Revisiting NO2 as Protecting Group of Arginine in Solid-Phase Peptide Synthesis

 ,
,  and
and 
Abstract
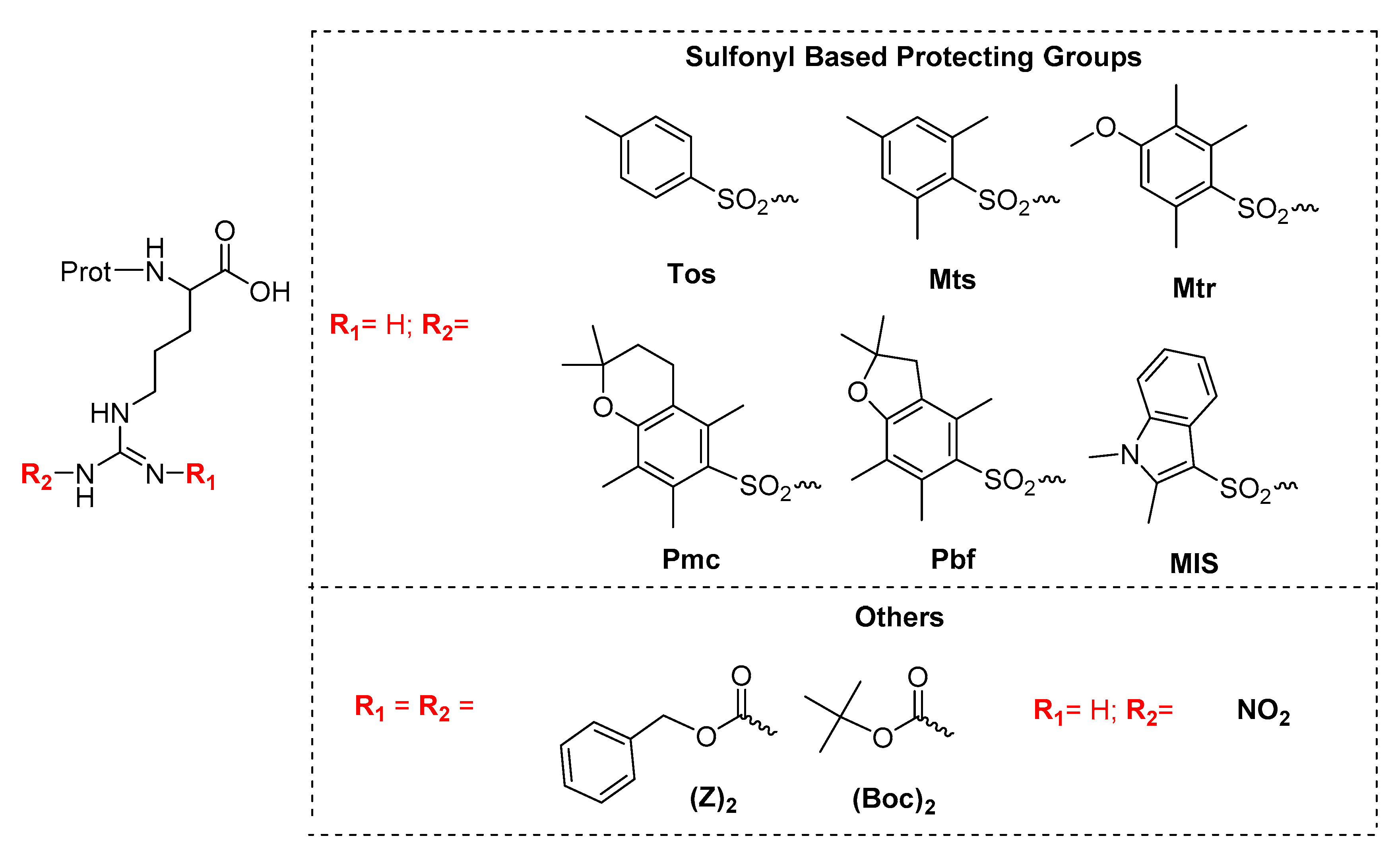
1. Introduction
2. Results and Discussion
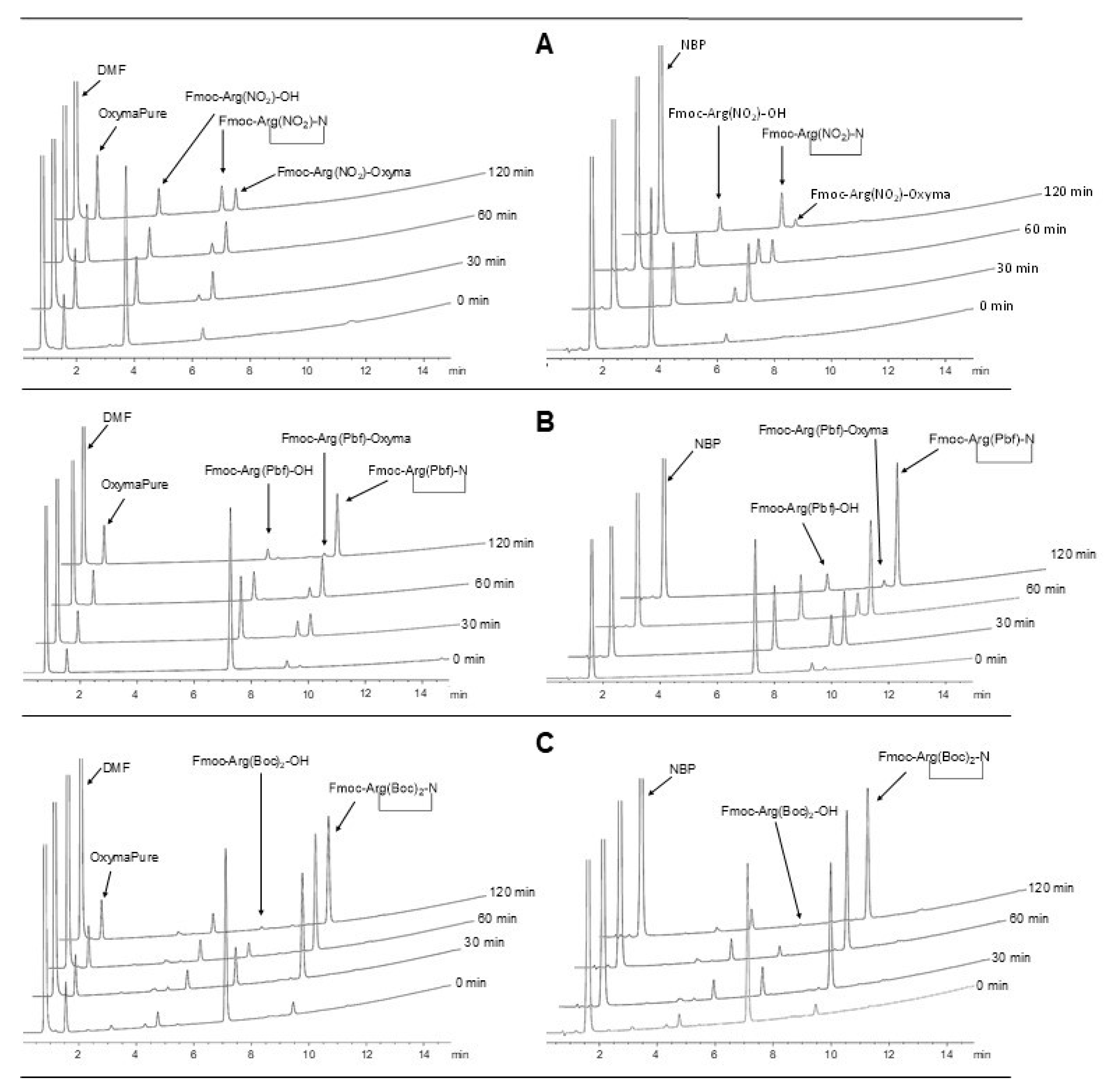
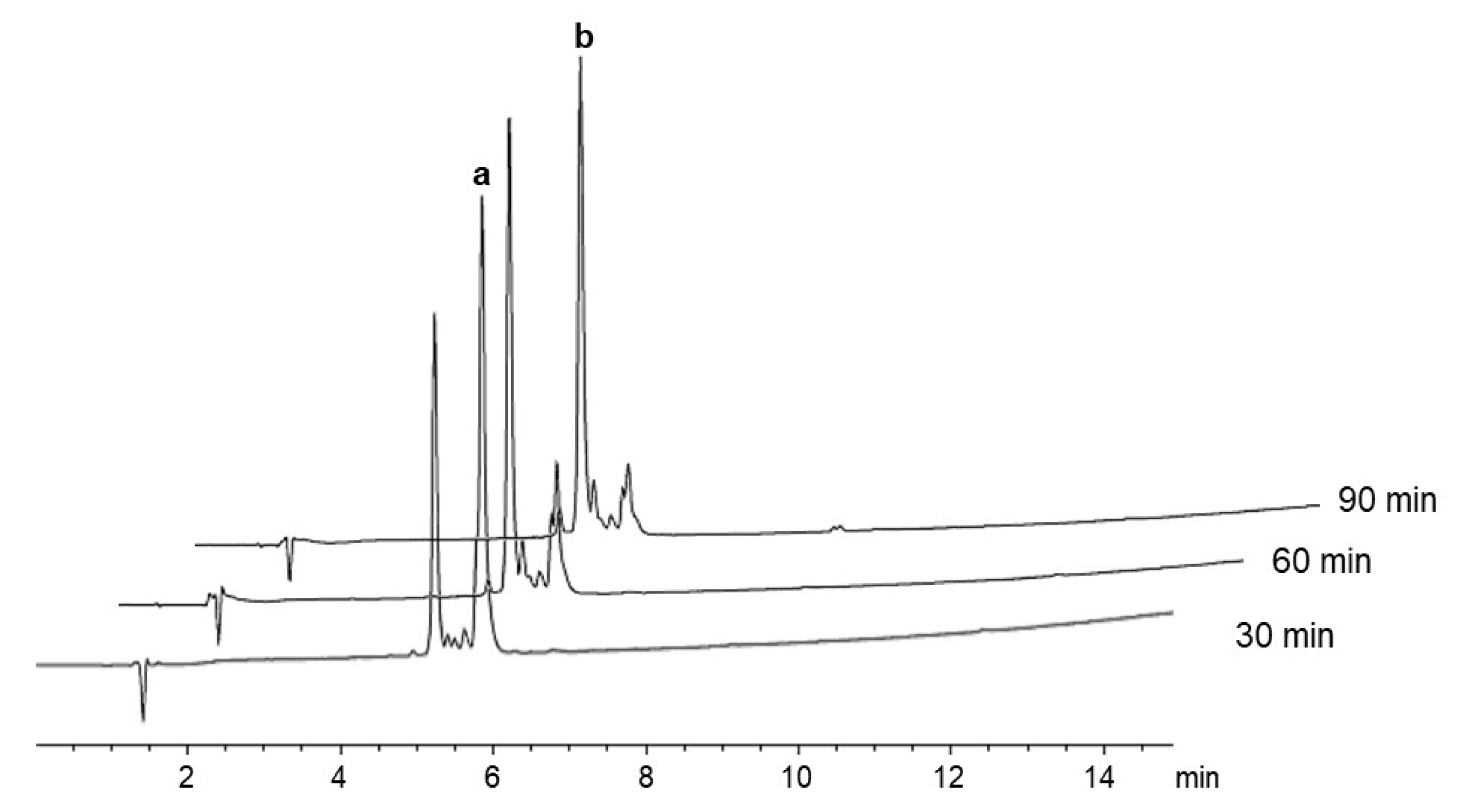
2.1. Stability of Fmoc-Arg(x)-OH in Solution
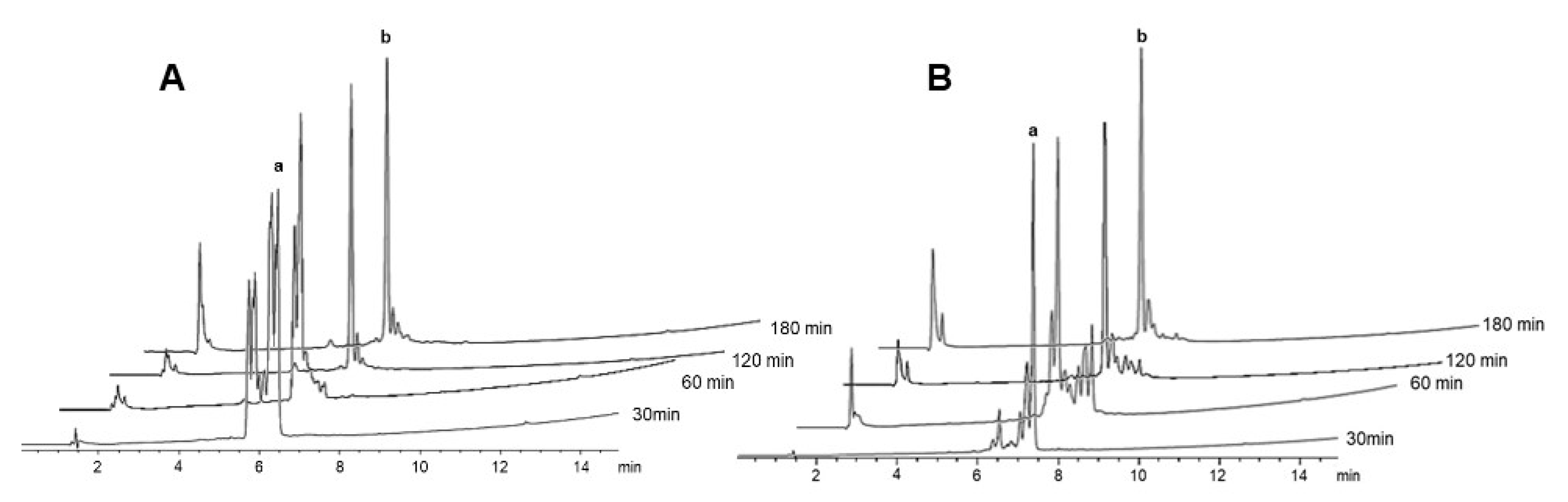
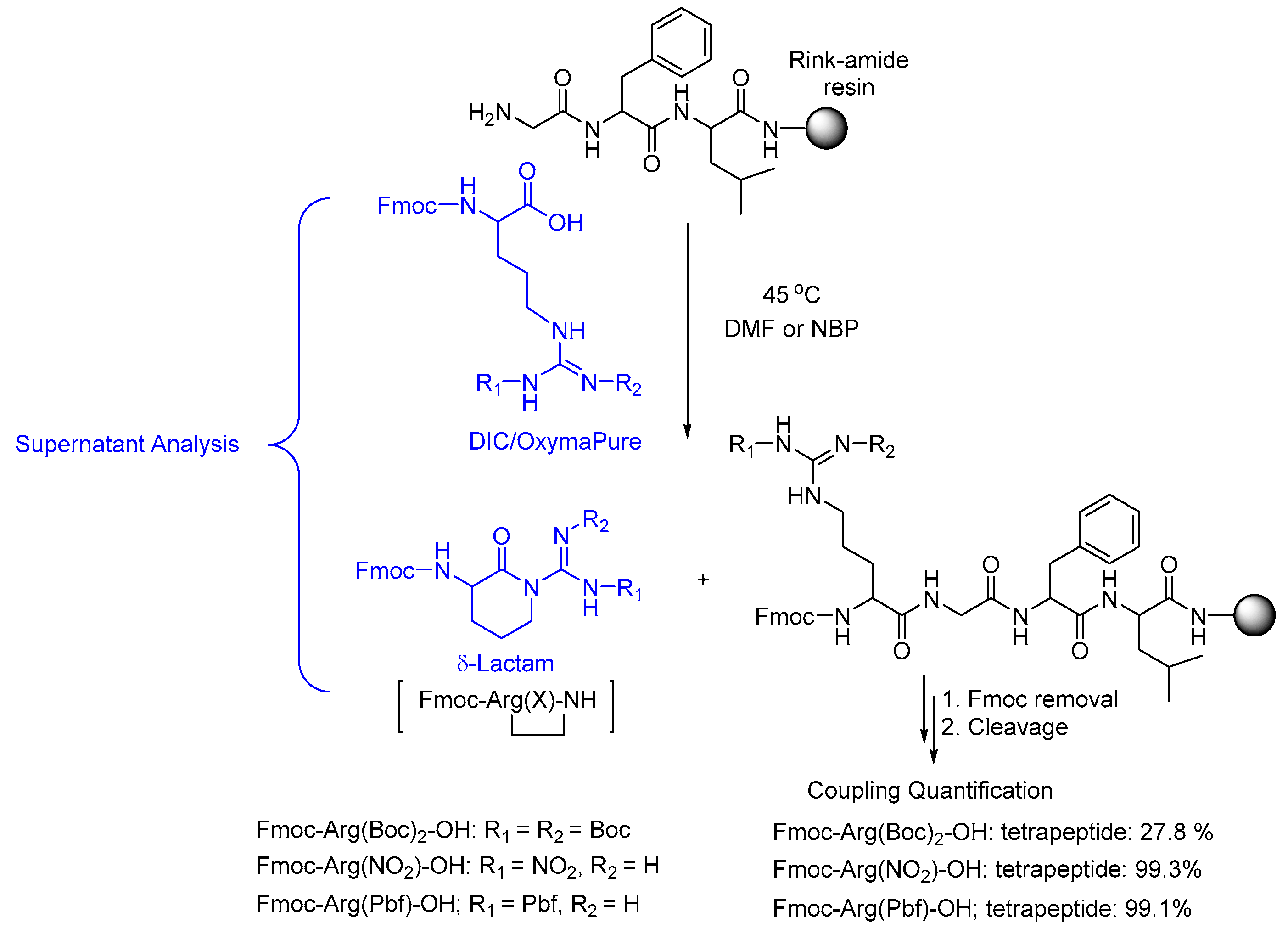
2.2. δ-Lactam Formation
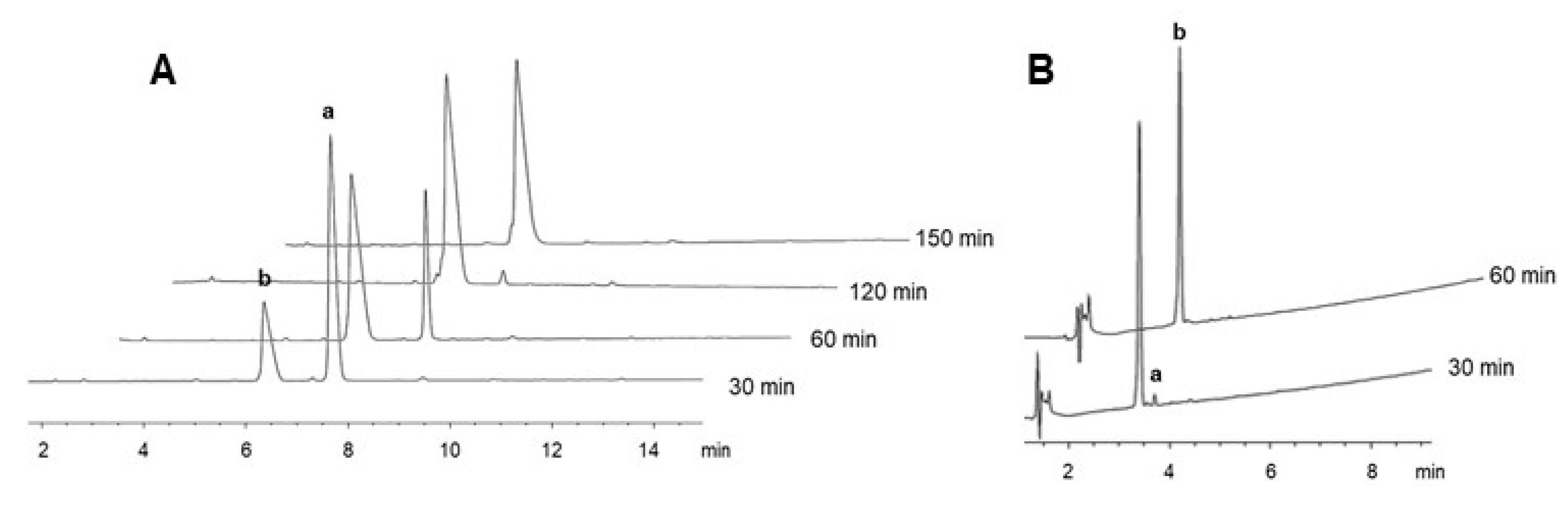
2.3. On-Resin Removal of NO2 from the Arg Side-Chain
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Peptide Synthesis
4.3. Stability Study Procedure
4.4. δ-Lactam Formation
4.5. Removal of the NO2 Group
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Futaki, S. Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv. Drug Deliv. Rev. 2005, 57, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Glasel, J.A. Chapter 1—Basic Physical Properties of Proteins and Nucleic Acids. In Introduction to Biophysical Methods for Protein and Nucleic Acid Research; Glasel, J.A., Deutscher, M.P., Eds.; Academic Press: San Diego, CA, USA, 1995; pp. 36–37. [Google Scholar]
- Fitch, C.A.; Platzer, G.; Okon, M.; Garcia-Moreno, B.E.; McIntosh, L.P. Arginine: Its pKa value revisited. Protein Sci. 2015, 24, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Hannon, C.L.; Anslyn, E.V. The Guanidinium Group: Its Biological Role and Synthetic Analogs. In Bioorganic Chemistry Frontiers; Dugas, H., Schmidtchen, F.P., Eds.; Springer: Berlin, Germany, 1993; Volume 3. [Google Scholar]
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2019 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2020, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O.; Alshaer, D.; de la Torre, B.G.; Albericio, F. 2017 FDA Peptide Harvest. Pharmaceuticals 2018, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.-D.; Won, H.-S.; Kim, J.-H.; Mishig-Ochir, T.; Lee, B.-J. Antimicrobial Peptides for Therapeutic Applications: A Review. Molecules 2012, 17, 12276–12286. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Govender, T.; Kruger, H.G.; de la Torre, B.G.; Albericio, F. Short AntiMicrobial Peptides (SAMPs) as a class of extraordinary promising therapeutic agents. J. Pept. Sci. 2016, 22, 438–451. [Google Scholar] [CrossRef]
- Takechi-Haraya, Y.; Saito, H.; Science, P. Current understanding of physicochemical mechanisms for cell membrane penetration of arginine-rich cell penetrating peptides: Role of glycosaminoglycan interactions. Curr. Protein Pept. Sci. 2018, 19, 623–630. [Google Scholar] [CrossRef]
- Moku, G.; Layek, B.; Trautman, L.; Putnam, S.; Panyam, J.; Prabha, S. Improving Payload Capacity and Anti-Tumor Efficacy of Mesenchymal Stem Cells Using TAT Peptide Functionalized Polymeric Nanoparticles. Cancers 2019, 11, 491. [Google Scholar] [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef]
- Yang, Y. Side Reactions in Peptide Synthesis; Academic Press: Oxford, UK, 2016; pp. 20–21. [Google Scholar]
- Rink, H.; Sieber, P.; Raschdorf, F. Conversion of NGurethane protected arginine to ornithine in peptide solid phase synthesis. Tetrahedron Lett. 1984, 25, 621–624. [Google Scholar] [CrossRef]
- Cezari, M.; Juliano, L. Studies on lactam formation during coupling procedures of N alpha-N omega-protected arginine derivatives. Pept. Res. 1996, 9, 88–91. [Google Scholar] [PubMed]
- Isidro Llobet, A.; Alvarez, M.; Albericio, F. Amino Acid-Protecting Groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef] [PubMed]
- Yajima, H.; Takeyama, M.; Kanaki, J.; Mitani, K. The mesitylene-2-sulphonyl group, an acidolytically removable N-protecting group for arginine. J. Chem. Soc. Chem. Commun. 1978, 482–483. [Google Scholar] [CrossRef]
- Fujino, M.; Wakimasu, M.; Kitada, C. Further Studies on the Use of Multi-substituted Benzenesulfonyl Groups for Protection of the Guanidino Function of Arginine. Chem. Pharm. Bull. 1981, 29, 2825–2831. [Google Scholar] [CrossRef][Green Version]
- Ramage, R.; Green, J. NG-2,2,5,7,8-pentamethylchroman-6-sulphonyl-L-arginine: A new acid labile derivative for peptide synthesis. Tetrahedron Lett. 1987, 28, 2287–2290. [Google Scholar] [CrossRef]
- Ramage, R.; Green, J.; Blake, A.J. An acid labile arginine derivative for peptide synthesis: NG-2,2,5,7,8-pentamethylchroman-6-sulphonyl-L-arginine. Tetrahedron 1991, 47, 6353–6370. [Google Scholar] [CrossRef]
- Carpino, L.A.; Shroff, H.; Triolo, S.A.; Mansour, E.-S.M.E.; Wenschuh, H.; Albericio, F. The 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl group (Pbf) as arginine side chain protectant. Tetrahedron Lett. 1993, 34, 7829–7832. [Google Scholar] [CrossRef]
- Verdini, A.S.; Lucietto, P.; Fossati, G.; Giordani, C. A facile preparation of Fmoc-Argω,ω′(Boc)2-OH and Z-Argω,ω′(Boc)2-OH, new arginine derivatives for peptide synthesis. Tetrahedron Lett. 1992, 33, 6541–6542. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Latassa, D.; Giraud, M.; Álvarez, M.; Albericio, F. 1, 2-Dimethylindole-3-sulfonyl (MIS) as protecting group for the side chain of arginine. Org. Biomol. Chem. 2009, 7, 2565–2569. [Google Scholar] [CrossRef][Green Version]
- de la Torre, B.G.; Kumar, A.; Alhassan, M.; Bucher, C.; Albericio, F.; Lopez, J. Successful development of a method for the incorporation of Fmoc-Arg(Pbf)-OH in solid-phase peptide synthesis using N-butylpyrrolidinone (NBP) as solvent. Green Chem. 2020, 22, 3162–3169. [Google Scholar] [CrossRef]
- Bergmann, M.; Zervas, L.; Rinke, H. Neues Verfahren zur Synthese von Peptiden des Arginins. Hoppe Seyler´s Z. Für Physiol. Chem. 1934, 224, 40–44. [Google Scholar] [CrossRef]
- Moroder, L.; Borin, G.; Marchiori, F.; Scoffone, E. Studies on cytochrome c. Part I. Synthesis of the protected hexadecapeptide (sequence 1–16) of Baker’s Yeast iso-1-cytochrome c. Biopolymers 1973, 12, 477–492. [Google Scholar] [CrossRef]
- Tadao, H.; Yukihiko, F.; Junzo, N. A New Method of Reducing Nitroarginine-peptide into Arginine-peptide, with Reference to the Synthesis of Poly-L-arginine Hydrochloride. Bull. Chem. Soc. Jpn. 1967, 40, 1205–1208. [Google Scholar] [CrossRef]
- Freidinger, R.M.; Hirschmann, R.; Veber, D.F. Titanium (III) as a selective reducing agent for nitroarginyl peptides: Synthesis of arginine vasotocin. J. Org. Chem. 1978, 43, 4800–4803. [Google Scholar] [CrossRef]
- Schafer, D.J.; Young, G.T.; Elliott, D.F.; Wade, R. Amino-acids and peptides. Part XXXII. A simplified synthesis of bradykinin by use of the picolyl ester method. J. Chem. Soc. C. 1971, 46–49. [Google Scholar] [CrossRef]
- Gros, C.; de Garilhe, M.P.; Costopanagiotis, A.; Schwyzer, R. Isolement à partir de l’hypophyse postérieure du tripeptide leucyl-arginyl-leucine et sa synthèse par une route nouvelle quant à l’incorporation de l’arginine. Helv. Chim. Acta 1961, 44, 2042–2048. [Google Scholar] [CrossRef]
- Bodanszky, M.; Martinez, J. Side reactions in peptide synthesis. Synthesis 1981, 1981, 333–356. [Google Scholar] [CrossRef]
- Lopez, J.; Pletscher, S.; Aemissegger, A.; Bucher, C.; Gallou, F. N-Butylpyrrolidinone as Alternative Solvent for Solid-Phase Peptide Synthesis. Org. Process Res. Dev. 2018, 22, 494–503. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Guasch-Camell, J.; Álvarez, M.; Albericio, F. p-Nitrobenzyloxycarbonyl (pNZ) as a Temporary Nα-Protecting Group in Orthogonal Solid-Phase Peptide Synthesis—Avoiding Diketopiperazine and Aspartimide Formation. Eur. J. Org. Chem. 2005, 2005, 3031–3039. [Google Scholar] [CrossRef]
- Kiselyov, A.S.; Smith II, L.; Armstrong, R.W. Solid Support Synthesis of Polysubstituted Tetrahydroquinolines via Three-Component Condensation Catalyzed by Yb(OTf)3. Tetrahedron 1998, 54, 5089–5096. [Google Scholar] [CrossRef]
- Schwarz, M.K.; Tumelty, D.; Gallop, M.A. Solid-Phase Synthesis of 3,5-Disubstituted 2,3-Dihydro-1,5-benzothiazepin-4(5H)-ones. J. Org. Chem. 1999, 64, 2219–2231. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
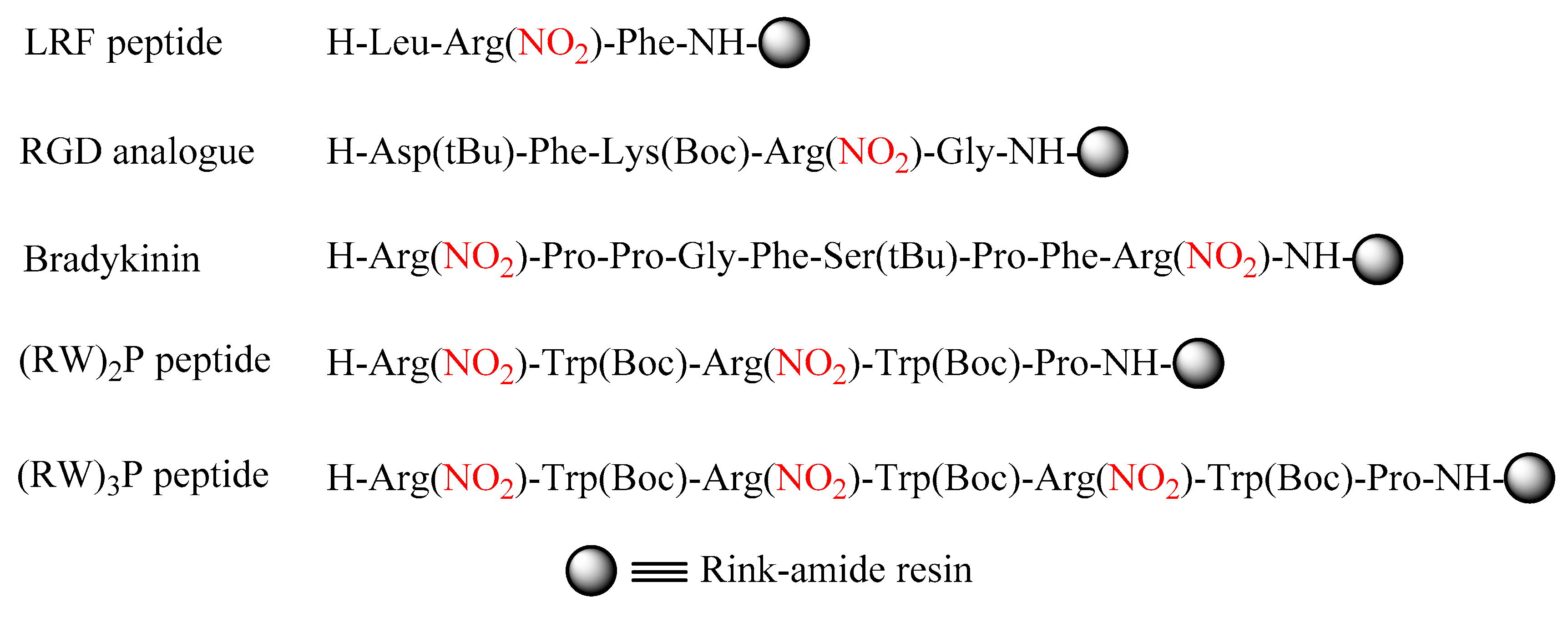{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0 h | 1 h | 24 h | 48 h | 10 d | 15 d | 20 d | 30 d | |
|---|---|---|---|---|---|---|---|---|
| Fmoc-Arg(Boc)2-OH, DMF | 88.8 | 88.6 | 86.9 | 85.0 | 77.6 | 65.1 | 58.5 | 51.2 |
| Fmoc-Arg(Boc)2-OH, NBP | 88.8 | 88.4 | 85.8 | 83.5 | 71.8 | 62.0 | 52.2 | 37.7 |
| Fmoc-Arg(NO2)-OH (DMF or NBP) 1 | 100 | 100 | 100 | 100 | 100 | |||
| Fmoc-Arg(Pbf)-OH (DMF or NBP) 1 | 100 | 100 | 100 | 100 | 100 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhassan, M.; Kumar, A.; Lopez, J.; Albericio, F.; de la Torre, B.G. Revisiting NO2 as Protecting Group of Arginine in Solid-Phase Peptide Synthesis. Int. J. Mol. Sci. 2020, 21, 4464. https://doi.org/10.3390/ijms21124464
Alhassan M, Kumar A, Lopez J, Albericio F, de la Torre BG. Revisiting NO2 as Protecting Group of Arginine in Solid-Phase Peptide Synthesis. International Journal of Molecular Sciences. 2020; 21(12):4464. https://doi.org/10.3390/ijms21124464
Chicago/Turabian StyleAlhassan, Mahama, Ashish Kumar, John Lopez, Fernando Albericio, and Beatriz G. de la Torre. 2020. "Revisiting NO2 as Protecting Group of Arginine in Solid-Phase Peptide Synthesis" International Journal of Molecular Sciences 21, no. 12: 4464. https://doi.org/10.3390/ijms21124464
APA StyleAlhassan, M., Kumar, A., Lopez, J., Albericio, F., & de la Torre, B. G. (2020). Revisiting NO2 as Protecting Group of Arginine in Solid-Phase Peptide Synthesis. International Journal of Molecular Sciences, 21(12), 4464. https://doi.org/10.3390/ijms21124464