Productive Cross-Talk with the Microenvironment: A Critical Step in Ovarian Cancer Metastasis




Abstract
:1. Introduction
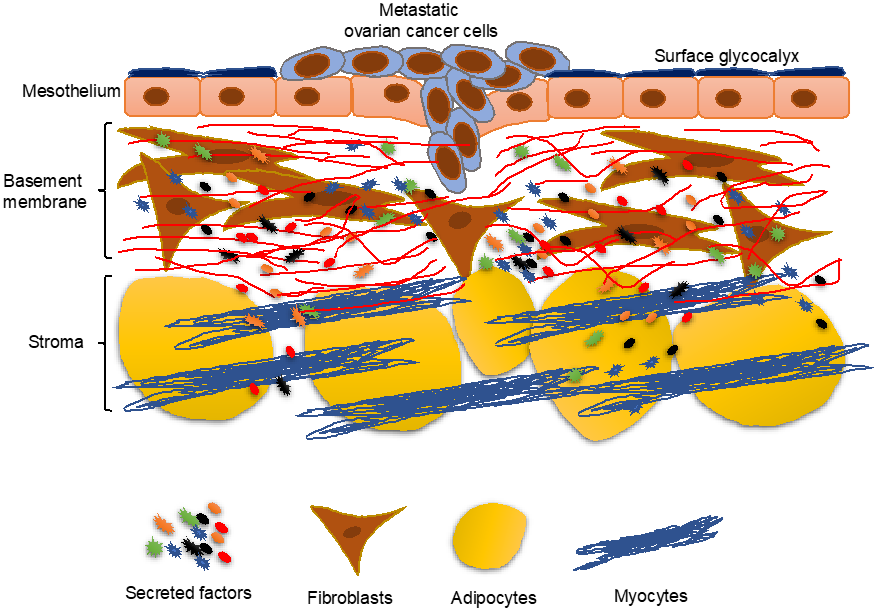
2. Overview of the Mesothelium
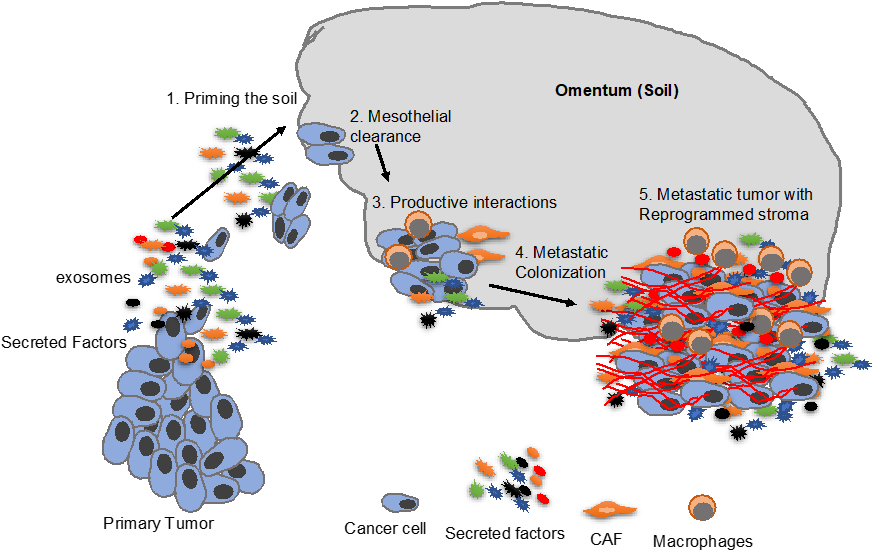
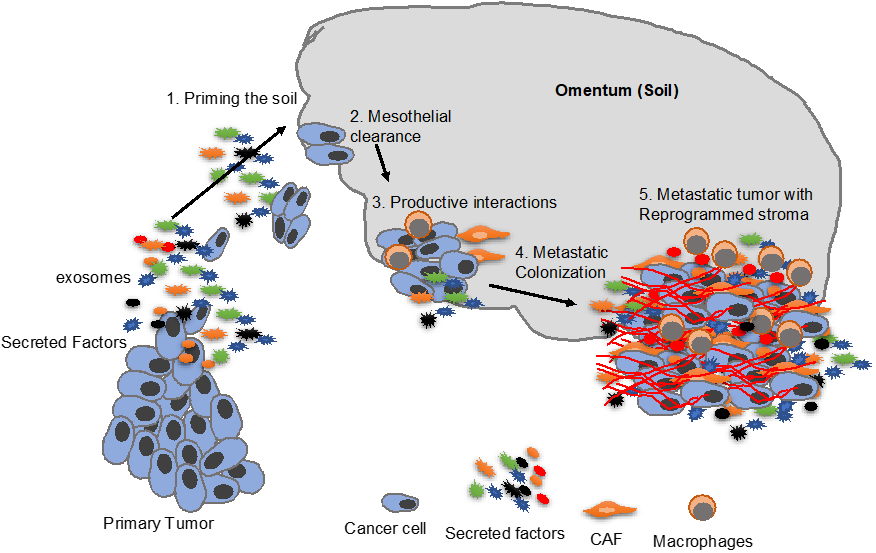
3. Priming the “Soil”
4. Cancer Cells Induce Changes in the Mesothelium
4.1. Fibronectin Secretion
4.2. Tissue Plasminogen Activator Inhibitor Type 1 (PAI-1) Induction
5. Mesothelial Clearance
6. Mesothelial Mesenchymal Transition (MMT)
7. Metastatic Colonization
8. Fibroblasts in Metastatic Colonization
9. Role of Adipocytes in Metastatic Colonization
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Grossman, D.C.; Curry, S.J.; Owens, D.K.; Barry, M.J.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W., Jr.; Kemper, A.R.; Krist, A.H.; Kurth, A.E.; et al. Screening for Ovarian Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 319, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Kaku, T.; Ogawa, S.; Kawano, Y.; Ohishi, Y.; Kobayashi, H.; Hirakawa, T.; Nakano, H. Histological classification of ovarian cancer. Med. Electron. Microsc. 2003, 36, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Prat, J. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynaecol. Obstet. 2014, 124, 1–5. [Google Scholar] [CrossRef]
- Visvanathan, K.; Shaw, P.; May, B.J.; Bahadirli-Talbott, A.; Kaushiva, A.; Risch, H.; Narod, S.; Wang, T.L.; Parkash, V.; Vang, R.; et al. Fallopian Tube Lesions in Women at High Risk for Ovarian Cancer: A Multicenter Study. Cancer Prev. Res. 2018, 11, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Eckert, M.A.; Pan, S.; Hernandez, K.M.; Loth, R.M.; Andrade, J.; Volchenboum, S.L.; Faber, P.; Montag, A.; Lastra, R.; Peter, M.E.; et al. Genomics of Ovarian Cancer Progression Reveals Diverse Metastatic Trajectories Including Intraepithelial Metastasis to the Fallopian Tube. Cancer Discov. 2016, 6, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.S.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, Z.; Xu, S.; Li, X.; Yang, X.; Jin, P.; Liu, Y.; Zhou, X.; Zhang, T.; Gong, C.; et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J. Exp. Med. 2019, 216, 688–703. [Google Scholar] [CrossRef]
- Pradeep, S.; Kim, S.W.; Wu, S.Y.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.V.; Kim, S.J.; Choi, H.J.; Bischoff, F.Z.; et al. Hematogenous metastasis of ovarian cancer: Rethinking mode of spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef]
- Blackburn, S.C.; Stanton, M.P. Anatomy and physiology of the peritoneum. Semin. Pediatr. Surg. 2014, 23, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, S.E.; Prele, C.M.; Lansley, S.M.; Herrick, S.E. The origin of regenerating mesothelium: A historical perspective. Int. J. Artif. Organs 2007, 30, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, S.E.; Wilkosz, S. Structure and function of mesothelial cells. Cancer Treat. Res. 2007, 134, 1–19. [Google Scholar] [PubMed]
- Mutsaers, S.E.; Prele, C.M.; Pengelly, S.; Herrick, S.E. Mesothelial cells and peritoneal homeostasis. Fertil. Steril. 2016, 106, 1018–1024. [Google Scholar] [CrossRef]
- Jonjic, N.; Peri, G.; Bernasconi, S.; Sciacca, F.L.; Colotta, F.; Pelicci, P.; Lanfrancone, L.; Mantovani, A. Expression of adhesion molecules and chemotactic cytokines in cultured human mesothelial cells. J. Exp. Med. 1992, 176, 1165–1174. [Google Scholar] [CrossRef]
- Warn, R.; Harvey, P.; Warn, A.; Foley-Comer, A.; Heldin, P.; Versnel, M.; Arakaki, N.; Daikuhara, Y.; Laurent, G.J.; Herrick, S.E.; et al. HGF/SF induces mesothelial cell migration and proliferation by autocrine and paracrine pathways. Exp. Cell Res. 2001, 267, 258–266. [Google Scholar] [CrossRef]
- Foley-Comer, A.J.; Herrick, S.E.; Al-Mishlab, T.; Prele, C.M.; Laurent, G.J.; Mutsaers, S.E. Evidence for incorporation of free-floating mesothelial cells as a mechanism of serosal healing. J. Cell Sci. 2002, 115, 1383–1389. [Google Scholar]
- Kenny, H.A.; Nieman, K.M.; Mitra, A.K.; Lengyel, E. The first line of intra-abdominal metastatic attack: Breaching the mesothelial cell layer. Cancer Discov. 2011, 1, 100–102. [Google Scholar] [CrossRef]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.A.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar]
- Langley, R.R.; Fidler, I.J. The seed and soil hypothesis revisited—The role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef] [PubMed]
- Hart, I.R.; Talmadge, J.E.; Fidler, I.J. Metastatic behavior of a murine reticulum cell sarcoma exhibiting organ-specific growth. Cancer Res. 1981, 41, 1281–1287. [Google Scholar] [PubMed]
- Coffman, L.G.; Burgos-Ojeda, D.; Wu, R.; Cho, K.; Bai, S.; Buckanovich, R.J. New models of hematogenous ovarian cancer metastasis demonstrate preferential spread to the ovary and a requirement for the ovary for abdominal dissemination. Transl. Res. 2016, 175, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Marini, F.C.; Watson, K.; Zwezdaryk, K.J.; Dembinski, J.L.; LaMarca, H.L.; Tomchuck, S.L.; Honer zu Bentrup, K.; Danka, E.S.; Henkle, S.L.; et al. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3806–3811. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef]
- Yasumoto, K.; Koizumi, K.; Kawashima, A.; Saitoh, Y.; Arita, Y.; Shinohara, K.; Minami, T.; Nakayama, T.; Sakurai, H.; Takahashi, Y.; et al. Role of the CXCL12/CXCR4 axis in peritoneal carcinomatosis of gastric cancer. Cancer Res. 2006, 66, 2181–2187. [Google Scholar] [CrossRef]
- Sawada, K.; Mitra, A.K.; Radjabi, A.R.; Bhaskar, V.; Kistner, E.O.; Tretiakova, M.; Jagadeeswaran, S.; Montag, A.; Becker, A.; Kenny, H.A.; et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008, 68, 2329–2339. [Google Scholar] [CrossRef]
- Kenny, H.A.; Kaur, S.; Coussens, L.M.; Lengyel, E. The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J. Clin. Investig. 2008, 118, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Sawada, K.; Tiwari, P.; Mui, K.; Gwin, K.; Lengyel, E. Ligand-independent activation of c-Met by fibronectin and alpha(5)beta(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene 2011, 30, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Almokadem, S.; Belani, C.P. Volociximab in Cancer. Expert Opin. Biol. Ther. 2012, 12, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Bell-McGuinn, K.M.; Matthews, C.M.; Ho, S.N.; Barve, M.; Gilbert, L.; Penson, R.T.; Lengyel, E.; Palaparthy, R.; Gilder, K.; Vassos, A.; et al. A phase II, single-arm study of the anti-alpha5beta1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol. Oncol. 2011, 121, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Mikula-Pietrasik, J.; Sosinska, P.; Ksiazek, K. Resveratrol inhibits ovarian cancer cell adhesion to peritoneal mesothelium in vitro by modulating the production of alpha5beta1 integrins and hyaluronic acid. Gynecol. Oncol. 2014, 134, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Wang, W.; He, G.; Kuick, R.D.; Gossner, G.; Kueck, A.S.; Wahl, H.; Opipari, A.W.; Liu, J.R. Resveratrol inhibits ovarian tumor growth in an in vivo mouse model. Cancer 2016, 122, 722–729. [Google Scholar] [CrossRef]
- Lang, F.; Qin, Z.; Li, F.; Zhang, H.; Fang, Z.; Hao, E. Apoptotic Cell Death Induced by Resveratrol Is Partially Mediated by the Autophagy Pathway in Human Ovarian Cancer Cells. PLoS ONE 2015, 10, e0129196. [Google Scholar] [CrossRef]
- Zhong, L.X.; Li, H.; Wu, M.L.; Liu, X.Y.; Zhong, M.J.; Chen, X.Y.; Liu, J.; Zhang, Y. Inhibition of STAT3 signaling as critical molecular event in resveratrol-suppressed ovarian cancer cells. J. Ovarian Res. 2015, 8, 25. [Google Scholar] [CrossRef]
- Howells, L.M.; Berry, D.P.; Elliott, P.J.; Jacobson, E.W.; Hoffmann, E.; Hegarty, B.; Brown, K.; Steward, W.P.; Gescher, A.J. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases—Safety, pharmacokinetics, and pharmacodynamics. Cancer Prev. Res. 2011, 4, 1419–1425. [Google Scholar] [CrossRef]
- Hirashima, Y.; Kobayashi, H.; Suzuki, M.; Tanaka, Y.; Kanayama, N.; Terao, T. Transforming growth factor-beta1 produced by ovarian cancer cell line HRA stimulates attachment and invasion through an up-regulation of plasminogen activator inhibitor type-1 in human peritoneal mesothelial cells. J. Biol. Chem. 2003, 278, 26793–26802. [Google Scholar] [CrossRef]
- Dasari, S.; Pandhiri, T.; Haley, J.; Lenz, D.; Mitra, A.K. A Proximal Culture Method to Study Paracrine Signaling Between Cells. JoVE 2018. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.K.; Tancioni, I.; Lawson, C.; Miller, N.L.; Jean, C.; Chen, X.L.; Uryu, S.; Kim, J.; Tarin, D.; Stupack, D.G.; et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin. Exp. Metastasis 2013, 30, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, E.; Sawada, K.; Nakamura, K.; Yoshimura, A.; Kinose, Y.; Kodama, M.; Hashimoto, K.; Mabuchi, S.; Makino, H.; Morii, E.; et al. Plasminogen activator inhibitor-1 is an independent prognostic factor of ovarian cancer and IMD-4482, a novel plasminogen activator inhibitor-1 inhibitor, inhibits ovarian cancer peritoneal dissemination. Oncotarget 2017, 8, 89887–89902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Dorn, J.; Napieralski, R.; Walch, A.; Diersch, S.; Kotzsch, M.; Ahmed, N.; Hooper, J.D.; Kiechle, M.; Schmitt, M.; et al. Plasmin(ogen) serves as a favorable biomarker for prediction of survival in advanced high-grade serous ovarian cancer. Biol. Chem. 2017, 398, 765–773. [Google Scholar] [CrossRef]
- Van Dam, P.A.; Coelho, A.; Rolfo, C. Is there a role for urokinase-type plasminogen activator inhibitors as maintenance therapy in patients with ovarian cancer? Eur. J. Surg. Oncol. 2017, 43, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Niedbala, M.J.; Crickard, K.; Bernacki, R.J. Interactions of human ovarian tumor cells with human mesothelial cells grown on extracellular matrix. An in vitro model system for studying tumor cell adhesion and invasion. Exp. Cell Res. 1985, 160, 499–513. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Pettengell, R.; Nasiri, N.; Kalia, V.; Dalgleish, A.G.; Barton, D.P. Characteristics and growth patterns of human peritoneal mesothelial cells: Comparison between advanced epithelial ovarian cancer and non-ovarian cancer sources. J. Soc. Gynecol. Investig. 1999, 6, 333–340. [Google Scholar] [CrossRef]
- Davidowitz, R.A.; Selfors, L.M.; Iwanicki, M.P.; Elias, K.M.; Karst, A.; Piao, H.; Ince, T.A.; Drage, M.G.; Dering, J.; Konecny, G.E.; et al. Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J. Clin. Investig. 2014, 124, 2611–2625. [Google Scholar] [CrossRef]
- Vaksman, O.; Trope, C.; Davidson, B.; Reich, R. Exosome-derived miRNAs and ovarian carcinoma progression. Carcinogenesis 2014, 35, 2113–2120. [Google Scholar] [CrossRef]
- Matte, I.; Lane, D.; Laplante, C.; Garde-Granger, P.; Rancourt, C.; Piche, A. Ovarian cancer ascites enhance the migration of patient-derived peritoneal mesothelial cells via cMet pathway through HGF-dependent and -independent mechanisms. Int. J. Cancer 2015, 137, 289–298. [Google Scholar] [CrossRef]
- Lee, Y.C.; Tsai, Y.S.; Hung, S.Y.; Lin, T.M.; Lin, S.H.; Liou, H.H.; Liu, H.C.; Chang, M.Y.; Wang, H.H.; Ho, L.C.; et al. Shorter daily dwelling time in peritoneal dialysis attenuates the epithelial-to-mesenchymal transition of mesothelial cells. BMC Nephrol. 2014, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Mo, M.; Lara-Pezzi, E.; Selgas, R.; Ramirez-Huesca, M.; Dominguez-Jimenez, C.; Jimenez-Heffernan, J.A.; Aguilera, A.; Sanchez-Tomero, J.A.; Bajo, M.A.; Alvarez, V.; et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N. Engl. J. Med. 2003, 348, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, P.; Jimenez-Heffernan, J.A.; Rynne-Vidal, A.; Perez-Lozano, M.L.; Gilsanz, A.; Ruiz-Carpio, V.; Reyes, R.; Garcia-Bordas, J.; Stamatakis, K.; Dotor, J.; et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J. Pathol. 2013, 231, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matte, I.; Lane, D.; Laplante, C.; Rancourt, C.; Piche, A. Profiling of cytokines in human epithelial ovarian cancer ascites. Am. J. Cancer Res. 2012, 2, 566–580. [Google Scholar] [PubMed]
- Milliken, D.; Scotton, C.; Raju, S.; Balkwill, F.; Wilson, J. Analysis of chemokines and chemokine receptor expression in ovarian cancer ascites. Clin. Cancer Res. 2002, 8, 1108–1114. [Google Scholar]
- Graves, L.E.; Ariztia, E.V.; Navari, J.R.; Matzel, H.J.; Stack, M.S.; Fishman, D.A. Proinvasive properties of ovarian cancer ascites-derived membrane vesicles. Cancer Res. 2004, 64, 7045–7049. [Google Scholar] [CrossRef]
- Choi, D.S.; Park, J.O.; Jang, S.C.; Yoon, Y.J.; Jung, J.W.; Choi, D.Y.; Kim, J.W.; Kang, J.S.; Park, J.; Hwang, D.; et al. Proteomic analysis of microvesicles derived from human colorectal cancer ascites. Proteomics 2011, 11, 2745–2751. [Google Scholar] [CrossRef]
- Freedman, R.S.; Deavers, M.; Liu, J.; Wang, E. Peritoneal inflammation—A microenvironment for Epithelial Ovarian Cancer (EOC). J. Transl. Med. 2004, 2, 23. [Google Scholar] [CrossRef]
- Matte, I.; Lane, D.; Bachvarov, D.; Rancourt, C.; Piche, A. Role of malignant ascites on human mesothelial cells and their gene expression profiles. BMC Cancer 2014, 14, 288. [Google Scholar] [CrossRef] [PubMed]
- Demir, A.Y.; Groothuis, P.G.; Dunselman, G.A.; Schurgers, L.; Evers, J.L.; de Goeij, A.F. Molecular characterization of soluble factors from human menstrual effluent that induce epithelial to mesenchymal transitions in mesothelial cells. Cell Tissue Res. 2005, 322, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Cabrera, M. Mesenchymal Conversion of Mesothelial Cells Is a Key Event in the Pathophysiology of the Peritoneum during Peritoneal Dialysis. Adv. Med. 2014, 2014, 473134. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.A.; Shin, K.S.; Kim, J.H.; Kim, Y.I.; Chung, S.S.; Park, S.H.; Kim, Y.L.; Kang, D.H. HGF and BMP-7 ameliorate high glucose-induced epithelial-to-mesenchymal transition of peritoneal mesothelium. J. Am. Soc. Nephrol. 2009, 20, 567–581. [Google Scholar] [CrossRef]
- Nakamura, M.; Ono, Y.J.; Kanemura, M.; Tanaka, T.; Hayashi, M.; Terai, Y.; Ohmichi, M. Hepatocyte growth factor secreted by ovarian cancer cells stimulates peritoneal implantation via the mesothelial-mesenchymal transition of the peritoneum. Gynecol. Oncol. 2015, 139, 345–354. [Google Scholar] [CrossRef]
- Manickam, N.; Patel, M.; Griendling, K.K.; Gorin, Y.; Barnes, J.L. RhoA/Rho kinase mediates TGF-beta1-induced kidney myofibroblast activation through Poldip2/Nox4-derived reactive oxygen species. Am. J. Physiol. Ren. Physiol. 2014, 307, F159–F171. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, X.; Liu, Y.; Yi, B.; Yu, X. Epithelial-mesenchymal transition of rat peritoneal mesothelial cells via Rhoa/Rock pathway. In Vitro Cell. Dev. Biol. Anim. 2011, 47, 165–172. [Google Scholar] [CrossRef]
- Kenny, H.A.; Chiang, C.Y.; White, E.A.; Schryver, E.M.; Habis, M.; Romero, I.L.; Ladanyi, A.; Penicka, C.V.; George, J.; Matlin, K.; et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J. Clin. Investig. 2014, 124, 4614–4628. [Google Scholar] [CrossRef] [Green Version]
- Rikitake, Y.; Liao, J.K. Rho GTPases, statins, and nitric oxide. Circ. Res. 2005, 97, 1232–1235. [Google Scholar] [CrossRef]
- Chang, T.I.; Kang, H.Y.; Kim, K.S.; Lee, S.H.; Nam, B.Y.; Paeng, J.; Kim, S.; Park, J.T.; Yoo, T.H.; Kang, S.W.; et al. The effect of statin on epithelial-mesenchymal transition in peritoneal mesothelial cells. PLoS ONE 2014, 9, e109628. [Google Scholar] [CrossRef]
- Elmore, R.G.; Ioffe, Y.; Scoles, D.R.; Karlan, B.Y.; Li, A.J. Impact of statin therapy on survival in epithelial ovarian cancer. Gynecol. Oncol. 2008, 111, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, A.; Kikuchi, N.; Osada, R.; Wang, C.; Hayashi, A.; Nikaido, T.; Konishi, I. Overexpression of RhoA enhances peritoneal dissemination: RhoA suppression with Lovastatin may be useful for ovarian cancer. Cancer Sci. 2008, 99, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Ning, L.; Huang, Y.; Liu, Y.; Zhang, W.; Hu, Y.; Lang, J.; Yang, J. Statin use and survival outcomes in endocrine-related gynecologic cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 41508–41517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Z.; He, W.; Xu, L.; Wang, J.; Shi, J.; Sheng, M. Effects of Astragaloside IV Against the TGF-beta1-Induced Epithelial-to-Mesenchymal Transition in Peritoneal Mesothelial Cells by Promoting Smad 7 Expression. Cell. Physiol. Biochem. 2015, 37, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Su, L.; Chen, D.; Zheng, W.; Liu, Y. Astragaloside IV Induced miR-134 Expression Reduces EMT and Increases Chemotherapeutic Sensitivity by Suppressing CREB1 Signaling in Colorectal Cancer Cell Line SW-480. Cell. Physiol. Biochem. 2017, 43, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.H.; Shin, H.S.; Sun Choi, H.; Ryu, E.S.; Jin Kim, M.; Ki Min, S.; Lee, J.H.; Kook Lee, H.; Kim, K.H.; Kang, D.H. Effects of dexamethasone on the TGF-beta1-induced epithelial-to-mesenchymal transition in human peritoneal mesothelial cells. Lab. Investig. 2013, 93, 194–206. [Google Scholar] [CrossRef]
- Yanaranop, M.; Chaithongwongwatthana, S. Intravenous versus oral dexamethasone for prophylaxis of paclitaxel-associated hypersensitivity reaction in patients with primary ovarian, fallopian tube and peritoneal cancer: A double-blind randomized controlled trial. Asia Pac. J. Clin. Oncol. 2016, 12, 289–299. [Google Scholar] [CrossRef]
- Sorbe, B.; Hallen, C. Betamethasone-dixyrazine versus betamethasone-metoclopramide as antiemetic treatment of cisplatin-doxorubicin-induced nausea in ovarian carcinoma patients. Eur. J. Gynaecol. Oncol. 1991, 12, 31–37. [Google Scholar]
- Melhem, A.; Yamada, S.D.; Fleming, G.F.; Delgado, B.; Brickley, D.R.; Wu, W.; Kocherginsky, M.; Conzen, S.D. Administration of glucocorticoids to ovarian cancer patients is associated with expression of the anti-apoptotic genes SGK1 and MKP1/DUSP1 in ovarian tissues. Clin. Cancer Res. 2009, 15, 3196–3204. [Google Scholar] [CrossRef]
- Loureiro, J.; Sandoval, P.; del Peso, G.; Gonzalez-Mateo, G.; Fernandez-Millara, V.; Santamaria, B.; Bajo, M.A.; Sanchez-Tomero, J.A.; Guerra-Azcona, G.; Selgas, R.; et al. Tamoxifen ameliorates peritoneal membrane damage by blocking mesothelial to mesenchymal transition in peritoneal dialysis. PLoS ONE 2013, 8, e61165. [Google Scholar] [CrossRef]
- Paleari, L.; Gandini, S.; Provinciali, N.; Puntoni, M.; Colombo, N.; DeCensi, A. Clinical benefit and risk of death with endocrine therapy in ovarian cancer: A comprehensive review and meta-analysis. Gynecol. Oncol. 2017, 146, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.K. Ovarian Cancer Metastasis: A Unique Mechanism of Dissemination. In Tumor Metastasis; InTech: Rijeka, Croatia, 2016. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.K.; Chiang, C.Y.; Tiwari, P.; Tomar, S.; Watters, K.M.; Peter, M.E.; Lengyel, E. Microenvironment-induced downregulation of miR-193b drives ovarian cancer metastasis. Oncogene 2015, 34, 5923–5932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomar, S.; Plotnik, J.P.; Haley, J.; Scantland, J.; Dasari, S.; Sheikh, Z.; Emerson, R.; Lenz, D.; Hollenhorst, P.C.; Mitra, A.K. ETS1 induction by the microenvironment promotes ovarian cancer metastasis through focal adhesion kinase. Cancer Lett. 2018, 414, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Gutierrez-Hartmann, A. Molecular mechanisms of ETS transcription factor-mediated tumorigenesis. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 522–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, A.; Watson, D.K. ETS transcription factors and their emerging roles in human cancer. Eur. J. Cancer 2005, 41, 2462–2478. [Google Scholar] [CrossRef]
- Dasari, S.; Fang, Y.; Mitra, A.K. Cancer Associated Fibroblasts: Naughty Neighbors That Drive Ovarian Cancer Progression. Cancers 2018, 10, 406. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics 2018, 18, e1700167. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blazenovic, I.; et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 2017, 8, 924. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, D.R.; Plastino, J. Invading, Leading and Navigating Cells in Caenorhabditis elegans: Insights into Cell Movement in Vivo. Genetics 2018, 208, 53–78. [Google Scholar] [CrossRef]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef]
- Mayor, R.; Etienne-Manneville, S. The front and rear of collective cell migration. Nat. Rev. Mol. Cell Biol. 2016, 17, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Ollila, S.; Domenech-Moreno, E.; Laajanen, K.; Wong, I.P.; Tripathi, S.; Pentinmikko, N.; Gao, Y.; Yan, Y.; Niemela, E.H.; Wang, T.C.; et al. Stromal Lkb1 deficiency leads to gastrointestinal tumorigenesis involving the IL-11-JAK/STAT3 pathway. J. Clin. Investig. 2018, 128, 402–414. [Google Scholar] [CrossRef]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef] [Green Version]
- Onoue, T.; Uchida, D.; Begum, N.M.; Tomizuka, Y.; Yoshida, H.; Sato, M. Epithelial-mesenchymal transition induced by the stromal cell-derived factor-1/CXCR4 system in oral squamous cell carcinoma cells. Int. J. Oncol. 2006, 29, 1133–1138. [Google Scholar] [CrossRef]
- Soon, P.S.; Kim, E.; Pon, C.K.; Gill, A.J.; Moore, K.; Spillane, A.J.; Benn, D.E.; Baxter, R.C. Breast cancer-associated fibroblasts induce epithelial-to-mesenchymal transition in breast cancer cells. Endocr. Relat. Cancer 2013, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [PubMed]
- Thibault, B.; Castells, M.; Delord, J.P.; Couderc, B. Ovarian cancer microenvironment: Implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev. 2014, 33, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef]
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.; Helou, Y.; Batlle, R.; Liu, X.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019, 29, 141–155. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Lehtonen, A.; Himanen, P.; Saraste, M.; Niittymaki, K.; Marniemi, J. Double-blind comparison of the effects of long-term treatment with doxazosin or atenolol on serum lipoproteins. Br. J. Clin. Pharmacol. 1986, 21 (Suppl. 1), 77s–81s. [Google Scholar] [CrossRef]
- Miranda, F.; Mannion, D.; Liu, S.; Zheng, Y.; Mangala, L.S.; Redondo, C.; Herrero-Gonzalez, S.; Xu, R.; Taylor, C.; Chedom, D.F.; et al. Salt-Inducible Kinase 2 Couples Ovarian Cancer Cell Metabolism with Survival at the Adipocyte-Rich Metastatic Niche. Cancer Cell 2016, 30, 273–289. [Google Scholar] [CrossRef]
- Tang, J.; Pulliam, N.; Ozes, A.; Buechlein, A.; Ding, N.; Keer, H.; Rusch, D.; O’Hagan, H.; Stack, M.S.; Nephew, K.P. Epigenetic Targeting of Adipocytes Inhibits High-Grade Serous Ovarian Cancer Cell Migration and Invasion. Mol. Cancer Res. 2018, 16, 1226–1240. [Google Scholar] [CrossRef] [Green Version]
- Schumann, T.; Adhikary, T.; Wortmann, A.; Finkernagel, F.; Lieber, S.; Schnitzer, E.; Legrand, N.; Schober, Y.; Nockher, W.A.; Toth, P.M.; et al. Deregulation of PPARbeta/delta target genes in tumor-associated macrophages by fatty acid ligands in the ovarian cancer microenvironment. Oncotarget 2015, 6, 13416–13433. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.A.; Rybalko, V.Y.; Bigelow, C.E.; Lugade, A.A.; Foster, T.H.; Frelinger, J.G.; Lord, E.M. Preferential attachment of peritoneal tumor metastases to omental immune aggregates and possible role of a unique vascular microenvironment in metastatic survival and growth. Am. J. Pathol. 2006, 169, 1739–1752. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.; Krishnan, V.; Schoof, M.; Rodriguez, I.; Theriault, B.; Chekmareva, M.; Rinker-Schaeffer, C. Milky spots promote ovarian cancer metastatic colonization of peritoneal adipose in experimental models. Am. J. Pathol. 2013, 183, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.X.; Magovern, C.J.; Mack, C.A.; Budenbender, K.T.; Ko, W.; Rosengart, T.K. Vascular endothelial growth factor is the major angiogenic factor in omentum: Mechanism of the omentum-mediated angiogenesis. J. Surg. Res. 1997, 67, 147–154. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| S.No | Secreted Factors | Sources | References |
|---|---|---|---|
| 1 | Transforming growth factor-β (TGF-β) | Mesothelial cells | [12,16] |
| 2 | Transforming growth factor-β (TGF-β) | Cancer cells | [71] |
| 3 | Platelet-derived growth factor (PDGF) | Mesothelial cells | [12,16] |
| 4 | Fibroblast growth factor (FGF) | Mesothelial cells | [12,16] |
| 5 | Hepatocyte growth factor (HGF) | Mesothelial cells | [12,16] |
| 6 | Keratinocyte growth factor (KGF) | Mesothelial cells | [12,16] |
| 7 | Epithelial growth factors (EGFs) | Mesothelial cells | [12,16] |
| 8 | Cytokines like IL6, stroma derived factor 1 (SDF1), leucine leucine 37 (LL 37) | Cancer cells | [27,28] |
| 9 | Fibronectin | Mesothelial cells | [71] |
| 10 | Matrix metalloprotease 9 (MMP 9) | Fibroblasts | [30,97] |
| 11 | Plasminogen activator inhibitor type 1(PAI-1) | Mesothelial cells | [43] |
| 12 | Urokinase plasminogen activator (uPA) | [53] | |
| 13 | Cytokines (angiogenin, angiopoietin-2, GRO, ICAM-1, IL-6, IL-6R, IL-8, IL-10, leptin, MCP-1, MIF NAP-2, osteprotegerin (OPG), RANTES, TIMP-2 and UPAR were elevated in most malignant ascites) | Malignant ascites | [53] |
| 14 | Chemokines (CCL2, -3, -4, -5, -8, and -22) | Malignant ascites | [60] |
| 15 | Chemokines (IL-6, IL-11) | Cancer associated fibroblasts | [101] |
| 16 | Exosomes/Membrane vesicles/microvesicles | Malignant ascites | [61,62] |
| 17 | Fatty acid binding protein 4 (FABP4) | Adipocytes | [110] |
| 18 | Adipokines (IL-8, IL-6), monocyte chemoattractant protein-1 and adiponectin | Adipocytes | [110] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd El Aziz, M.A.; Agarwal, K.; Dasari, S.; Mitra, A.K. Productive Cross-Talk with the Microenvironment: A Critical Step in Ovarian Cancer Metastasis. Cancers 2019, 11, 1608. https://doi.org/10.3390/cancers11101608
Abd El Aziz MA, Agarwal K, Dasari S, Mitra AK. Productive Cross-Talk with the Microenvironment: A Critical Step in Ovarian Cancer Metastasis. Cancers. 2019; 11(10):1608. https://doi.org/10.3390/cancers11101608
Chicago/Turabian StyleAbd El Aziz, Mohamed A., Komal Agarwal, Subramanyam Dasari, and Anirban K. Mitra. 2019. "Productive Cross-Talk with the Microenvironment: A Critical Step in Ovarian Cancer Metastasis" Cancers 11, no. 10: 1608. https://doi.org/10.3390/cancers11101608
APA StyleAbd El Aziz, M. A., Agarwal, K., Dasari, S., & Mitra, A. K. (2019). Productive Cross-Talk with the Microenvironment: A Critical Step in Ovarian Cancer Metastasis. Cancers, 11(10), 1608. https://doi.org/10.3390/cancers11101608