Novel Flavonoid Glycosides of Quercetin from Leaves and Flowers of Gaiadendron punctatum G.Don. (Violeta de Campo), used by the Saraguro Community in Southern Ecuador, Inhibit α-Glucosidase Enzyme




Abstract
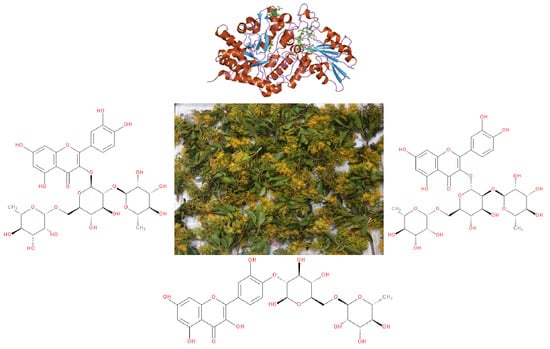
1. Introduction
2. Results
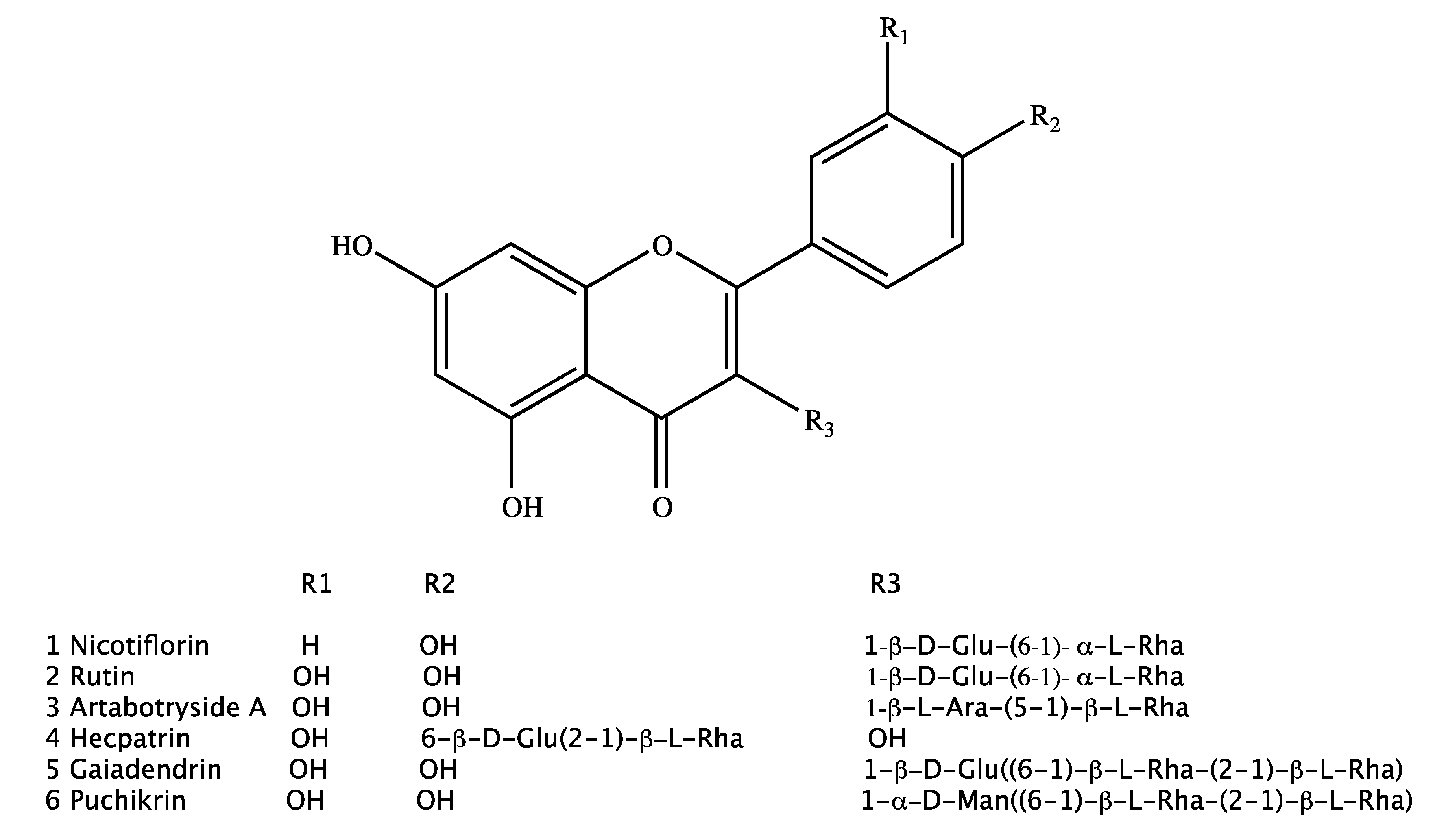
2.1. Isolation of Compounds
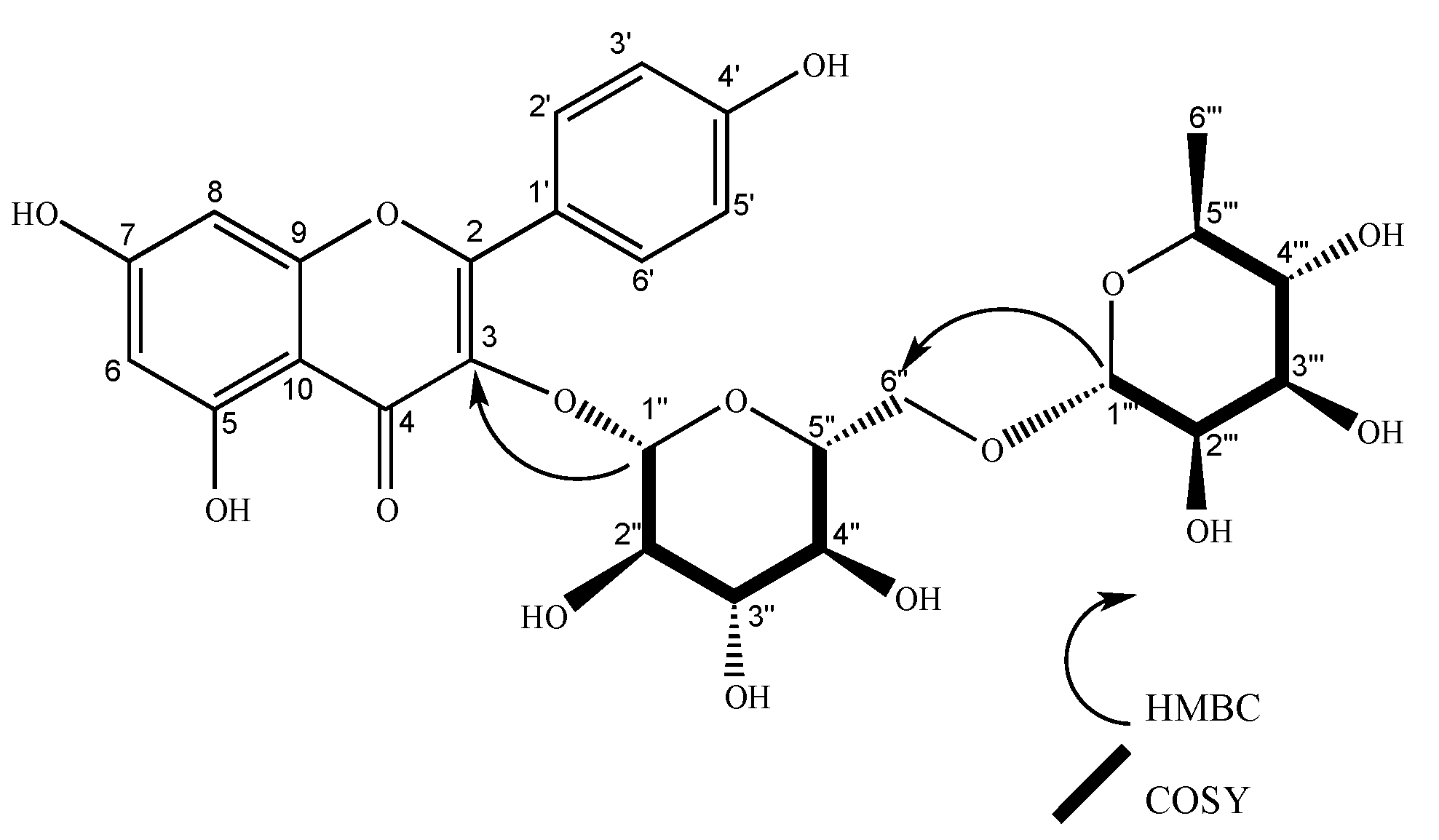
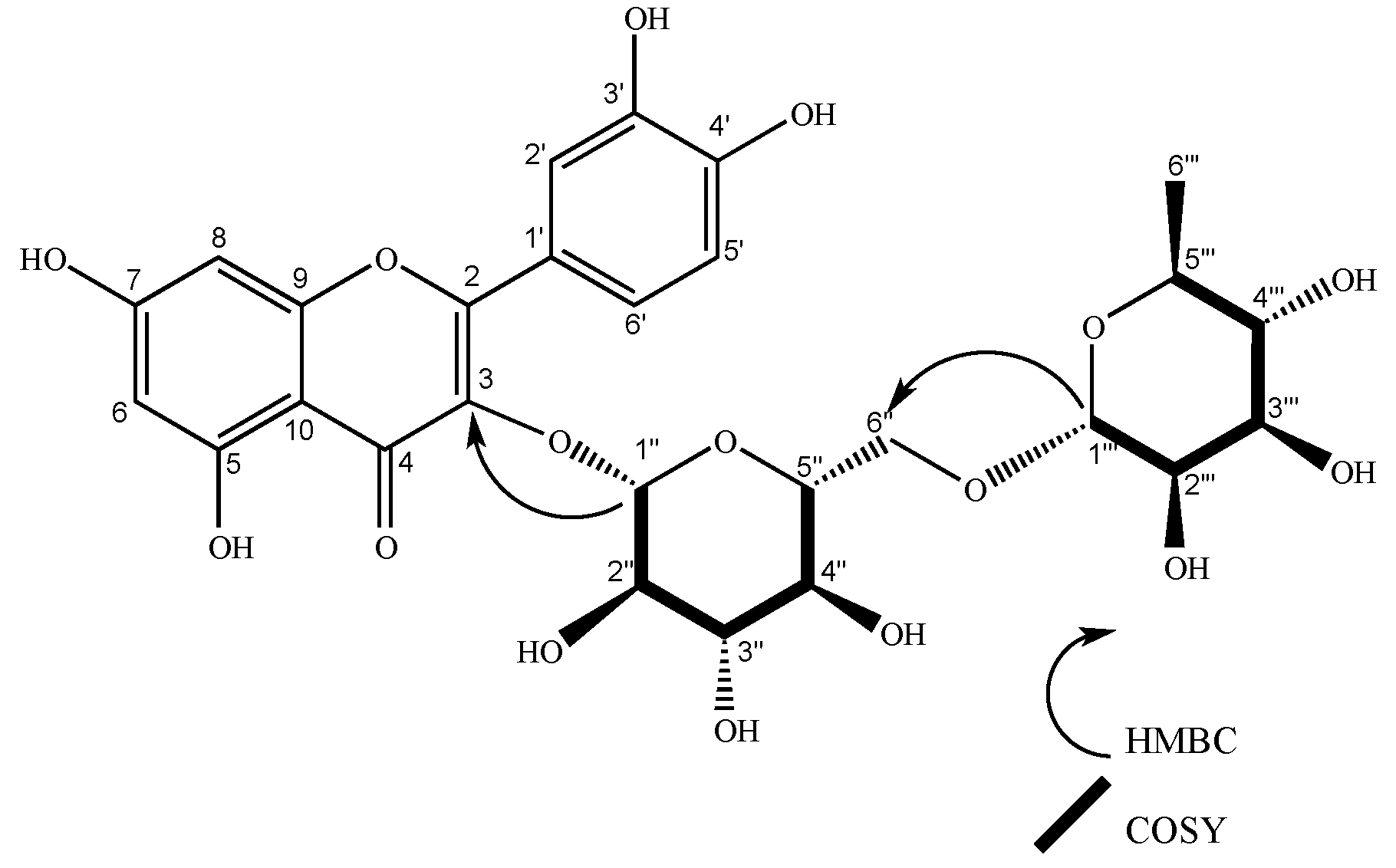
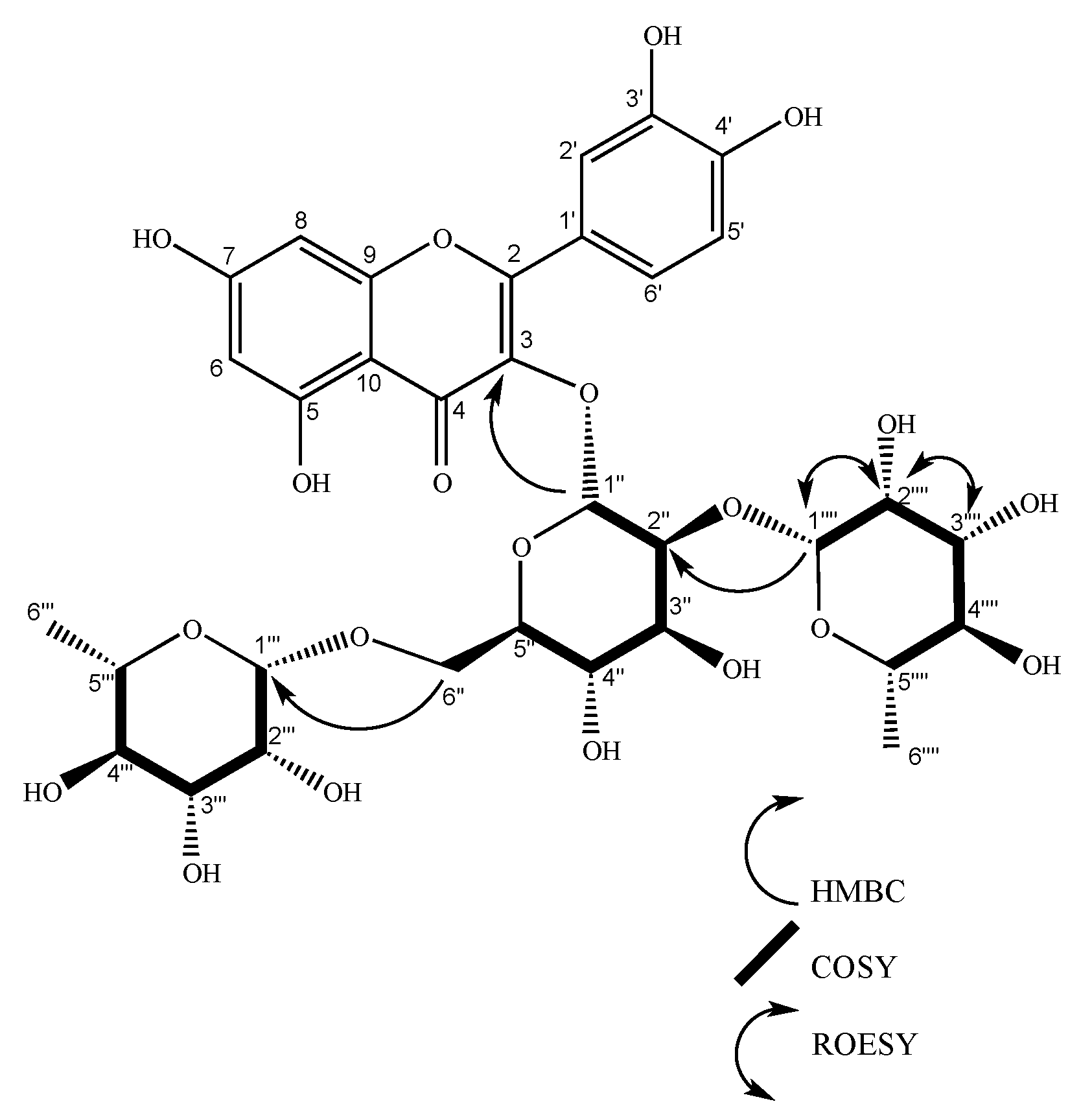
2.2. Characterization of Isolated Compounds
2.3. Analysis of Antimicrobial Activity
2.4. Inhibitory Activity of the α-Glucosidase Enzyme
Kinetic Analysis of Pure Compounds
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Collection of Plant Material and Obtaining Extracts
4.3. Dechlorophyllation and Fractionation of Hydroalcoholic Extracts of Leaves and Flowers
4.4. Purification of Secondary Metabolites
4.5. Characterization of the Obtained Metabolites
4.6. Hydrolysis and Derivatization of the New Compounds
4.7. Biological Activity
4.7.1. Antimicrobial Activity
4.7.2. α-Glucosidase Inhibition Assay
4.7.3. Kinetic Analysis of Pure Compounds
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Finerman, D. A matter of life and death: Health care change in an andean community. Soc. Sci. Med. 1984, 18, 329–334. [Google Scholar] [CrossRef]
- Davis, C.C.; Anderson, W.R.; Wurdack, K.J. Gene transfer from a parasitic flowering plant to a fern. Proc. R. Soc. B Boil. Sci. 2005, 272, 2237–2242. [Google Scholar] [CrossRef]
- Tene, V.; Malagón, O.; Finzi, P.V.; Vidari, G.; Armijos, C.; Zaragoza, T. An ethnobotanical survey of medicinal plants used in Loja and Zamora-Chinchipe, Ecuador. J. Ethnopharmacol. 2007, 111, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Andrade, J.M.; Mosquera, H.L.; Armijos, C. Ethnobotany of Indigenous Saraguros: Medicinal Plants Used by Community Healers “Hampiyachakkuna” in the San Lucas Parish, Southern Ecuador. BioMed Res. Int. 2017, 2017, 1–20. [Google Scholar] [CrossRef]
- Vidal-Russell, R.; Nickrent, D.L. The first mistletoes: Origins of aerial parasitism in Santalales. Mol. Phylogenet. Evol. 2008, 47, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.A.; Calvin, C.L. An origin of aerial branch parasitism in the mistletoe family, Loranthaceae. Am. J. Bot. 2006, 93, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, A.; Nishida, K.; Davis, D. Systematics, host plants, and life histories of three new Phyllocnistis species from the central highlands of Costa Rica (Lepidoptera, Gracillariidae, Phyllocnistinae). ZooKeys 2009, 27, 7–30. [Google Scholar] [CrossRef]
- Jorgensen, P.; León-Yánez, S. Catalogue of the Vascular Plants of Ecuador; Missouri Botanical Garden Press: St. Louis, MO, USA, 1999. [Google Scholar]
- Di Pasquale, G.; Marziano, M.; Impagliazzo, S.; Lubritto, C.; De Natale, A.; Bader, M.Y. The Holocene treeline in the northern Andes (Ecuador): First evidence from soil charcoal. Palaeogeogr. Palaeoclim. Palaeoecol. 2008, 259, 17–34. [Google Scholar] [CrossRef]
- Beck, E.; Bendix, J.; Kottke, I.; Makeschin, F.; Mosandl, R. Gradients in a Tropical Mountain Ecosystem of Ecuador; Springer: Berlin/Heidelberg, Germany, 2007; Volume 198. [Google Scholar]
- Orfali, R.; Perveen, S.; Siddiqui, N.A.; Alam, P.; Alhowiriny, T.A.; Al-Taweel, A.M.; Al-Yahya, S.; Ameen, F.; Majrashi, N.; Alluhayb, K.; et al. Pharmacological Evaluation of Secondary Metabolites and Their Simultaneous Determination in the Arabian Medicinal Plant Plicosepalus curviflorus Using HPTLC Validated Method. J. Anal. Methods Chem. 2019, 2019, 7435909. [Google Scholar] [CrossRef]
- Muhammad, K.J.; Jamil, S.; Basar, N. Phytochemical study and biological activities of Scurrula parasitica L (Loranthaceae) leaves. J. Res. Pharm. 2019, 23, 522–531. [Google Scholar] [CrossRef]
- Atun, S.; A’Yun, Z.Q.; Lutfia, N.; Handayani, S. Phytochemical and antioxidant evaluation of ethanol extract leaves of dendrophthoe falcata (loranthaceae) hemiparasitic on melia azedarach host tree. J. Physics. Conf. Ser. 2019, 1156, 012011. [Google Scholar] [CrossRef]
- Rajachan, O.-A.; Hongtanee, L.; Chalermsaen, K.; Kanokmedhakul, K.; Kanokmedhakul, S. Bioactive galloyl flavans from the stems of Helixanthera parasitica. J. Asian Nat. Prod. Res. 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nag, M.; Kar, A.J.C.; Mukherjee, K.P. RP-HPLC analysis of methanol extract of Viscum articulatum e A plant from Ayurveda. J. Ayurveda Integr. Med. 2018, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Ouachinou, J.-A.; Dassou, G.; Idohou, R.; Adomou, A.; Yédomonhan, H. National inventory and usage of plant-based medicine to treat gastrointestinal disorders with cattle in Benin (West Africa). S. Afr. J. Bot. 2019, 122, 432–446. [Google Scholar] [CrossRef]
- Ricco, M.V.; Bari, M.L.; Bagnato, F.; Cornacchioli, C.; Laguia-Becher, M.; Spairani, L.U.; Posadaz, A.; Dobrecky, C.; Ricco, R.A.; Wagner, M.L.; et al. Establishment of callus—cultures of the Argentinean mistletoe, Ligaria cuneifolia (R et P.) Tiegh (Loranthaceae) and screening of their polyphenolic content. Plant Cell Tissue Organ Cult. 2019, 138, 167–180. [Google Scholar] [CrossRef]
- Liu, R.; Su, B.; Huang, F.; Ru, M.; Zhang, H.; Qin, Z.; Li, Y.; Zhu, K. Identification and analysis of cardiac glycosides in Loranthaceae parasites Taxillus chinensis (DC.) Danser and Scurrula parasitica Linn. and their host Nerium indicum Mill. J. Pharm. Biomed. Anal. 2019, 174, 450–459. [Google Scholar] [CrossRef]
- Lu, B.; Huang, Y.; Chen, Z.; Ye, J.; Xu, H.; Chen, W.; Long, X. Niosomal Nanocarriers for Enhanced Skin Delivery of Quercetin with Functions of Anti-Tyrosinase and Antioxidant. Molecules 2019, 24, 2322. [Google Scholar] [CrossRef]
- Rutkowska, M.; Olszewska, M.A.; Kolodziejczyk-czepas, J.; Nowak, P.; Owczarek, A. Sorbus domestica Leaf Extracts and Their Activity Markers: Antioxidant Potential and Synergy Effects in Scavenging Assays of Multiple Oxidants. Molecules 2019, 24, 2289. [Google Scholar] [CrossRef]
- Cui, H.; Lu, T.; Wang, M.; Zou, X.; Zhang, Y.; Yang, X.; Dong, Y.; Zhou, H. Flavonoids from Morus alba L. Leaves: Optimization of Extraction by Response Surface Methodology and Comprehensive Evaluation of Their Antioxidant, Antimicrobial, and Inhibition of α-Amylase Activities through Analytical Hierarchy Process. Molecules 2019, 24, 2398. [Google Scholar] [CrossRef]
- Kamkaew, N.; Paracha, T.U.; Ingkaninan, K.; Waranuch, N.; Chootip, K. Vasodilatory Effects and Mechanisms of Action of Bacopa monnieri Active Compounds on Rat Mesenteric Arteries. Molecules 2019, 24, 2243. [Google Scholar] [CrossRef]
- Al-hamoud, G.A. Four New Flavonoids Isolated from the Aerial Parts of Cadaba routundifolia Forssk (Qaddab). Molecules 2019, 24, 2167. [Google Scholar]
- Gomes, N.G.M.; Oliveira, A.P.; Cunha, D.; Pereira, D.M.; Valentão, P.; Pinto, E.; Araújo, L.; Andrade, P.B. Flavonoid Composition of Salacia senegalensis (Lam.) DC.; Leaves, Evaluation of Antidermatophytic Effects, and Potential Amelioration of the Associated Inflammatory Response. Molecules 2019, 24, 2530. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Dong, Y.; Xie, Y.; Wang, L.; Wang, J.; Liu, Y.; Zhao, L.; Cao, F. Effects of β-glucosidase and α-rhamnosidase on the Contents of Flavonoids, Ginkgolides, and Aroma Components in Ginkgo Tea Drink. Molecules 2019, 24, 2009. [Google Scholar] [CrossRef] [PubMed]
- Sarbu, L.G.; Shova, S.; Peptanariu, D.; Sandu, I.A.; Birsa, L.M.; Bahrin, L.G. The Cytotoxic Properties of Some Tricyclic 1,3-Dithiolium Flavonoids. Molecules 2019, 24, 2459. [Google Scholar] [CrossRef]
- Ibáñez, S.; Ruiz, G.; Sauvain, M. Evaluación de la Flora en el Valle de Zongo Contra Leishmania y Chagas, Rev. Boliv. Química 2009, 26, 1–11. [Google Scholar]
- Andrade, M.; Armijos, C.; Malagón, O.; Lucero, H. Plantas Medicinales Silvestres Empleadas por la Etnia Saraguro en la Parroquia San Lucas, Provincia de Loja-Ecuador, 1st ed.; Editorial UTPL: Loja, Ecuador, 2009. [Google Scholar]
- De la Torre, L.; Navarrete, H.; Muriel, P.; Macia, M.J.; Balslev, H. Enciclopedia de las Plantas Útiles del Ecuador, 1st ed.; Herbario QCA de la Escuela de Ciencias Biológicas de la Pontificia Universidad Católica del Ecuador & Herbario AAU del Departamento de Ciencias Biológicas de la Universidad de Aarhus: Quito, Ecuador, 2008. [Google Scholar]
- Ríos, M.; Koziol, M.J.; Borgtoft, H.; Granda, G. Plantas Útiles del Ecuador. Aplicaciones, Retos y Perspectivas, 1st ed.; Abya-Yala: Quito, Ecuador, 2007. [Google Scholar]
- De Oliveira, D.M.; Siqueira, E.P.; Nunes, Y.R.; Cota, B.B. Flavonoids from leaves of Mauritia flexuosa. Rev. Bras. Farm. 2013, 23, 614–620. [Google Scholar] [CrossRef]
- Şerban, G.; Pop, I.A.; Horvath, T.; Bota, S. The isolation and identification of rutin from pharmaceutical products. Analele Univ Din Oradea Fasc Ecotoxicologie, Zooteh Si Tehnol Ind Aliment 2016, 15, 107–114. [Google Scholar]
- Khan, M.; Artabotryside, A. A constituent from Descurainia sophia (L.) induces cell death in U87 glioma cells through apoptosis and cell cycle arrest at G2/M phase. J. Med. Plants Res. 2012, 6, 3754–3765. [Google Scholar]
- Agrawal, P.K. NMR Spectroscopy in the structural elucidation of oligosaccharides and glycosides. Phytochemistry 1992, 31, 3307–3330. [Google Scholar] [CrossRef]
- Selvaraj, K.; Chowdhury, R.; Bhattacharjee, C. Isolation and Structural Elucidation of Flavonoids From Aquatic Fern Azolla Microphylla and Evaluation of Free Radical Scavenging Activity. Int. J. Pharm. Pharm. Sci. 2013, 5, 743–749. [Google Scholar]
- Turgut-Kara, N. Identification and Production Of Phenolic Nicotiflorin In Astragalus Chrysochlorus Callus. FARMACIA 2018, 66, 558–562. [Google Scholar] [CrossRef]
- Dehaghani, Z.A.; Asghari, G.; Dinani, M.S. Isolation and Identification of Nicotiflorin and Narcissin from the Aerial Parts of Peucedanum aucheri Boiss. J. Agric. Sci. Technol. A 2017, 7, 45–51. [Google Scholar]
- DiNicolantonio, J.J.; Bhutani, J.; O’Keefe, J.H. Acarbose: Safe and effective for lowering postprandial hyperglycaemia and improving cardiovascular outcomes. Open Hear. 2015, 2, 000327. [Google Scholar] [CrossRef]
- Habtemariam, S. A-Glucosidase Inhibitory Activity of Kaempferol-3-O-rutinoside. Nat. Prod. Commun. 2011, 6, 201–203. [Google Scholar]
- Li, Y.Q.; Zhou, F.C.; Gao, F.; Bian, J.S.; Shan, F. Comparative Evaluation of Quercetin, Isoquercetin and Rutin as Inhibitors of α-Glucosidase. J. Agric. Food Chem. 2009, 57, 11463–11468. [Google Scholar] [CrossRef]
- Dong, H.-Q.; Li, M.; Zhu, F.; Liu, F.-L.; Huang, J.-B. Inhibitory potential of trilobatin from Lithocarpus polystachyus Rehd against α-glucosidase and α-amylase linked to type 2 diabetes. Food Chem. 2012, 130, 261–266. [Google Scholar] [CrossRef]
- Gao, H.; Huang, Y.-N.; Gao, B.; Xu, P.-Y.; Inagaki, C.; Kawabata, J. α-Glucosidase inhibitory effect by the flower buds of Tussilago farfara L. Food Chem. 2008, 106, 1195–1201. [Google Scholar] [CrossRef]
- Ruiz-Matute, A.I.; Hernandez-Hernandez, O.; Rodríguez-Sánchez, S.; Sanz, M.L.; Martínez-Castro, I. Derivatization of carbohydrates for GC and GC–MS analyses. J. Chromatogr. B 2011, 879, 1226–1240. [Google Scholar] [CrossRef]
- Lai, Z.; Fiehn, O. Mass spectral fragmentation of trimethylsilylylated small molecules. Mass Spectrom. Rev. 2018, 37, 245–257. [Google Scholar] [CrossRef]
- Rojas-Escudero, E.; Alarcón-Jiménez, A.L.; Elizalde-Galván, P.; Rojo-Callejas, F. Optimization of carbohidrate silylation for gas chromatography. J. Chromatogr. A. 2004, 1027, 117–120. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Diekema, D.J. Progress in Antifungal Susceptibility Testing of Candida spp. by Use of Clinical and Laboratory Standards Institute Broth Microdilution Methods, 2010 to 2012. J. Clin. Microbiol. 2012, 50, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhang, Y.; Cheng, Y.; Wang, Y. Rapid screening and identification of α-glucosidase inhibitors from mulberry leaves using enzyme-immobilized magnetic beads coupled with HPLC/MS and NMR. Biomed. Chromatogr. 2013, 27, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Yilmazer-Musa, M.; Griffith, A.M.; Michels, A.J.; Schneider, E.; Frei, B. Grape Seed and Tea Extracts and Catechin 3-Gallates Are Potent Inhibitors of α-Amylase and α-Glucosidase Activity. J. Agric. Food Chem. 2012, 60, 8924–8929. [Google Scholar] [CrossRef] [PubMed]
- Rouzbehan, S.; Moein, S.; Homaei, A.; Moein, M.R. Kinetics of α-glucosidase inhibition by different fractions of three species of Labiatae extracts: A new diabetes treatment model. Pharm. Boil. 2017, 55, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Jeong, Y.-K.; Wang, M.-H.; Lee, W.-Y.; Rhee, H.-I. Inhibitory effect of pine extract on α-glucosidase activity and postprandial hyperglycemia. Nutrition 2005, 21, 756–761. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hecpatrin (4) | Gaiadendrin (5) | Puchikrin (6) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | 13C | 1H | COSY | HMBC | NOESY | 13C | 1H | COSY | HMBC | ROESY | 13C | 1H | COSY | HMBC | ROESY |
| 4 | 179.33 | 179.25 | 8 | 177.84 | 8 | ||||||||||
| 7 | 166.05 | 165.84 | 6, 8 | 164.17 | 6, 8 | ||||||||||
| 5 | 163.19 | 163.11 | 6 | 161.71 | 6 | ||||||||||
| 9 | 158.41 | 2’ | 158.89 | 6’, 2’ | 157.49 | 6’ | |||||||||
| 2 | 158.36 | 158.44 | 8 | 157 | 8 | ||||||||||
| 4’ | 149.58 | 6’’, 2’ | 149.54 | 6’, 2’, 5’ | 148.11 | 5’, 2’, 6’ | |||||||||
| 3’ | 146.03 | 145.91 | 6’, 2’, 5’ | 144.48 | 5’, 2’, 6’ | ||||||||||
| 3 | 134.53 | 134.42 | 1’’ | 133.01 | 1´´ | ||||||||||
| 1’ | 123.47 | 5’ | 123.52 | 6’, 2’, 5’ | 122.08 | 5’, 2’, 6’ | |||||||||
| 6’ | 123.21 | 7.60 (d, J = 2.2 Hz, 1H) | 5’ | 2’ | 123.44 | 7.62 – 7.59 (m, 2H), | 5’ | 5’ | 122.01 | 7.64 – 7.57 (m, 2H), | 5’ | 5’ | |||
| 2’ | 117.16 | 7.62 (d, J = 0.8 Hz, 1H) | 6’ | 117.39 | 115.96 | ||||||||||
| 5’ | 115.97 | 6.87 (d, J = 8.7 Hz, 1H) | 6’ | 2’ | 116.04 | 6.87 (dd, J = 8.0, 0.7 Hz, 1H), | 2’, 6’ | 6’, 2’, 5’ | 114.6 | 6.87 (d, J = 8.2 Hz, 1H) | 2’, 6’ | 2’, 6’ | |||
| 10 | 105.84 | 8 | 105.83 | 6, 8 | 104.46 | 6, 8 | |||||||||
| 1’’’’ | 102.63 | 5.23 (d, J = 1.6 Hz, 1H), | 2’’’’ | 2’’, 5’’’’ | 5’’’’, 2’’ | 101.21 | 5.22 (d, J = 1.7 Hz, 1H) | 2’’’’ | 2’’ | 2’’’’ | |||||
| 1’’’ | 102.65 | 5.23 (d, J = 1.6 Hz, 1H) | 2’’’ | 2’’ | 2’’’, 2’’ | 102.24 | 4.51 (d, J = 1.6 Hz, 1H), | 2’’’ | 6´’, 5’’’ | 5’’’, 6’’ | 100.81 | 4.51 (d, J = 1.7 Hz, 1H) | 2’’’ | 5’’’, 6´´ | |
| 1’’ | 100.31 | 5.75 (d, J = 7.7 Hz, 1H) | 2’’ | 2’’ | 5’’ | 100.47 | 5.59 (d, J = 7.6 Hz, 1H), | 2’’ | 3, 2’’ | 99.04 | 5.59 (d, J = 7.7 Hz, 1H) | 2´´ | 3, 2’’ | ||
| 6 | 99.79 | 6.18 (d, J = 2.1 Hz, 1H) | 8 | 99.81 | 6.18 (d, J = 2.1 Hz, 1H), | 8 | 98.29 | 6.19 (d, J = 2.1 Hz, 1H) | 8 | ||||||
| 8 | 94.58 | 6.37 (d, J = 2.1 Hz, 1H) | 6 | 94.73 | 6.37 (d, J = 2.1 Hz, 1H), | 6 | 93.24 | 6.37 (d, J = 2.1 Hz, 1H) | 6 | ||||||
| 2’’ | 80.1 | 3.66 (dd, J = 7.7, 9.14 Hz, 1H) | 1’’ | 1’’’, 5’’ | 80.02 | 3.65 (dd, J = 9.2, 7.6 Hz, 1H), | 1’’, 3’’ | 1’’’’, 3’’ | 78.59 | 3.64 (dd, J = 9.2, 7.6 Hz, 1H) | 1´´,3´´ | 1´´´´ | |||
| 4’’’ | 74.05 | 3.34 (d, J = 1.5 Hz, 1H), | 3’’’ | 6’’’ | 78.9 | 3.56 (d, J = 8.7 Hz, 1H), | 77.47 | 3.55 (d, J = 8.7 Hz, 1H) | 4’’’’ | ||||||
| 4’’ | 78.35 | 3.26 – 3.19 (m, 1H) | 3’’, 5’’ | 77.06 | 3.33 (q, J = 2.4, 1.9 Hz, 1H) | 5’’, | 75.65 | 3.34 – 3.32 (m, 1H) | 6’’, 5’’ | ||||||
| 4’’’’ | 74.04 | 3.36 (d, J = 3.7 Hz, 1H) | 5’’’’, 3’’’’ | 2’’’’, 6’’’’ | 5’’’’ | 72.61 | 3.39 – 3.35 (m, 1H) | 5’’’’, 3’’’’ | 6’’’’ | ||||||
| 3’’’ | 72.29 | 3.76 (t, J = 3.2 Hz, 1H) | 4’’’, 2’’’ | 73.86 | 3.26 – 3.21 (m, 1H) | 5’’’, 2’’’, 6’’’ | 72.42 | 3.24 (d, J = 6.6 Hz, 1H) | 3’’’, | 6’’’ | |||||
| 2’’’’ | 72.39 | 4.01 (dd, J = 3.4, 1.7 Hz, 1H), | 1’’’’, 3’’’’ | 1’’’’ | 5’’’’ | 70.96 | 4.01 (dd, J = 3.4, 1.7 Hz, 1H) | 1’’’’, 3’’’’ | 1’’’’ | 3’’’’, 1’’’’ | |||||
| 3’’’’ | 72.28 | 3.81 (d, J = 3.4 Hz, 1H) | 2’’’’ | 2’’’’ | 70.85 | 3.78 (d, J = 3.4 Hz, 1H) | 4’’’’, 2’’’’ | 1’’’’, 2’’’’ | |||||||
| 3’’ | 71.69 | 3.36 (t, J = 9.3 Hz, 1H) | 4’’ | 72.24 | 3.50 (dd, J = 9.5, 3.4 Hz, 1H) | 5’’, 2’’ | 70.81 | 3.49 (dd, J = 9.5, 3.4 Hz, 1H) | 2’’ | ||||||
| 2’’’ | 72.4 | 4.00 (dd, J = 3.4, 1.7 Hz, 1H) | 1’’’, 3’’’ | 1’’’ | 72.12 | 3.59 (dd, J = 3.4, 1.6 Hz, 1H) | 1’’’, | 1’’’ | 70.7 | 3.59 (dd, J = 3.4, 1.7 Hz, 1H) | |||||
| 5’’ | 78.95 | 3.56 (td, J = 8.9, 1.7 Hz, 1H) | 4’’, 6’’ | 2’’ | 71.85 | 3.29 – 3.26 (m, 1H) | 3’’ | 6’’, 3’’ | 70.42 | 3.30 – 3.26 (m, 1H) | 3’’ | ||||
| 5’’’’ | 69.95 | 4.09 (dd, J = 9.7, 6.2 Hz, 1H) | 1’’’’,4’’’’, 6 ’’’’ | 6’’’’, 1’’’’, 4’’’’ | 2’’’’, 4’’’’ | 68.51 | 4.08 (dd, J = 9.6, 6.2 Hz, 1H) | 6’’’’, 4’’’’ | 1’’’’, 4’’’’, 6’’’’ | 1’’’’, 2’’’’ | |||||
| 5’’’ | 69.96 | 4.07 – 4.02 (m, 1H) | 6’’’ | 6’’’, 1’’’ | 69.71 | 3.41 (d, J = 6.2 Hz, 1H) | 6’’’ | 6’’’, 1’’’, 3’’’,2’’’ | 68.28 | 3.41 (d, J = 6.2 Hz, 1H) | 6’’’ | 6’’’, 1’’’ | |||
| 6’’ | 62.55 | 3.72 (d, J = 2.2 Hz, 1H), 3.55 (m, 1H) | 5’’ | 4’ | 68.26 | 3.84 (d, J = 1.4 Hz, 1H), 3.39 (d, J = 6.5 Hz, 1H) | 1’’’, 4’’, 5’’ | 66.84 | 3.83 (d, J = 1.5 Hz, 1H), 3.39 (m, 1H) | 1´´´ | |||||
| 6’’’ | 17.46 | 0.97 (d, J = 6.2 Hz, 3H) | 5’’’ | 5’’’ | 17.81 | 1.08 (d, J = 6.2 Hz, 3H) | 5’’’, 4’’’’ | 5’’’ | 2’’’ | 16,37 | 1.08 (d, J = 6.2 Hz, 3H) | 5’’’ | 3’’’ | ||
| 6’’’’ | 17.5 | 1.01 (d, J = 6.2 Hz, 3H) | 5’’’’ | 5’’’’ | 4’’’’, 5’’’’ | 16.07 | 1.00 (d, J = 6.2 Hz, 3H) | 5’’’’ | 4’’’’ | ||||||
| Sample | Micrococcus luteus | Candida albicans | Staphylococcus aureus | Escherichia coli | Enterococcus faecalis |
|---|---|---|---|---|---|
| Leaf extract | 500 | >4000 | 1000 | >4000 | 1000 |
| Flower extract | 2000 | >4000 | >4000 | >4000 | >4000 |
| Hecpatrin | >327.59 | >327.59 | >327.59 | >327.59 | >327.59 |
| Rutin | >327.59 | >327.59 | >327.59 | >327.59 | >327.59 |
| Gaiadendrin | >264.32 | >264.32 | >264.32 | >264.32 | >264.32 |
| Puchikrin | >264.32 | >264.32 | >264.32 | >264.32 | >264.32 |
| Nicotiflorin | >336.41 | >336.41 | >336.41 | >336.41 | >336.41 |
| Gentamicin (1000 μg/mL) | <0.39 | - | 0.78 | 125 | |
| Penicillin (250 μg/mL) | 12.5 | - | <0.097 | - | 6.25 |
| Amphotericin B (200 μg/mL) | - | <0.075 | - | - |
| No. | Sample | α-Glucosidase IC50 (SE) (mg/mL, µM†) |
|---|---|---|
| 1 | Leaves extract | 2.13 (0.07) |
| 2 | Flowers extract | 51.89 (4.49) |
| 3 | Hecpatrin | 9.66 (1.04) † |
| 4 | Rutin | 7.53 (1.08) † |
| 5 | Gaiadendrin | 16.87 (1.02) † |
| 6 | Puchikrin | NA †* |
| 7 | Nicotiflorin | 159.09 (9.54) † |
| 8 | Acarbose | 300–700 † |
| Compound | Dose (μM) | V. max (SE) (nM.min−1) | Km (μM) | Inhibition Type | Ki |
|---|---|---|---|---|---|
| No inhibitor | 0 | 697.7 (67.26) | 985.6 (226.5) | ||
| Hecpatrin | 27.3 | 62.0 (13.41) | 1107.0 (545.8) | C | 3.92 (1.48) |
| 13.65 | 412.0 (159.3) | 1587.0 (573.0) | |||
| 6.82 | 680.0 (159.1) | 1492.0 (710.0) | |||
| Rutin | 27.3 | 184.6 (47.97) | 1351.0 (741.6) | NC | 10.53 (1.07) |
| 13.65 | 242.7 (20.23) | 624.9 (148.1) | |||
| 6.82 | 583.0 (46.54) | 1489.0 (241.6) | |||
| Gaiadendrin | 27.53 | 138.2 (11.40) | 881.5 (11.40) | NC | 12.29(1.58) |
| 20.65 | 250.1 (30.26) | 1099.0 (30.26) | |||
| 13.77 | 328.7 (24.77) | 676.4 (8.86) | |||
| Nicotiflorin | 280.48 | 170.3 (14.63) | 756.2 (171.5) | NC | 124.6 (8.65) |
| 140.24 | 379.0 (44.98) | 1297.0 (330.3) | |||
| 70.12 | 540.8 (29.47) | 1360.0 (156.2) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cedeño, H.; Espinosa, S.; Andrade, J.M.; Cartuche, L.; Malagón, O. Novel Flavonoid Glycosides of Quercetin from Leaves and Flowers of Gaiadendron punctatum G.Don. (Violeta de Campo), used by the Saraguro Community in Southern Ecuador, Inhibit α-Glucosidase Enzyme. Molecules 2019, 24, 4267. https://doi.org/10.3390/molecules24234267
Cedeño H, Espinosa S, Andrade JM, Cartuche L, Malagón O. Novel Flavonoid Glycosides of Quercetin from Leaves and Flowers of Gaiadendron punctatum G.Don. (Violeta de Campo), used by the Saraguro Community in Southern Ecuador, Inhibit α-Glucosidase Enzyme. Molecules. 2019; 24(23):4267. https://doi.org/10.3390/molecules24234267
Chicago/Turabian StyleCedeño, Héctor, Sandra Espinosa, José Miguel Andrade, Luis Cartuche, and Omar Malagón. 2019. "Novel Flavonoid Glycosides of Quercetin from Leaves and Flowers of Gaiadendron punctatum G.Don. (Violeta de Campo), used by the Saraguro Community in Southern Ecuador, Inhibit α-Glucosidase Enzyme" Molecules 24, no. 23: 4267. https://doi.org/10.3390/molecules24234267
APA StyleCedeño, H., Espinosa, S., Andrade, J. M., Cartuche, L., & Malagón, O. (2019). Novel Flavonoid Glycosides of Quercetin from Leaves and Flowers of Gaiadendron punctatum G.Don. (Violeta de Campo), used by the Saraguro Community in Southern Ecuador, Inhibit α-Glucosidase Enzyme. Molecules, 24(23), 4267. https://doi.org/10.3390/molecules24234267