Google Scholar] [CrossRef]
- Chiang, N.; Libreros, S.; Norris, P.C.; de la Rosa, X.; Serhan, C.N. Maresin 1 activates LGR6 receptor promoting phagocyte immunoresolvent functions. J. Clin. Investig. 2019, 129, 5294–5311. [Google Scholar] [CrossRef]
- Hansen, T.V.; Vik, A.; Serhan, C.N. The Protectin Family of Specialized Pro-resolving Mediators: Potent Immunoresolvents Enabling Innovative Approaches to Target Obesity and Diabetes. Front. Pharmacol. 2019, 9, 1582. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| MEDIATOR | PRECURSOR | SPECIES: MODEL | SOURCE | CELL TYPE/TISSUE | REPORTED ACTIONS | REFERENCES |
|---|---|---|---|---|---|---|
| PRORESOLVING MEDIATORS | ||||||
| RVD1 | DHA | M: DIO | En, Ex | ATMs, Peritoneal macrophages |
| [139,162] |
| M: db/db mice | Ex | ATMs |
| [163] | ||
| M: DIO H: lean and obese | En, Ex | Epididymal AT, BMDM, adipocytes, PBMC |
| [162] | ||
| H: lean and obese ± T2D and THP-1 cells | En, Ex | Omental AT and THP-1 macrophages |
| [164] | ||
| RVD2 | DHA | M: DIO H: Lean and obese | Ex | Epididymal AT |
| [162] |
| RVE1 | EPA | M: ob/ob mice | Ex | Epididymal AT |
| [165] |
| PD1 | DHA | M: ob/ob mice | Ex | Epididymal AT |
| [165] |
| MARESIN 1 | DHA | M: DIO and ob/ob mice and H: lean subjects | Ex | Epididymal AT, subcutaneous AT |
| [166] |
| LXA4 | AA | M: DIO | Ex | Epididymal AT |
| [162,167] |
| M: DIO and 3T3-L1, J774 cells | Ex | ATMs, 3T3-L1 adipocytes, J774 macrophages |
| [167] | ||
| M: Standard diet and 3T3-L1, J774 cells | Ex | Perigonadal AT and 3T3-L1 adipocytes, J774 macrophages |
| [168] | ||
| PGD2 | AA | M: DIO + aP2-Cre/L-PGDSflox/flox mice, DIO and ob/ob mice and H: obese subjects | En, Ex | ATM, epididymal AT, subcutaneous AT, BMDM |
| [169,170] |
| PGE2 | AA | H: Lean and obese and 3T3-L1 cells | En, Ex | Omental AT, primary adipocytes, 3T3-L1 adipocytes |
| [157] |
| M: DIO + CREBLysMKO mice | Ex | BMDM |
| [158] | ||
| PRO-INFLAMMATORY MEDIATORS | ||||||
| PGE2 | AA | H: lean and obese, M: db/db mice, R: DIO and 3T3-L1, RAW cells | En | SVF, Primary adipocytes, 3T3-L1 adipocytes, RAW macrophages |
| [159] |
| LTB4 | AA | M: DIO and U937, 3T3-L1 cells | En, Ex | Peritoneal macrophages, 3T3-L1 adipocytes, |
| [171] |
| LTD4 | AA | M: DIO and ob/ob mice | En, Ex | Epididymal AT |
| [172] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahik, V.D.; Frisdal, E.; Le Goff, W. Rewiring of Lipid Metabolism in Adipose Tissue Macrophages in Obesity: Impact on Insulin Resistance and Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 5505. https://doi.org/10.3390/ijms21155505
Dahik VD, Frisdal E, Le Goff W. Rewiring of Lipid Metabolism in Adipose Tissue Macrophages in Obesity: Impact on Insulin Resistance and Type 2 Diabetes. International Journal of Molecular Sciences. 2020; 21(15):5505. https://doi.org/10.3390/ijms21155505
Chicago/Turabian StyleDahik, Veronica D., Eric Frisdal, and Wilfried Le Goff. 2020. "Rewiring of Lipid Metabolism in Adipose Tissue Macrophages in Obesity: Impact on Insulin Resistance and Type 2 Diabetes" International Journal of Molecular Sciences 21, no. 15: 5505. https://doi.org/10.3390/ijms21155505
APA StyleDahik, V. D., Frisdal, E., & Le Goff, W. (2020). Rewiring of Lipid Metabolism in Adipose Tissue Macrophages in Obesity: Impact on Insulin Resistance and Type 2 Diabetes. International Journal of Molecular Sciences, 21(15), 5505. https://doi.org/10.3390/ijms21155505