Inactivation of APC Induces CD34 Upregulation to Promote Epithelial-Mesenchymal Transition and Cancer Stem Cell Traits in Pancreatic Cancer

 , and
, and 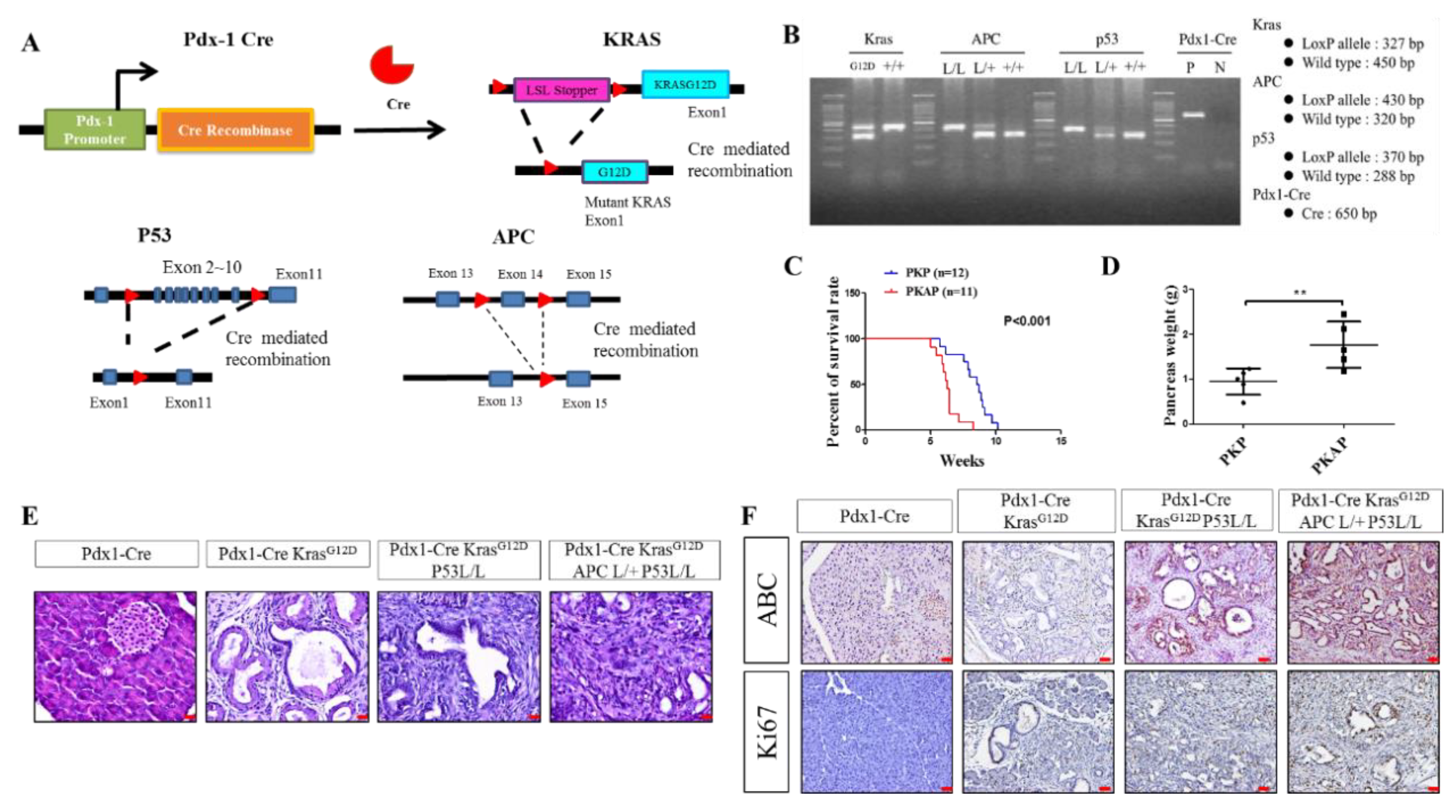{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
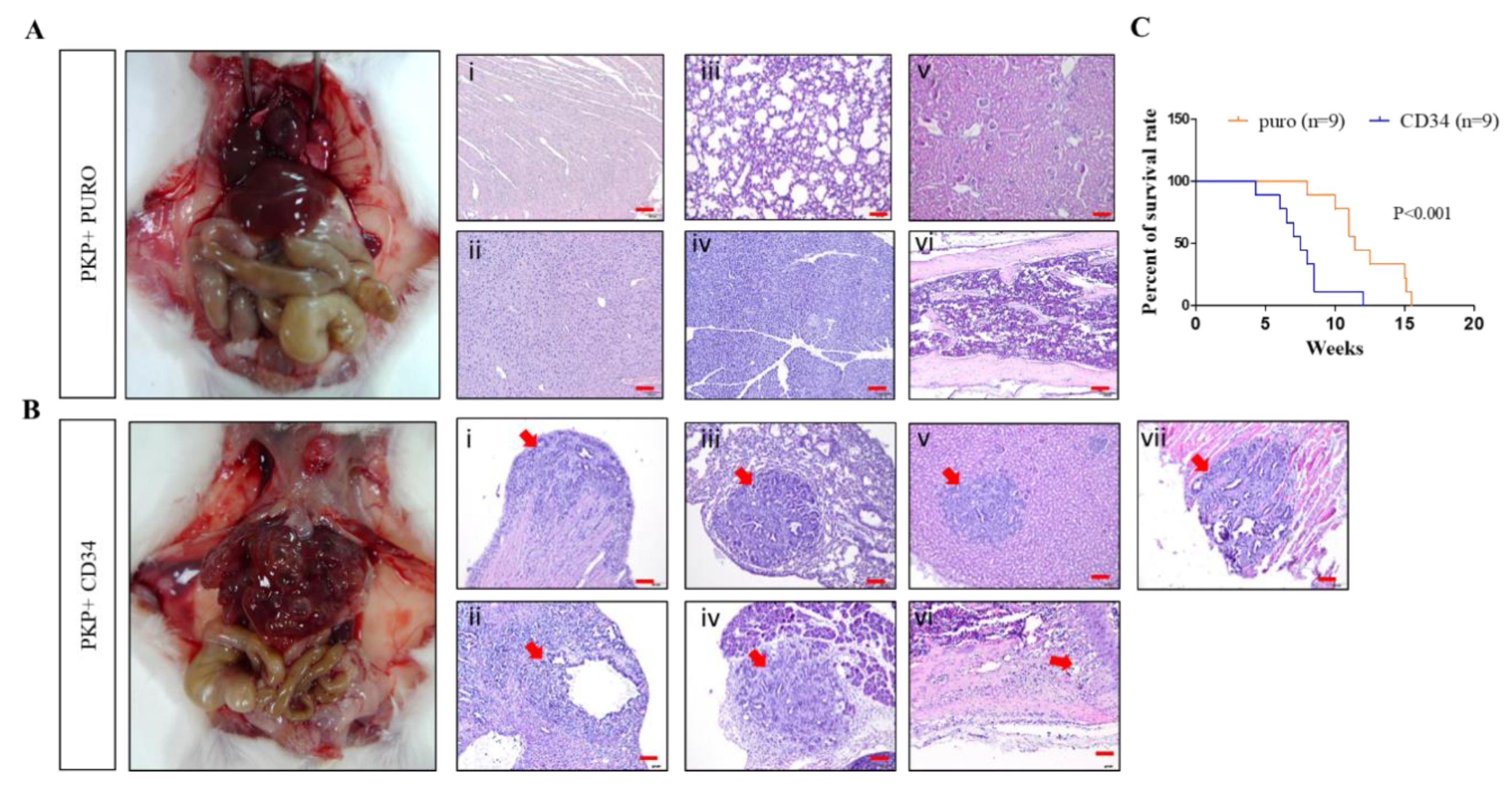
2.1. Genetically Engineered Mouse Models (GEMMs) Recapitulate the Loss of APC in PDAC to Evoke Metastatic PDAC Development
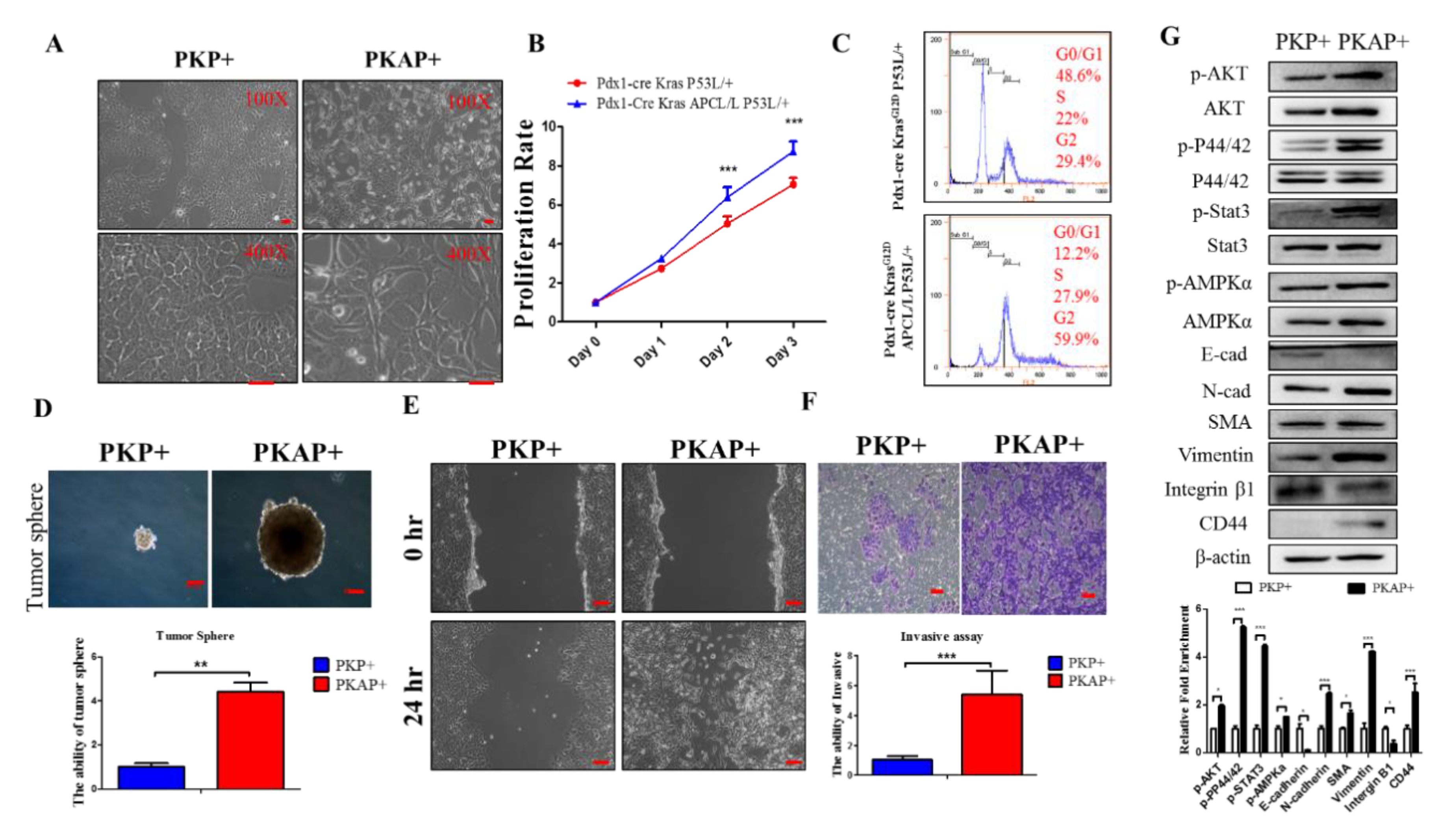
2.2. Inactivation of APC in PDAC Induced EMT to Increase PDAC Cell Tumorigenic and Migratory Abilities
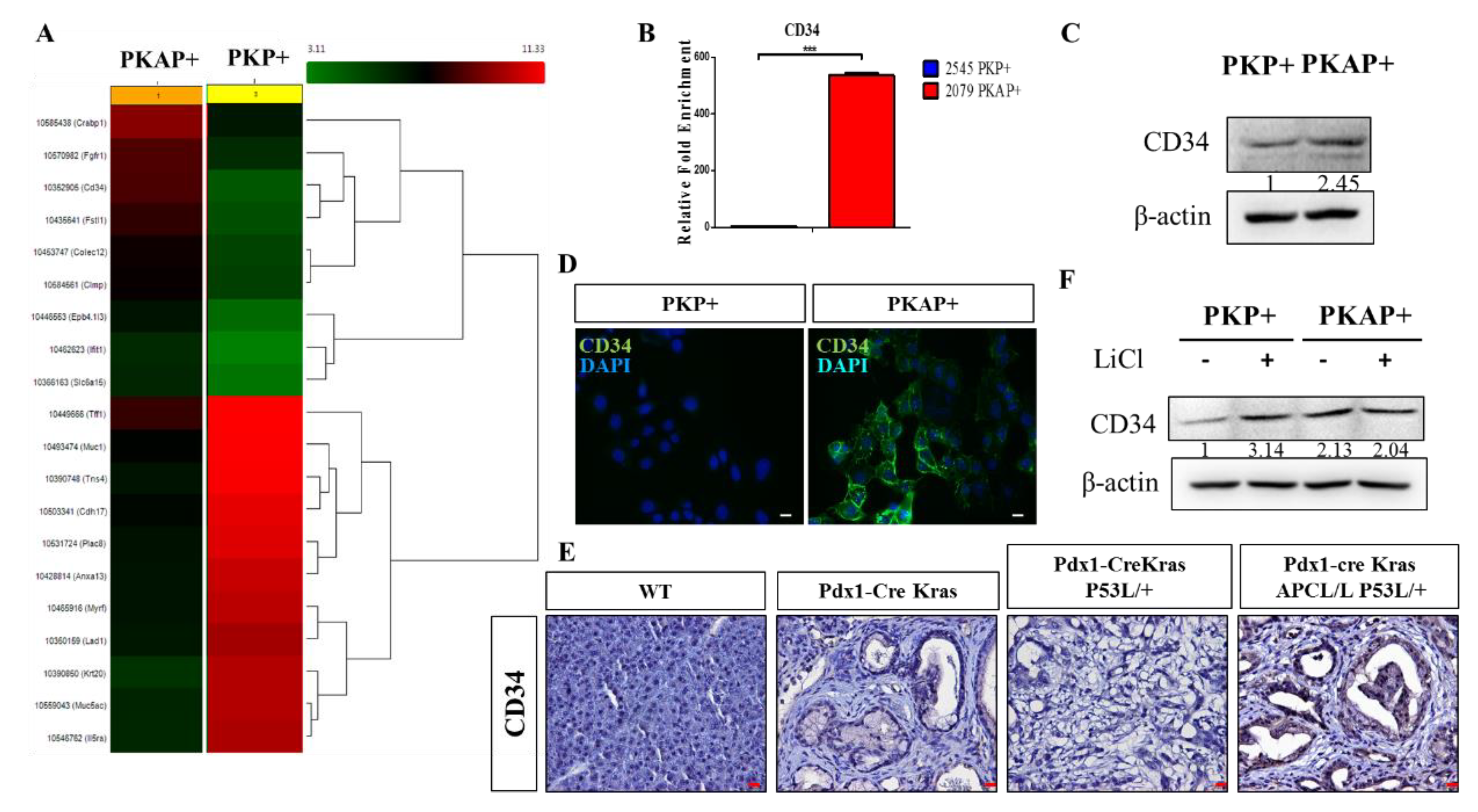
2.3. Activation of CD34 Pathway in PKAP+ PDAC Cells
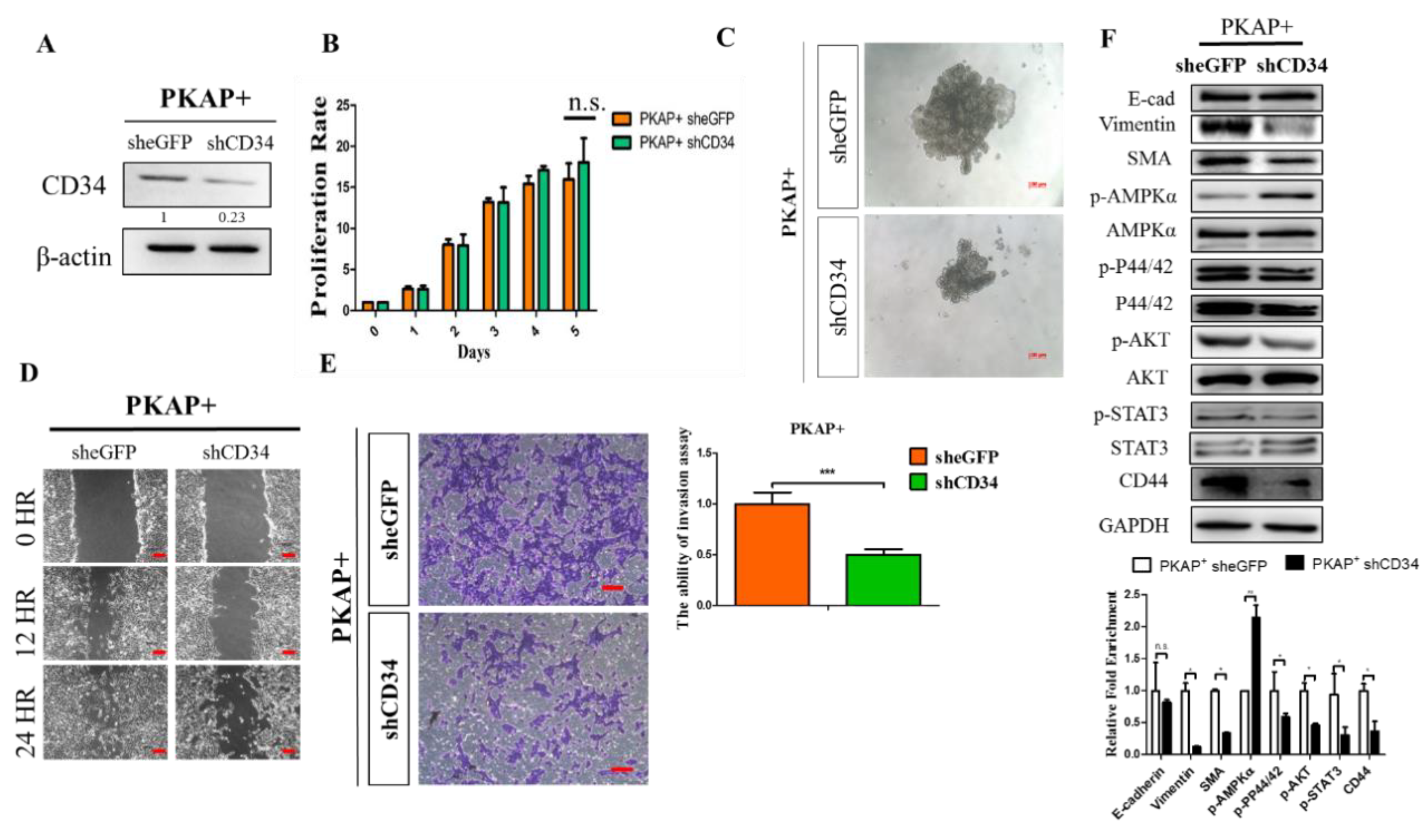
2.4. Knockdown of CD34 in PKAP+ Tumor Cells Reduces Cell Invasion and Migration
2.5. Overexpression CD34 Increases Tumorigenic and Cell Migration Abilities in PDAC
3. Discussion
4. Materials and Methods
4.1. Genetically Modified Mice and Mouse Genotyping
4.2. Immunohistochemistry (IHC) and Immunofluorescence (IF)
4.3. Western Blot Analysis
4.4. Real-Time–Quantitative PCR Analysis (RT–qPCR)
4.5. Cell Proliferation Assay
4.6. Colony Formation and Hanging Drop Assays
4.7. Wound Healing Assay
4.8. Flow Cytometry Analysis
4.9. Complementary DNA Microarray Analysis
4.10. Murine Primary PDAC Cell Culture, Cytokine and Inhibitor Treatment
4.11. Retroviral Production and Infection of Target Cells
4.12. Lentivirus Production and shRNA for Gene Knockdown
4.13. Intracardiac Injection of Metastatic Tumor Model
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PDAC | Pancreatic ductal adenocarcinoma |
| EMT | Epithelial–mesenchymal transition |
| IF | Immunofluorescence |
| IHC | Immunohistochemistry |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA: A Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Kong, D.; Sarkar, F.H. Pancreatic cancer: Understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Suker, M.; Beumer, B.R.; Sadot, E.; Marthey, L.; Faris, J.E.; Mellon, E.A.; El-Rayes, B.F.; Wang-Gilliam, A.; Lacy, J.; Moorcraft, S.Y.; et al. FOLFIRINOX for locally advanced pancreatic cancer: A systematic review and patient-level meta-analysis. Lancet Oncol. 2016, 17, 801–810. [Google Scholar] [CrossRef]
- Bosetti, C.; Bertuccio, P.; Negri, E.; La Vecchia, C.; Zeegers, M.P.; Boffetta, P. Pancreatic cancer: Overview of descriptive epidemiology. Mol. Carcinogenesis 2012, 51, 3–13. [Google Scholar] [CrossRef]
- Raimondi, S.; Maisonneuve, P.; Lowenfels, A.B. Epidemiology of pancreatic cancer: An overview. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 699–708. [Google Scholar] [CrossRef]
- Michaud, D.S.; Giovannucci, E.; Willett, W.C.; Colditz, G.A.; Stampfer, M.J.; Fuchs, C.S. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001, 286, 921–929. [Google Scholar] [CrossRef]
- Klein, A.P.; Hruban, R.H.; Brune, K.A.; Petersen, G.M.; Goggins, M. Familial pancreatic cancer. Cancer J. 2001, 7, 266–273. [Google Scholar]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef]
- Kuo, T.L.; Weng, C.C.; Kuo, K.K.; Chen, C.Y.; Wu, D.C.; Hung, W.C.; Cheng, K.H. APC haploinsufficiency coupled with p53 loss sufficiently induces mucinous cystic neoplasms and invasive pancreatic carcinoma in mice. Oncogene 2016, 35, 2223–2234. [Google Scholar] [CrossRef]
- Bardeesy, N.; DePinho, R.A. DePinho, Pancreatic cancer biology and genetics. Nat. Rev. Cancer 2002, 2, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Hornre, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef] [PubMed]
- Horii, A.; Nakatsuru, S.; Miyoshi, Y.; Ichii, S.; Nagase, H.; Kato, Y.; Yanagisawa, A.; Nakamura, Y. The APC gene, responsible for familial adenomatous polyposis, is mutated in human gastric cancer. Cancer Res. 1992, 52, 3231–3233. [Google Scholar] [PubMed]
- Miyoshi, Y.; Ando, H.; Nagase, H.; Nishisho, I.; Horii, A.; Miki, Y.; Mori, T.; Utsunomiya, J.; Baba, S.; Petersen, G. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc. Natl. Acad. Sci. USA 1992, 89, 4452–4456. [Google Scholar] [CrossRef] [PubMed]
- Horii, A.; Nakatsuru, S.; Miyoshi, Y.; Ichii, S.; Nagase, H.; Ando, H.; Yanagisawa, A.; Tsuchiya, E.; Kato, Y.; Nakamura, Y. Frequent somatic mutations of the APC gene in human pancreatic cancer. Cancer Res. 1992, 52, 6696–6698. [Google Scholar] [PubMed]
- McKie, A.B.; Lemoine, N.R.; Filipe, M.I. Abnormalities affecting the APC and MCC tumour suppressor gene loci on chromosome 5q occur frequently in gastric cancer but not in pancreatic cancer. Int. J. Cancer 1993, 55, 598–603. [Google Scholar] [CrossRef]
- Behrens, J.; Jerchow, B.A.; Würtele, M.; Grimm, J.; Asbrand, C.; Wirtz, R.; Kühl, M.; Wedlich, D.; Birchmeier, W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science 1998, 280, 596–599. [Google Scholar] [CrossRef]
- Neufeld, K.L.; Zhang, F.; Cullen, B.R.; White, R.L. APC-mediated downregulation of beta-catenin activity involves nuclear sequestration and nuclear export. EMBO Rep. 2000, 1, 519–523. [Google Scholar] [CrossRef]
- Huang, H.; Mahler-Araujo, B.M.; Sankila, A.; Chimelli, L.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. APC mutations in sporadic medulloblastomas. Am. J. Pathol. 2000, 156, 433–437. [Google Scholar] [CrossRef]
- Gocke, C.D.; Benko, F.A.; Kopreski, M.S.; McGARRITY, T.J. p53 and APC mutations are detectable in the plasma and serum of patients with colorectal cancer (CRC) or adenomas. Ann. N. Y. Acad. Sci. 2000, 906, 44–50. [Google Scholar] [CrossRef]
- White, B.D.; Chien, A.J.; Dawson, D.W. Dysregulation of Wnt/beta-catenin signaling in gastrointestinal cancers. Gastroenterology 2012, 142, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Driscoll, D.R.; De Jesus-Monge, W.E.; Klimstra, D.S.; Lewis, B.C. Activated wnt signaling in stroma contributes to development of pancreatic mucinous cystic neoplasms. Gastroenterology 2014, 146, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.I.; Jones, K.T. Non-canonical function of spindle assembly checkpoint proteins after APC activation reduces aneuploidy in mouse oocytes. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Kuipers, J.; Rosenberg, C.; Smits, R.; Kielman, M.; Gaspar, C.; van Es, J.H.; Breukel, C.; Wiegant, J.; Giles, R.H. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat. Cell. Biol. 2001, 3, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Smits, R.; Clevers, H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef]
- Gaspar, C.; Franken, P.; Molenaar, L.; Breukel, C.; van der Valk, M.; Smits, R.; Fodde, R. A targeted constitutive mutation in the APC tumor suppressor gene underlies mammary but not intestinal tumorigenesis. PLoS Genet. 2009, 5, e1000547. [Google Scholar] [CrossRef]
- Kuraguchi, M.; Wang, X.P.; Bronson, R.T.; Rothenberg, R.; Ohene-Baah, N.Y.; Lund, J.J.; Kucherlapati, M.; Maas, R.L.; Kucherlapati, R. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS Genet. 2006, 2, e146. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Hanahan, D. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef]
- Weng, C.C.; Hawse, J.R.; Subramaniam, M.; Chang, V.H.S.; Yu, W.C.Y.; Hung, W.C.; Chen, L.-T.; Cheng, K.H. KLF10 loss in the pancreas provokes activation of SDF-1 and induces distant metastases of pancreatic ductal adenocarcinoma in the Kras(G12D) p53(flox/flox) model. Oncogene 2017, 36, 5532–5543. [Google Scholar] [CrossRef]
- Kuo, T.L.; Cheng, K.H.; Shan, Y.S.; Chen, L.T.; Hung, W.C. beta-catenin-activated autocrine PDGF/Src signaling is a therapeutic target in pancreatic cancer. Theranostics 2019, 9, 324–336. [Google Scholar] [CrossRef] [PubMed]
- SStanger, B.Z.; Stiles, B.; Lauwers, G.Y.; Bardeesy, N.; Mendoza, M.; Wang, Y.; Greenwood, A.; Cheng, K.; McLaughlin, M.; Brown, D. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell 2005, 8, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Fackler, M.J.; Civin, C.I.; May, W.S. CD34: Structure, biology, and clinical utility. Blood 1996, 87, 1–13. [Google Scholar] [CrossRef]
- Majdic, O.; Stockl, J.; Pickl, W.F.; Bohuslav, J.; Strobl, H.; Scheinecker, C.; Knapp, W. Signaling and induction of enhanced cytoadhesiveness via the hematopoietic progenitor cell surface molecule CD34. Blood 1994, 83, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Champseix, C.; Marechal, V.; Khazaal, I.; Schwartz, O.; Fournier, S.; Schlegel, N.; Heard, J.M. A cell surface marker gene transferred with a retroviral vector into CD34+ cord blood cells is expressed by their T-cell progeny in the SCID-hu thymus. Blood 1996, 88, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Galy, A.; Rudraraju, S.; Baynes, R.; Klein, J. Recovery of lymphocyte and dendritic cell subsets after autologous CD34+ cell transplantation. Bone Marrow Transpl. 2000, 25, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Mohle, R.; Bautz, F.; Rafii, S.; Moore, M.A.; Brugger, W.; Kanz, L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood 1998, 91, 4523–4530. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Canque, B.; Gluckman, J.C. Human dendritic cell differentiation pathway from CD34+ hematopoietic precursor cells. Blood 1996, 87, 535–544. [Google Scholar] [CrossRef]
- Arakawa, A.; Soh, S.; Chakraborty, S.; Scardino, P.T.; Wheeler, T.M. Prognostic significance of angiogenesis in clinically localized prostate cancer (staining for Factor VIII-related antigen and CD34 Antigen. Prostate Cancer Prostatic Dis. 1997, 1, 32–38. [Google Scholar] [CrossRef][Green Version]
- Ma, J.; Siegel, R.; Jemal, A. Pancreatic cancer death rates by race among US men and women, 1970–2009. J. Natl. Cancer Inst. 2013, 105, 1694–1700. [Google Scholar] [CrossRef]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.M.; Neoptolemos, J.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Wilentz, R.E.; Goggins, M.; Offerhaus, G.J.A.; Yeo, C.J.; Kern, S.E. Pathology of incipient pancreatic cancer. Ann. Oncol. 1999, 10 (Suppl. 4), 9–11. [Google Scholar] [CrossRef]
- Yasui, M.; Park, Y.D.; Okamura, T.; Chayama, K.; Yoshimoto, T.; Inoue, M.; Yagi, K.; Kawa, K. CD34+ progenitor cell transplantation from two HLA-mismatched healthy fathers to two infants with severe aplastic anemia. Int. J. Hematol. 1998, 67, 15–22. [Google Scholar] [CrossRef]
- George, A.A.; Franklin, J.; Kerkof, K.; Shah, A.J.; Price, M.; Tsark, E.; Bockstoce, D.; Yao, D.; Hart, N.; Carcich, S. Detection of leukemic cells in the CD34(+)CD38(-) bone marrow progenitor population in children with acute lymphoblastic leukemia. Blood 2001, 97, 3925–3930. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Cho, D.; Chung, I.J.; Cho, S.H.; Park, K.S.; Park, M.R.; Ryang, D.W.; Kim, H.J. CD34 expression is associated with poor clinical outcome in patients with acute promyelocytic leukemia. Am. J. Hematol. 2003, 73, 149–153. [Google Scholar] [CrossRef]
- Won, E.J.; Kim, H.R.; Park, R.Y.; Choi, S.Y.; Shin, J.H.; Suh, S.P.; Ryang, D.W.; Szardenings, M.; Shin, M.G. Direct confirmation of quiescence of CD34+CD38- leukemia stem cell populations using single cell culture, their molecular signature and clinicopathological implications. BMC Cancer 2015, 15, 217. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Omori, N.; Omori, M.; Evarts, R.P.; Teramoto, T.; Miller, M.J.; Hoang, T.N.; Thorgeirsson, S.S. Partial cloning of rat CD34 cDNA and expression during stem cell-dependent liver regeneration in the adult rat. Hepatology 1997, 26, 720–727. [Google Scholar] [CrossRef]
- Park, S.C.; Nguyen, N.T.; Eun, J.R.; Zhang, Y.; Jung, Y.J.; Tschudy-Seney, B.; Trotsyuk, A.; Lam, A.; Ramsamooj, B.; Zhang, Y.; et al. Identification of cancer stem cell subpopulations of CD34(+) PLC/PRF/5 that result in three types of human liver carcinomas. Stem Cells Dev. 2015, 24, 1008–1021. [Google Scholar] [CrossRef]
- Weng, C.C.; Hsieh, M.J.; Wu, C.C.; Lin, Y.C.; Shan, Y.S.; Hung, W.C.; Chen, L.-T.; Cheng, K.-H. Loss of the transcriptional repressor TGIF1 results in enhanced Kras-driven development of pancreatic cancer. Mol. Cancer 2019, 18, 96. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, M.J.; Chiu, T.-J.; Lin, Y.C.; Weng, C.-C.; Weng, Y.-T.; Hsiao, C.-C.; Cheng, K.-h. Inactivation of APC Induces CD34 Upregulation to Promote Epithelial-Mesenchymal Transition and Cancer Stem Cell Traits in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 4473. https://doi.org/10.3390/ijms21124473
Hsieh MJ, Chiu T-J, Lin YC, Weng C-C, Weng Y-T, Hsiao C-C, Cheng K-h. Inactivation of APC Induces CD34 Upregulation to Promote Epithelial-Mesenchymal Transition and Cancer Stem Cell Traits in Pancreatic Cancer. International Journal of Molecular Sciences. 2020; 21(12):4473. https://doi.org/10.3390/ijms21124473
Chicago/Turabian StyleHsieh, Mei Jen, Tai-Jan Chiu, Yu Chun Lin, Ching-Chieh Weng, Yu-Ting Weng, Chang-Chun Hsiao, and Kuang-hung Cheng. 2020. "Inactivation of APC Induces CD34 Upregulation to Promote Epithelial-Mesenchymal Transition and Cancer Stem Cell Traits in Pancreatic Cancer" International Journal of Molecular Sciences 21, no. 12: 4473. https://doi.org/10.3390/ijms21124473
APA StyleHsieh, M. J., Chiu, T.-J., Lin, Y. C., Weng, C.-C., Weng, Y.-T., Hsiao, C.-C., & Cheng, K.-h. (2020). Inactivation of APC Induces CD34 Upregulation to Promote Epithelial-Mesenchymal Transition and Cancer Stem Cell Traits in Pancreatic Cancer. International Journal of Molecular Sciences, 21(12), 4473. https://doi.org/10.3390/ijms21124473