
Novel Convenient Approach to the Solid-Phase Synthesis of Oligonucleotide Conjugates

 , ,
, ,
Abstract
1. Introduction
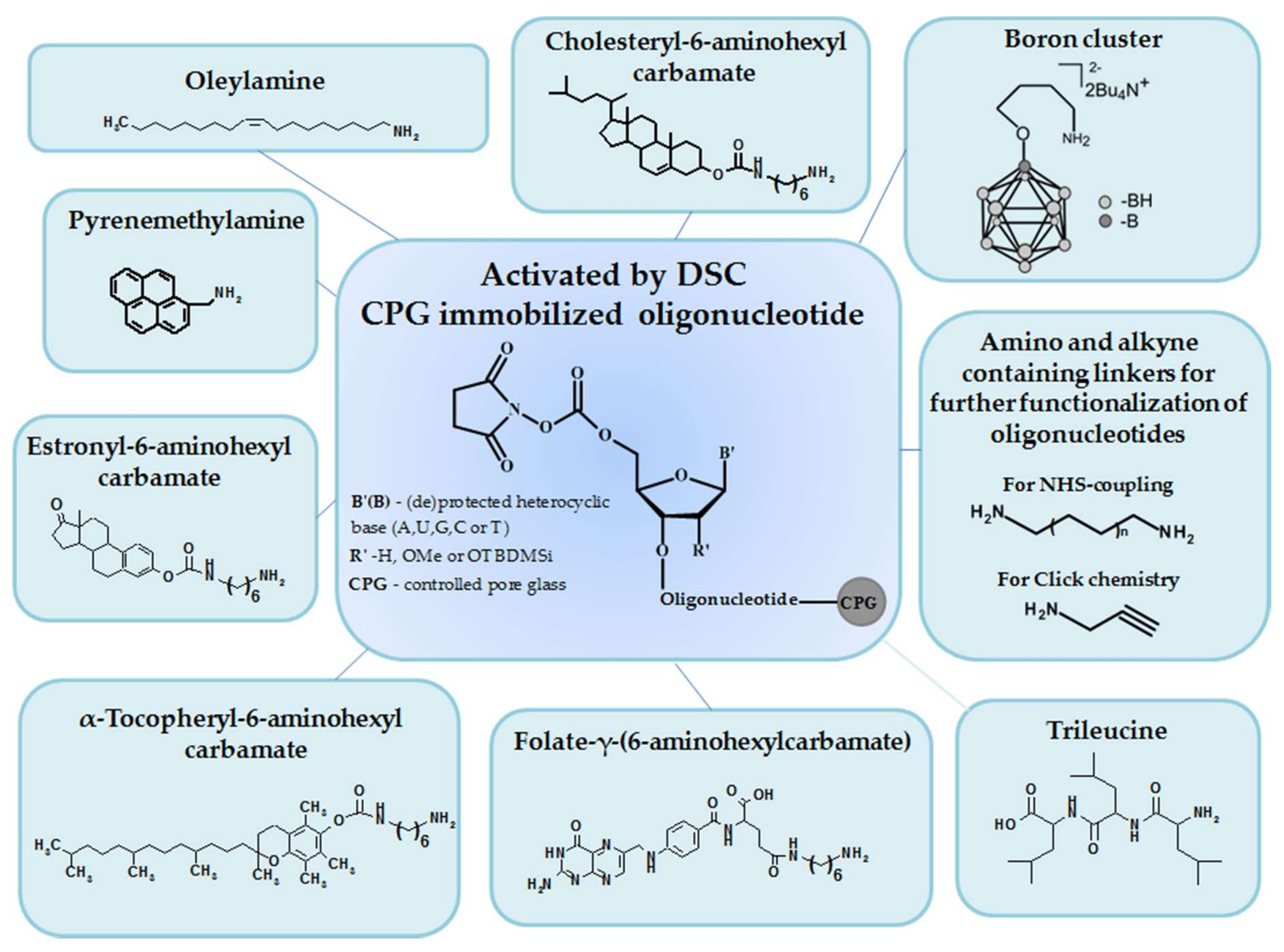
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Physical Measurements
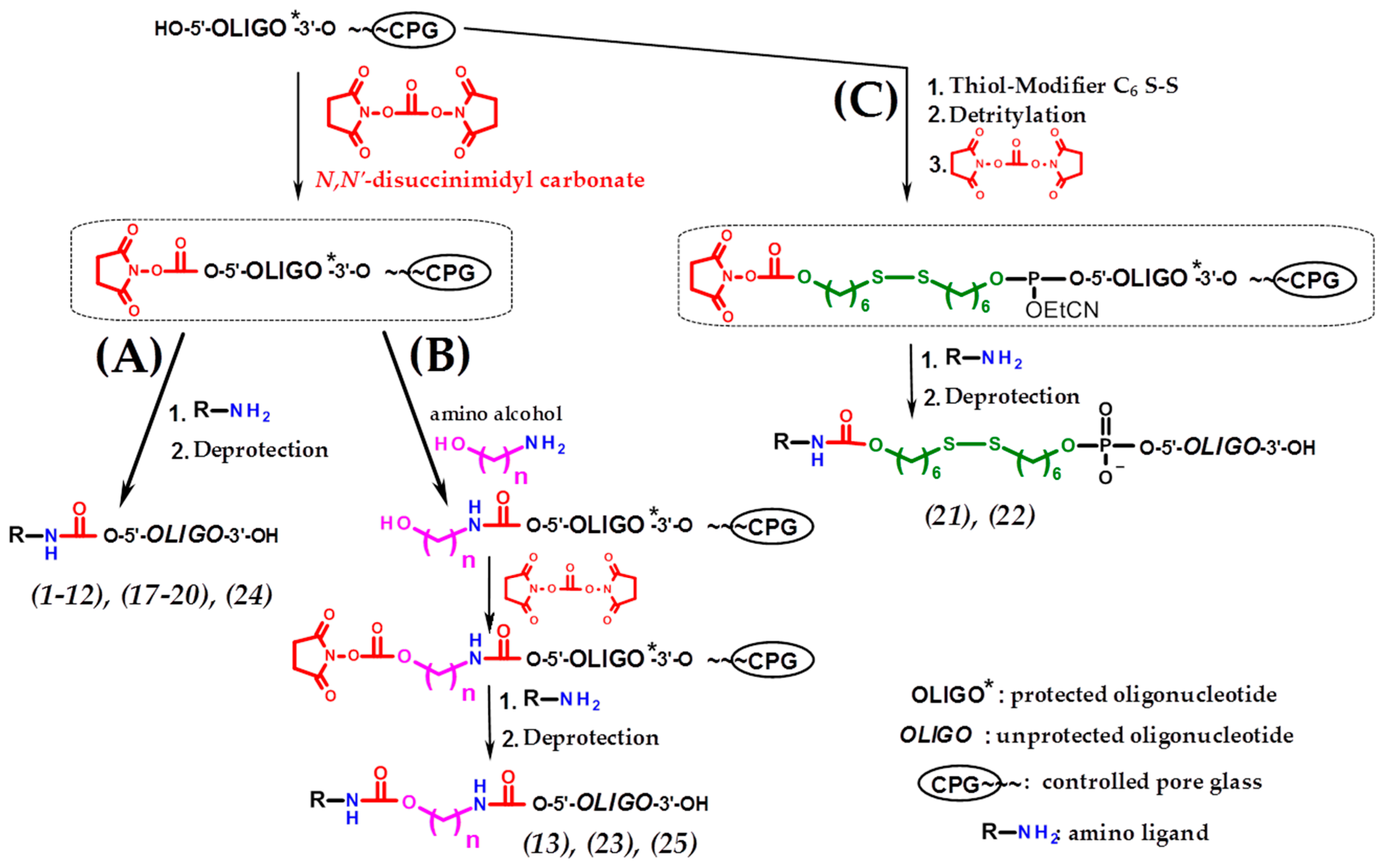
4.3. Synthesis of Polymer-Bound Oligonucleotides
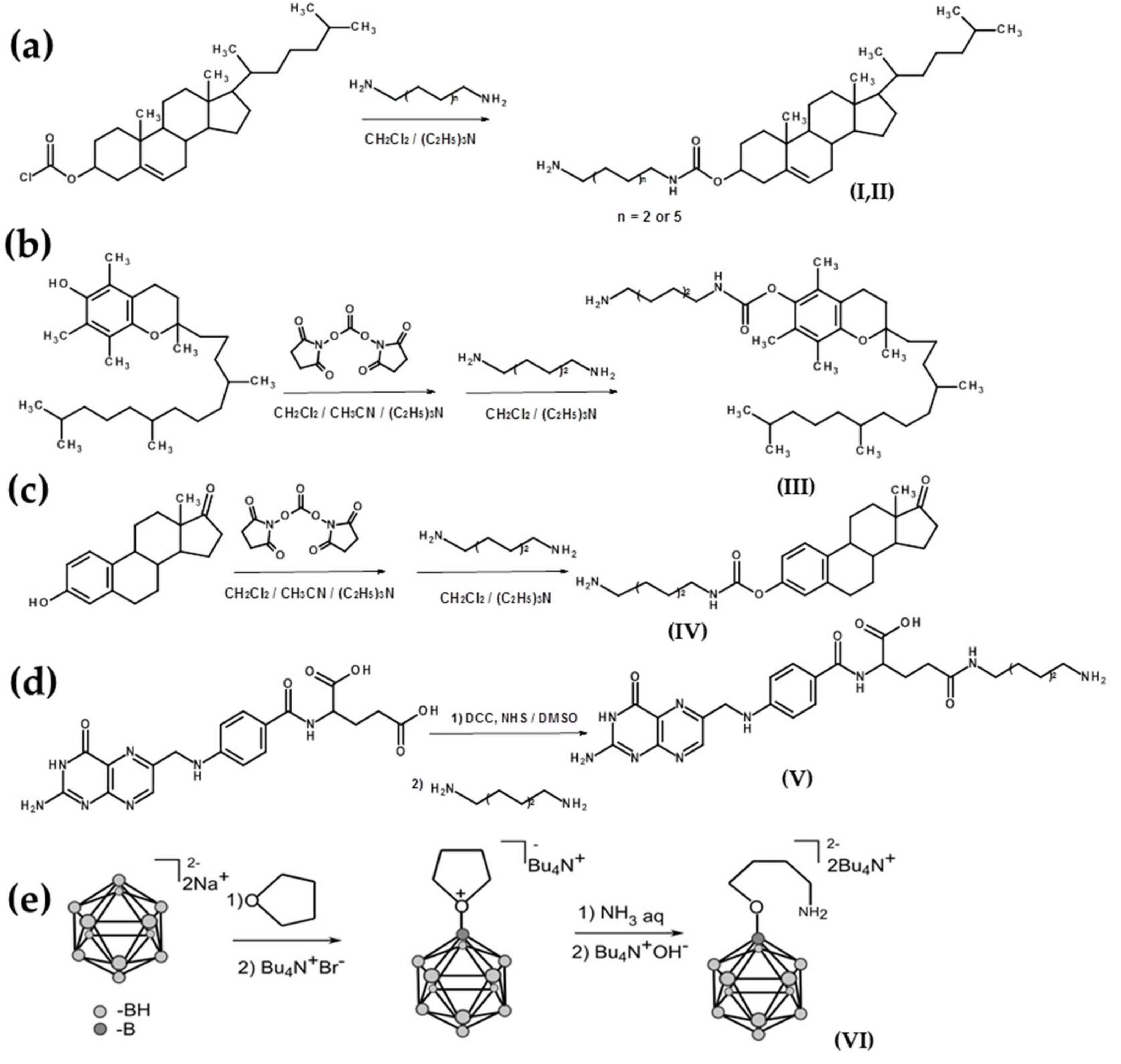
4.4. Preparation of Amino Compounds
4.5. Solid-Phase Synthesis of Oligonucleotide Conjugates
4.6. Deprotection and Isolation of the Conjugates
4.7. Synthesis of N-(2-Hydroxyethyl)phenazinium Conjugates (14, 15)
4.8. Synthesis of Cy3 Conjugate (16)
4.9. RP HPLC Analysis of the Oligonucleotide Conjugates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lönnberg, H. Solid-phase synthesis of oligonucleotide conjugates useful for delivery and targeting of potential nucleic acid therapeutics. Bioconjug. Chem. 2009, 20, 1065–1094. [Google Scholar] [CrossRef] [PubMed]
- Grijalvo, S.; Alagia, A.; Jorge, A.; Eritja, R. Covalent strategies for targeting messenger and non-coding RNAs: An updated review on siRNA, miRNA and antimiR conjugates. Genes (Basel) 2018, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Shevelev, G.Y.; Gulyak, E.L.; Lomzov, A.A.; Kuzhelev, A.A.; Krumkacheva, O.A.; Kupryushkin, M.S.; Tormyshev, V.M.; Fedin, M.V.; Bagryanskaya, E.G.; Pyshnyi, D.V. A Versatile approach to attachment of triarylmethyl labels to DNA for nanoscale structural EPR studies at physiological temperatures. J. Phys. Chem. B. 2018, 122, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Zatsepin, T.S.; Oretskaya, T.S. Synthesis and applications of oligonucleotide-carbohydrate conjugates. Chem. Biodivers. 2004, 1, 1401–1417. [Google Scholar] [CrossRef]
- Østergaard, M.E.; Jackson, M.; Low, A.E.; Chappell, A.G.; Lee, R.; Peralta, R.Q.; Yu, J.; Kinberger, G.A.; Dan, A.; Carty, R.; et al. Conjugation of hydrophobic moieties enhances potency of antisense oligonucleotides in the muscle of rodents and non-human primates. Nucl. Acids. Res. 2019, 47, 6045–6058. [Google Scholar] [CrossRef]
- Astakhova, I.K.; Pasternak, K.; Campbell, M.A.; Gupta, P.; Wengel, J. A locked nucleic acid-based nanocrawler: Designed and reversible movement detected by multicolor fluorescence. J. Am. Chem. Soc. 2013, 135, 2423–2426. [Google Scholar] [CrossRef]
- Taskova, M.; Madsen, C.S.; Jensen, K.J.; Hansen, L.H.; Vester, B.; Astakhova, K. Antisense oligonucleotides internally labeled with peptides show improved target recognition and stability to enzymatic degradation. Bioconjug. Chem. 2017, 28, 768–774. [Google Scholar] [CrossRef]
- Tai, W. Current aspects of siRNA bioconjugate for in vitro and in vivo delivery. Molecules 2019, 24, 2211. [Google Scholar] [CrossRef]
- Ming, X.; Laing, B. Bioconjugates for targeted delivery of therapeutic oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 81–89. [Google Scholar] [CrossRef]
- Jo, H.; Ban, C. Aptamer–nanoparticle complexes as powerful diagnostic and therapeutic tools. Exp. Mol. Med. 2016, 48, e230. [Google Scholar] [CrossRef]
- Craig, K.; Abrams, M.; Amiji, M. Recent preclinical and clinical advances in oligonucleotide conjugates. Expert Opin. Drug Deliv. 2018, 15, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Takakura, K.; Kawamura, A.; Torisu, Y.; Koido, S.; Yahagi, N.; Saruta, M. The clinical potential of oligonucleotide therapeutics against pancreatic cancer. Int. J. Mol. Sci. 2019, 20, 3331. [Google Scholar] [CrossRef] [PubMed]
- Capek, I. Dispersions based on noble metal nanoparticles-DNA conjugates. Adv. Colloid Interface Sci. 2011, 163, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Olejniczak, A.; Nawrot, B.; Leśnikowski, Z. DNA modified with boron–metal cluster complexes [M(C2B9H11)2]—synthesis, properties, and applications. Int. J. Mol. Sci. 2018, 19, 3501. [Google Scholar] [CrossRef] [PubMed]
- MacCulloch, T.; Buchberger, A.; Stephanopoulos, N. Emerging applications of peptide–oligonucleotide conjugates: Bioactive scaffolds, self-assembling systems, and hybrid nanomaterials. Org. Biomol. Chem. 2019, 17, 1668–1682. [Google Scholar] [CrossRef]
- Sergeeva, Z.A.; Lokhov, S.G.; Ven’yaminova, A.G. Oligo(2′-O-methylribonucleotides) and their derivatives. II. Synthesis and properties of oligo(2′-O-methylribonucleotides) modified with N-(3-hydroxyethyl)phanazinium and steroid groups at the 5′-terminus. Bioorg. Khim. 1996, 22, 916–922. [Google Scholar]
- Iglina, A.A.; Meschaninova, M.I.; Venyaminova, A.G. 5′-Lipophilic conjugates of oligonucleotides as components of cell delivery systems. Nucleic Acids Symp. Ser. 2009, 121–122. [Google Scholar] [CrossRef]
- Petrova, N.S.; Chernikov, I.V.; Meschaninova, M.I.; Dovydenko, I.S.; Venyaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Carrier-free cellular uptake and the gene-silencing activity of the lipophilic siRNAs is strongly affected by the length of the linker between siRNA and lipophilic group. Nucleic Acids Res. 2012, 40, 2330–2344. [Google Scholar] [CrossRef]
- Kye, M.; Lim, Y. Synthesis and purification of self-assembling peptide-oligonucleotide conjugates by solid-phase peptide fragment condensation. J. Pept. Sci. 2018, 24, e3092. [Google Scholar] [CrossRef]
- Farzan, V.M.; Ulashchik, E.A.; Martynenko-Makaev, Y.V.; Kvach, M.V.; Aparin, I.O.; Brylev, V.A.; Prikazchikova, T.A.; Maklakova, S.Y.; Majouga, A.G.; Ustinov, A.V.; et al. Automated solid-phase click synthesis of oligonucleotide conjugates: From small molecules to diverse N -acetylgalactosamine clusters. Bioconjug. Chem. 2017, 28, 2599–2607. [Google Scholar] [CrossRef]
- Hermanson, G. Bioconjugate Techniques; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 9780123705013. [Google Scholar]
- Zalipsky, S.; Seltzer, R.; Menon-Rudolph, S. Evaluation of a new reagent for covalent attachment of polyethylene glycol to proteins. Biotechnol. Appl. Biochem. 1992, 15, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Wachter, L.; Jablonski, J.A.; Ramachandran, K.L. A simple and efficient procedure for the synthesis of 5′-aminoalkyl oligodeoxynucleotides. Nucleic. Acids. Res. 1986, 14, 7985–7994. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shevelev, G.Y.; Krumkacheva, O.A.; Lomzov, A.A.; Kuzhelev, A.A.; Trukhin, D.V.; Rogozhnikova, O.Y.; Tormyshev, V.M.; Pyshnyi, D.V.; Fedin, M.V.; Bagryanskaya, E.G. Triarylmethyl labels: Toward improving the accuracy of EPR nanoscale distance measurements in DNAs. J. Phys. Chem. B. 2015, 119, 13641–13648. [Google Scholar] [CrossRef]
- Ballico, M.; Cogoi, S.; Drioli, S.; Bonora, G.M. Postsynthetic conjugation of biopolymers with high molecular mass poly(ethylene glycol): Optimization of a solution process tested on synthetic oligonucleotides. Bioconjug. Chem. 2003, 14, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, G.; Zalutsky, M.R. Improved synthesis of N-succinimidyl 4-[18F]fluorobenzoate and its application to the labeling of a monoclonal antibody fragment. Bioconjug. Chem. 1994, 5, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Tsang, E.M.W.; Wang, Y.A.; Peng, X.; Yu, H.-Z. Bioreactive surfaces prepared via the self-assembly of dendron thiols and subsequent dendrimer bridging reactions. Langmuir 2005, 21, 1858–1865. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ren, Y.; Pan, L.; Xu, H.-M. In vivo anti-tumor activity of polypeptide HM-3 modified by different polyethylene glycols (PEG). Int. J. Mol. Sci. 2011, 12, 2650–2663. [Google Scholar] [CrossRef]
- Diala, I.; Osada, A.; Maruoka, S.; Imanisi, T.; Murao, S.; Ato, T.; Ohba, H.; Fujii, M. Synthesis of phosphorothioate oligonucleotide–peptide conjugates by solid phase fragment condensation. Bioorg. Med. Chem. Lett. 2007, 17, 6576–6578. [Google Scholar] [CrossRef]
- Xu, Z.; Peng, J.; Yan, N.; Yu, H.; Zhang, S.; Liu, K.; Fang, Y. Simple design but marvelous performances: Molecular gels of superior strength and self-healing properties. Soft. Matter. 2013, 9, 1091–1099. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Doung, T.T.; McKee, S.P.; Thompson, W.J. N,N′-dissuccinimidyl carbonate: A useful reagent for alkoxycarbonylation of amines. Tetrahedron Lett. 1992, 33, 2781–2784. [Google Scholar] [CrossRef]
- Manoharan, M.; Kesavan, V.; Rajeev, K.G. Modified iRNA agents. U.S. Patent 20,050,107,325, 5 May 2019. [Google Scholar]
- Trindade, A.F.; Frade, R.F.M.; Maçôas, E.M.S.; Graça, C.; Rodrigues, C.A.B.; Martinho, J.M.G.; Afonso, C.A.M. “Click and go”: Simple and fast folic acid conjugation. Org. Biomol. Chem. 2014, 12, 3181–3190. [Google Scholar] [CrossRef] [PubMed]
- Sivaev, I.B.; Semioshkin, A.A.; Brellochs, B.; Sjöberg, S.; Bregadze, V.I. Synthesis of oxonium derivatives of the dodecahydro-closo-dodecaborate anion [B12H12]2−. Tetramethylene oxonium derivative of [B12H12]2− as a convenient precursor for the synthesis of functional compounds for boron neutron capture therapy. Polyhedron 2000, 19, 627–632. [Google Scholar] [CrossRef]
- Semioshkin, A.; Nizhnik, E.; Godovikov, I.; Starikova, Z.; Bregadze, V. Reactions of oxonium derivatives of [B12H12]2− with amines: Synthesis and structure of novel B12-based ammonium salts and amino acids. J. Organomet. Chem. 2007, 692, 4020–4028. [Google Scholar] [CrossRef]
- Volkov, A.A.; Kruglova, N.S.; Meschaninova, M.I.; Venyaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Selective protection of nuclease-sensitive sites in siRNA prolongs silencing effect. Oligonucleotides 2009, 19, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Kruglova, N.S.; Meschaninova, M.I.; Venyaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Cholesterol-modified anti-MDR1 small interfering RNA: Uptake and biological activity. Mol. Biol. 2010, 44, 254–261. [Google Scholar] [CrossRef]
- Ullah, I.; Chung, K.; Beloor, J.; Kim, J.; Cho, M.; Kim, N.; Lee, K.Y.; Kumar, P.; Lee, S.-K. Trileucine residues in a ligand-CPP-based siRNA delivery platform improve endosomal escape of siRNA. J. Drug. Target. 2017, 25, 320–329. [Google Scholar] [CrossRef]
- Singh, T.; Murthy, A.S.N.; Yang, H.-J.; Im, J. Versatility of cell-penetrating peptides for intracellular delivery of siRNA. Drug. Deliv. 2018, 25, 1996–2006. [Google Scholar] [CrossRef]
- Chernikov, I.V.; Vlassov, V.V.; Chernolovskaya, E.L. Current development of siRNA bioconjugates: From research to the clinic. Front. Pharmacol. 2019, 10, 444. [Google Scholar] [CrossRef]
- Chandela, A.; Ueno, Y. Systemic delivery of small interfering RNA therapeutics: Obstacles and advances. Rev. Agric. Sci. 2019, 7, 10–28. [Google Scholar] [CrossRef]
- Calabrese, G.; Daou, A.; Barbu, E.; Tsibouklis, J. Towards carborane-functionalised structures for the treatment of brain cancer. Drug Discov. Today 2018, 23, 63–75. [Google Scholar] [CrossRef]
- Lesnikowski, Z.J. Boron clusters − A new entity for DNA-oligonucleotide modification. European J. Org. Chem. 2003, 2003, 4489–4500. [Google Scholar] [CrossRef]
- Kaniowski, D.; Ebenryter-Olbińska, K.; Sobczak, M.; Wojtczak, B.; Janczak, S.; Leśnikowski, Z.; Nawrot, B. High Boron-loaded DNA-oligomers as potential boron neutron capture therapy and antisense oligonucleotide dual-action anticancer agents. Molecules 2017, 22, 1393. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, M.; Kiliszek, A.; Rypniewski, W.; Lesnikowski, Z.J.; Olejniczak, A.B. Nucleoside bearing boron clusters and their phosphoramidites – building blocks for modified oligonucleotide synthesis. New. J. Chem. 2015, 39, 1202–1221. [Google Scholar] [CrossRef]
- Bellon, L. Oligoribonucleotides with 2′-O-(tert-Butyldimethylsilyl) Groups. In Current Protocols in Nucleic Acid Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000; pp. 3.6.1–3.6.13. [Google Scholar]
- Lokhov, S.G.; Podyminogin, M.A.; Sergeev, D.S.; Sil’nikov, V.N.; Kutyavin, I.V.; Shishkin, G.V.; Zarytova, V.P. Synthesis and high stability of complementary complexes of N-(2-hydroxyethyl)phenazinium derivatives of oligonucleotides. Bioconjug. Chem. 1992, 3, 414–419. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Conjugate, 5′–3′ | RP HPLC Retention Time, min1 | Molecular Weight | Yield, %3 | |
|---|---|---|---|---|---|
| Calculated | Experimental2 | ||||
| 1 | CholL6–NH–C(O)–d(TTTTTTT) | 13.1 (+3.5) | 2623.2 | 2624.7 | 10.5 |
| 2 | CholL12–NH–C(O)–d(TTTTTTT) | 18.2 (+8.6) | 2707.0 | 2708.6 | 3.2 |
| 3 | Oleyl–NH–C(O)–d(TTTTTTT) | 12.2 (+2.6) | 2361.9 | 2361.3 | 18.2 |
| 4 | EstL6–NH–C(O)–d(TTTTTTT) | 12.3 (+2.7) | 2506.9 | 2507.6 | 3.4 |
| 5 | TocL6–NH–C(O)–d(TTTTTTT) | 11.1 (+1.5) | 2667.3 | 2668.2 | 2.8 |
| 6 | FolL6–NH–C(O)–d(TTTTTTT) | 11.8 (+2.2) | 2677.9 | 2676.2 | 16.1 |
| 7 | (Leu)3–NH–C(O)–d(TTTTTTT) | 12.5 (+2.9) | 2451.9 | 2352.1 | 25.4 |
| 8 | Pyr–NH–C(O)–d(TTTTTTT) | 15.9 (+6.3) | 2325.7 | 2325.8 | 18.6 |
| 9 | c–B12–NH–C(O)–d(TTTTTTT) | 11.2 (+1.6) | 2303.0 | 2301.6 | 7.1 |
| 10 | NH2L6–NH–C(O)–d(TTTTTTT) | 10.4 (+0.8) | 2210.6 | 2210.4 | 16.3 |
| 11 | NH2L12–NH–C(O)–d(TTTTTTT) | 14.3 (+4.7) | 2294.8 | 2293.4 | 15.1 |
| 12 | CH≡C–CH2–NH–C(O)–d(TTTTTTT) | 10.8 (+1.2) | 2149.5 | 2149.5 | 17.2 |
| 13 | Pyr–NH–C(O)–L3–NH–C(O)–d(TTTTTTT) | 17.0 (+7.4) | 2427.8 | 2426.1 | 7.5 |
| 14 | Phn–NHL6–NH–C(O)–d(TTTTTTT) | 13.2(+3.6) | n.d.5 | n.d.5 | n.d.5 |
| 15 | Phn–NHL12–NH–C(O)–d(TTTTTTT) | 17.2(+7.6) | n.d.5 | n.d.5 | n.d.5 |
| 16 | Cy3L–CH2–NH–C(O)–d(TTTTTTT) | n/a4 | n.d.5 | n.d.5 | n.d.5 |
| 17 | c–B12–NH–C(O)–d(ATACGTTAACGATCCTTCAC) | n/a4 | 6281.2 | 6282.0 | 5.1 |
| 18 | Oleyl–NH–C(O)–d(ATACGTTAACGATCCTTCAC) | n/a4 | 6330.4 | 6330.2 | 15.3 |
| 19 | (Leu)3–NH–C(O)–GGCUUmGACmAAGUUmGUmAUmAUmGG | n/a4 | 7221.7 | 7222.4 | 9.3 |
| 20 | CholL6–NH–C(O)–GGCUUmGACmAAGUUmGUmAUmAUmGG | n/a4 | 7394.2 | 7393.8 | 5.8 |
| 21 | (Leu)3–NH–C(O)–LSSL–GGCUUmGACmAAGUUmGUmAUmAUmGG | n/a4 | 7549.6 | 7551.9 | 3.9 |
| 22 | CholL6–NH–C(O)–LSSL–GGCUUmGACmAAGUUmGUmAUmAUmGG | n/a4 | 7722.5 | 7722.8 | 4.1 |
| 23 | CholL6–NH–C(O)–L12–NHC(O)–GGCUUmGACmAAGUUmGUmAUmAUmGG | n/a4 | 7620.5 | 7619.5 | 2.5 |
| 24 | Pyr–NH–C(O)–GmAmCmAmGmUmAmGmAmUmUmGmUmAmUmAmGm | n/a4 | 5972.1 | 5974.3 | 9.7 |
| 25 | Pyr–NH–C(O)–L3–NHC(O)–GmAmCmAmGmUmAmGmAmUmUmGmUmAmUmAmGm | n/a4 | 6074.2 | 6076.2 | 6.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meschaninova, M.I.; Novopashina, D.S.; Semikolenova, O.A.; Silnikov, V.N.; Venyaminova, A.G. Novel Convenient Approach to the Solid-Phase Synthesis of Oligonucleotide Conjugates. Molecules 2019, 24, 4266. https://doi.org/10.3390/molecules24234266
Meschaninova MI, Novopashina DS, Semikolenova OA, Silnikov VN, Venyaminova AG. Novel Convenient Approach to the Solid-Phase Synthesis of Oligonucleotide Conjugates. Molecules. 2019; 24(23):4266. https://doi.org/10.3390/molecules24234266
Chicago/Turabian StyleMeschaninova, Mariya I., Darya S. Novopashina, Olga A. Semikolenova, Vladimir N. Silnikov, and Alya G. Venyaminova. 2019. "Novel Convenient Approach to the Solid-Phase Synthesis of Oligonucleotide Conjugates" Molecules 24, no. 23: 4266. https://doi.org/10.3390/molecules24234266
APA StyleMeschaninova, M. I., Novopashina, D. S., Semikolenova, O. A., Silnikov, V. N., & Venyaminova, A. G. (2019). Novel Convenient Approach to the Solid-Phase Synthesis of Oligonucleotide Conjugates. Molecules, 24(23), 4266. https://doi.org/10.3390/molecules24234266