Methyl Jasmonate Reduces Inflammation and Oxidative Stress in the Brain of Arthritic Rats

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Induction of Arthritis
2.3. Experimental Design
2.4. Brain Homogenate Preparation and Mitochondria Isolation
2.5. Brain Oxidative Stress and Inflammatory Parameters
2.6. Mitochondrial Transmembrane Potential (ΔΨm) and Enzyme Activities
2.7. Brain Hexokinase (HK) Activity
2.8. Statistical Analysis
3. Results
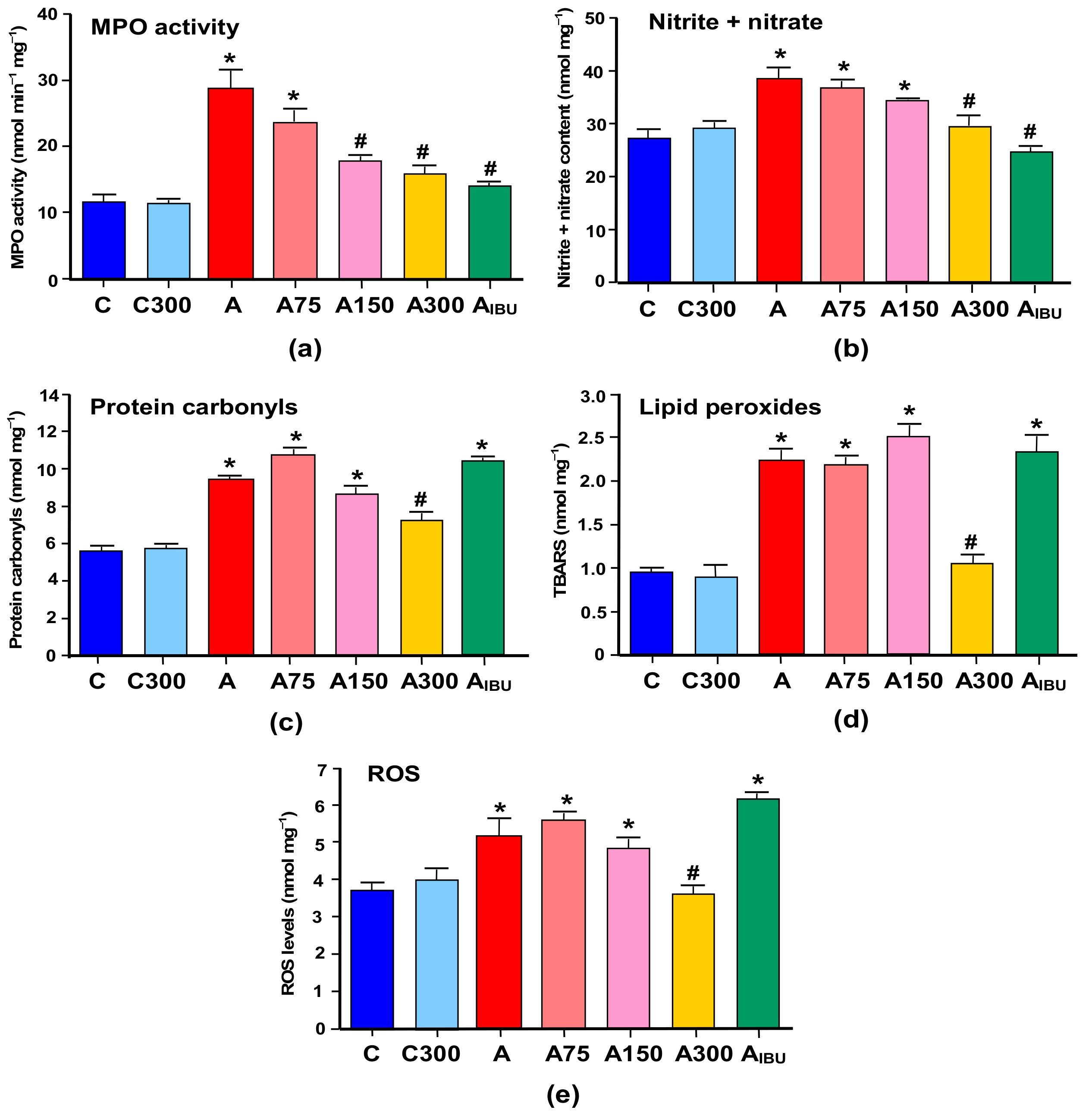
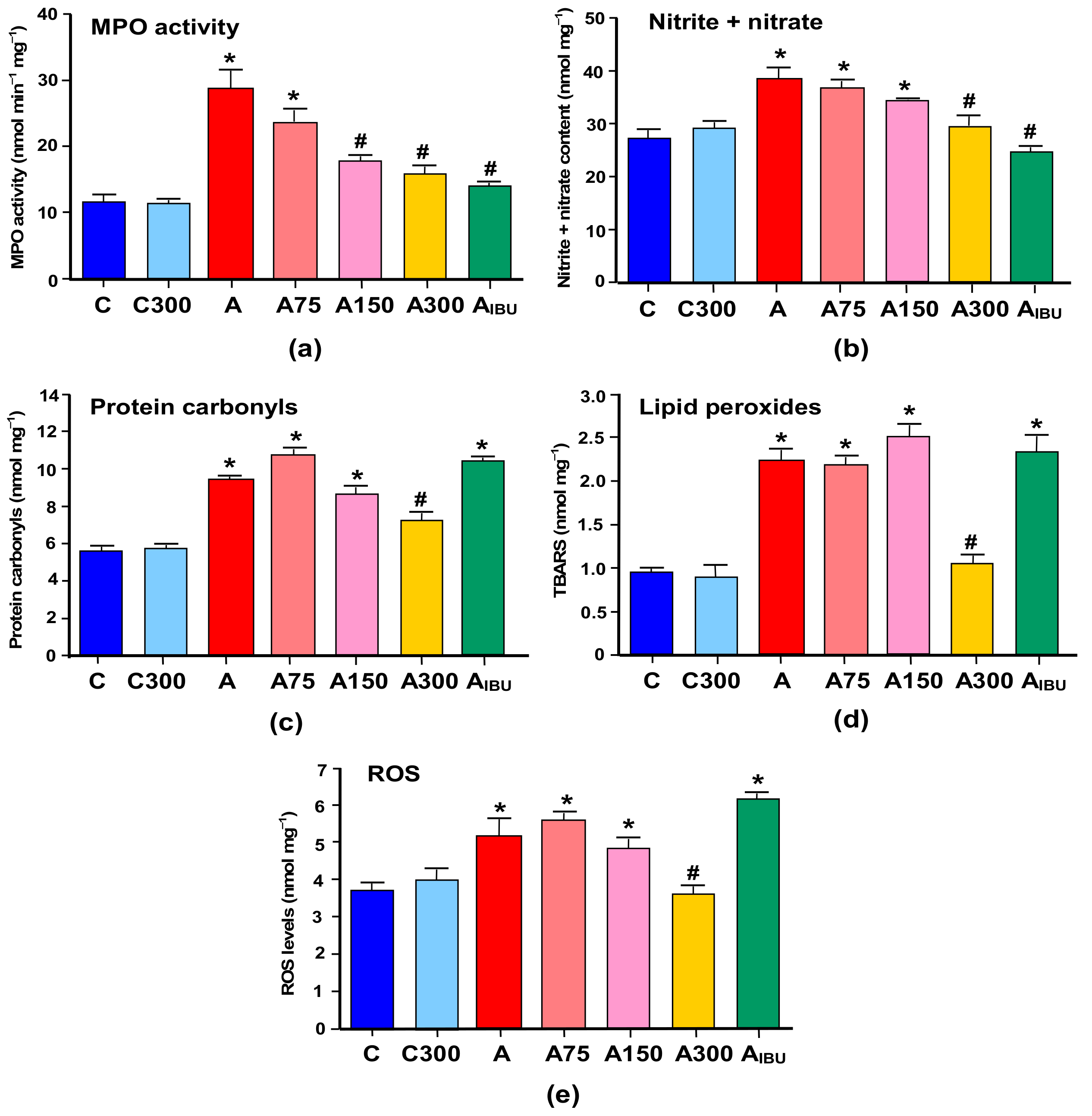
3.1. Indicators of Inflammation and Oxidative Stress
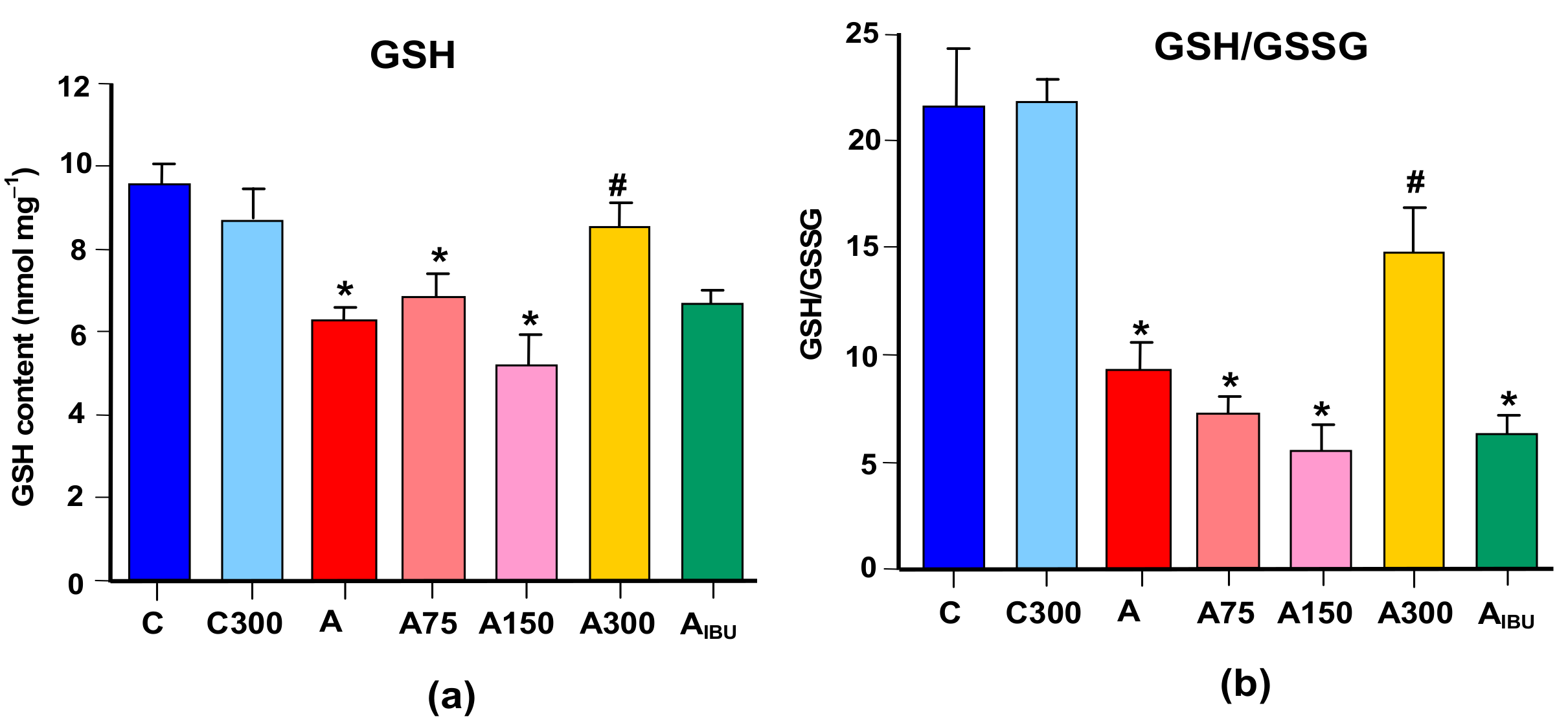
3.2. GSH Levels
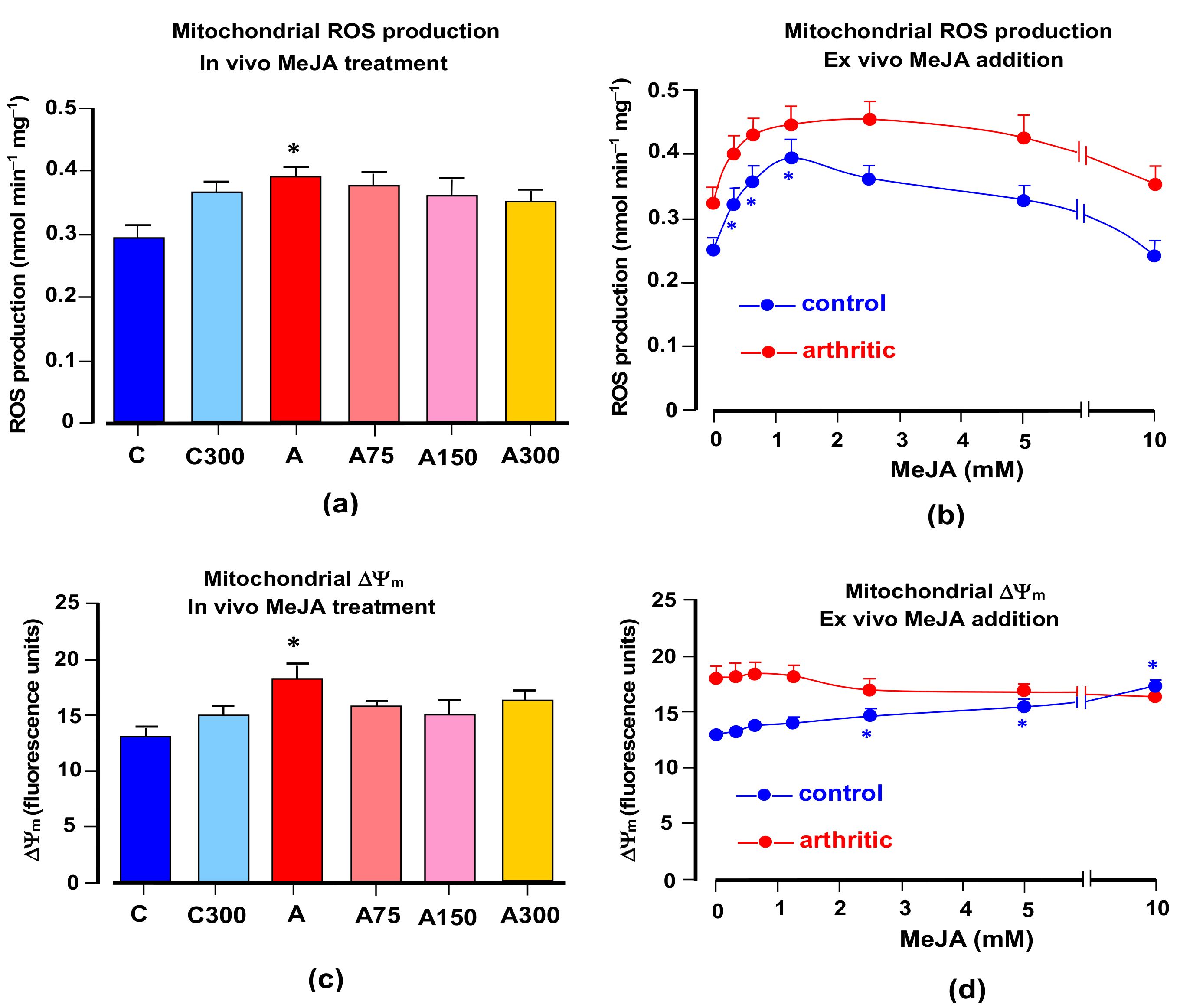
3.3. Mitochondrial ROS Generation and Membrane Potential
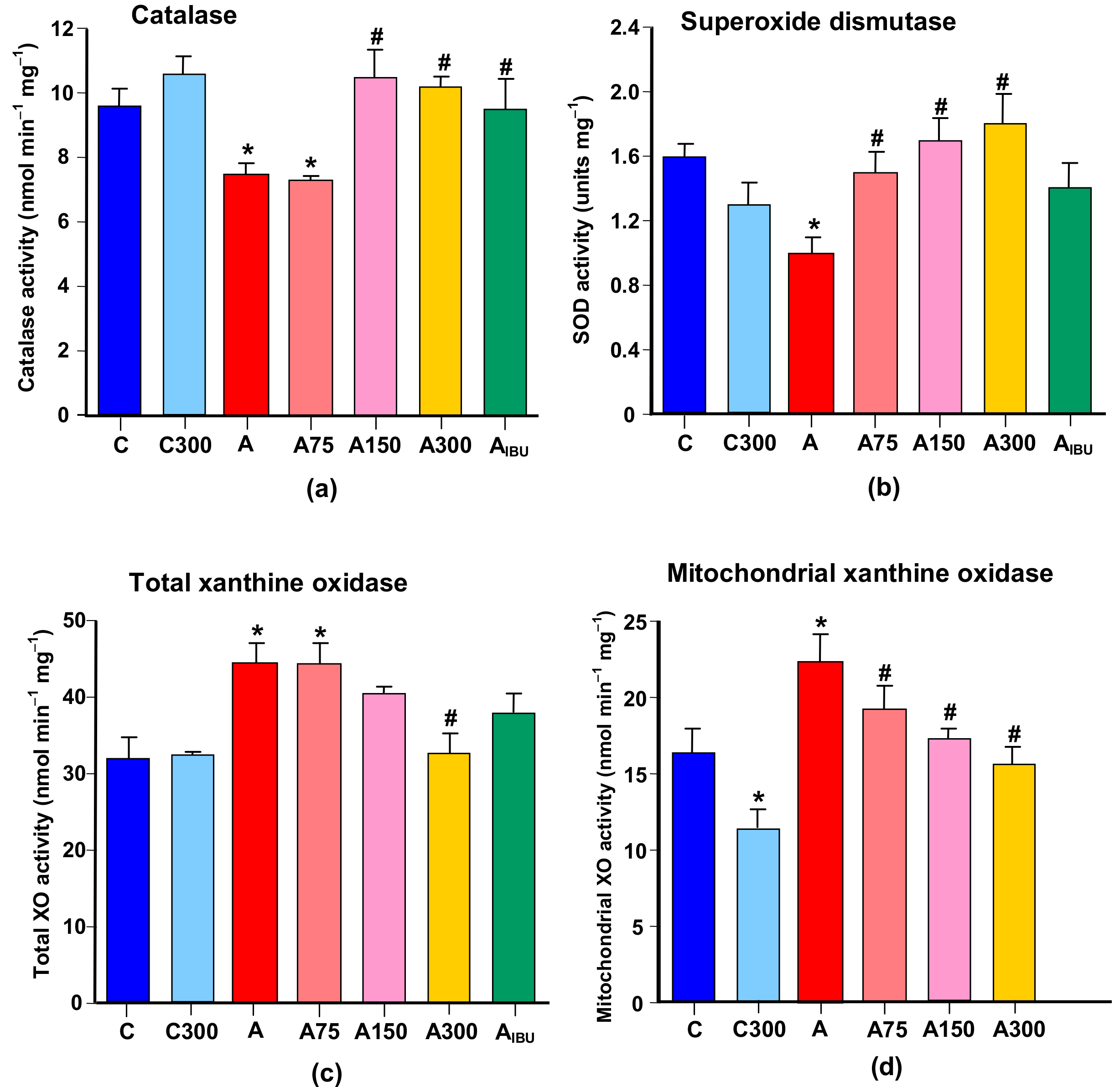
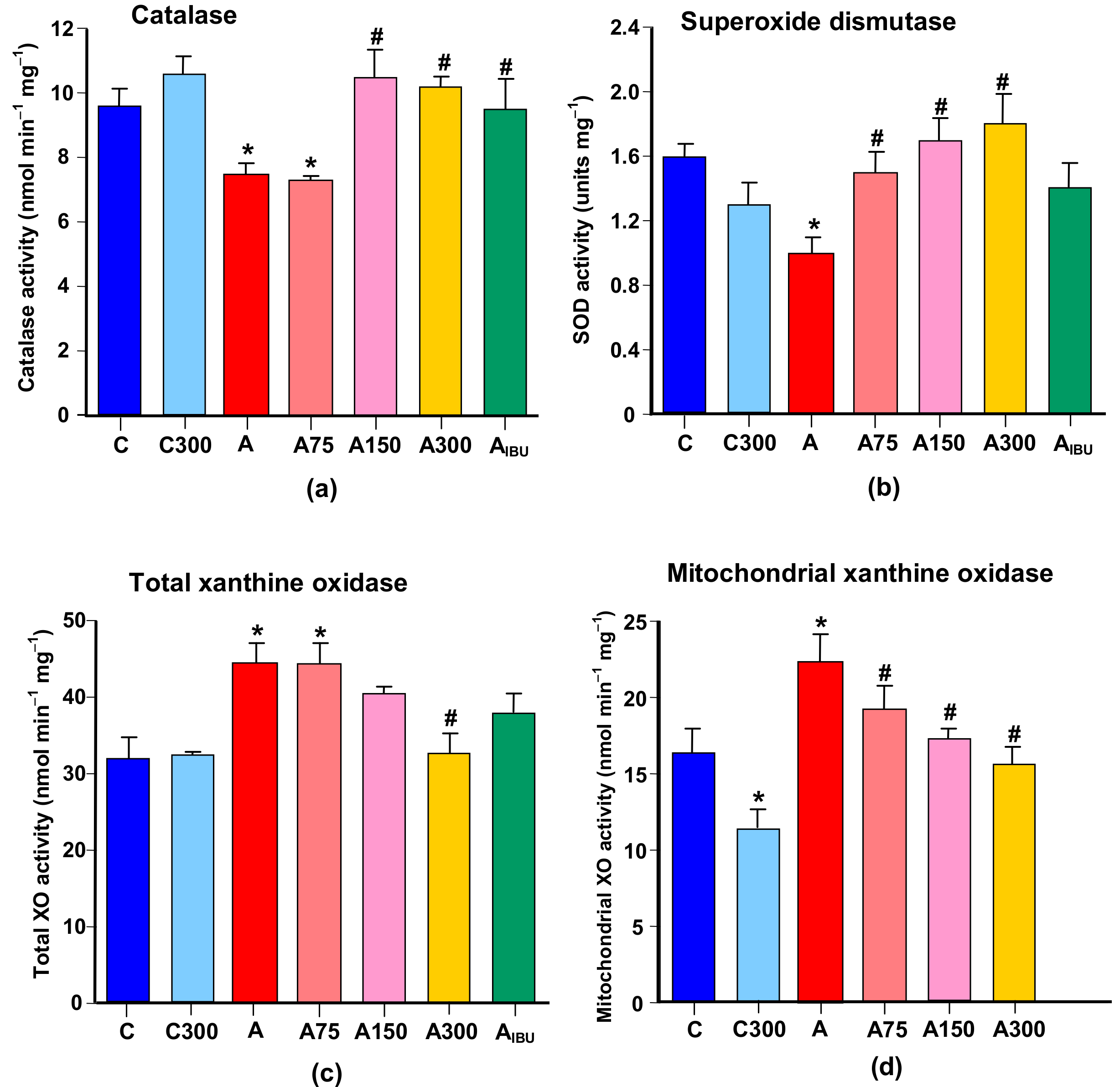
3.4. Antioxidant and Prooxidant Enzyme Activities
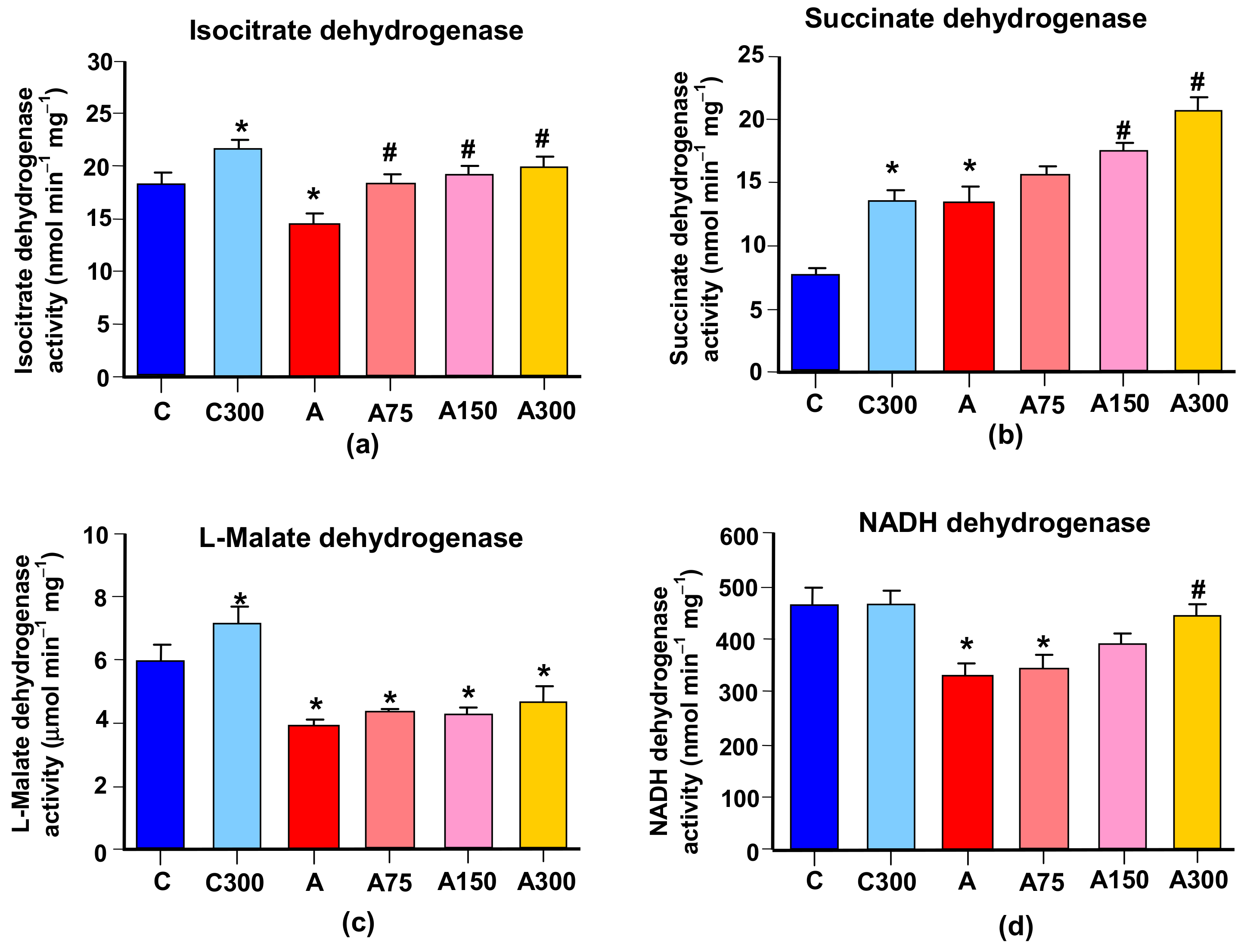
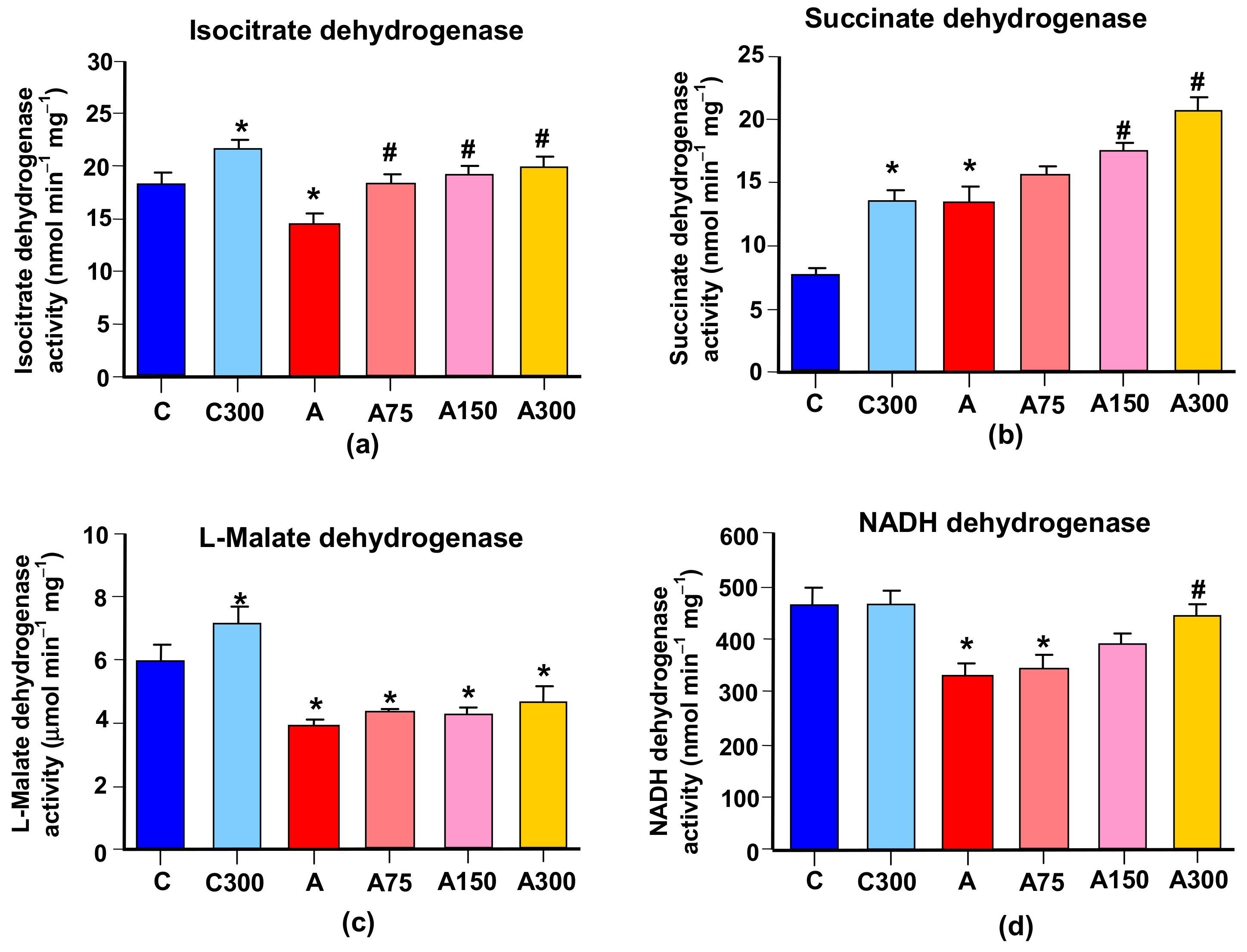
3.5. Mitochondrial Enzymes Linked to Oxidative Metabolism
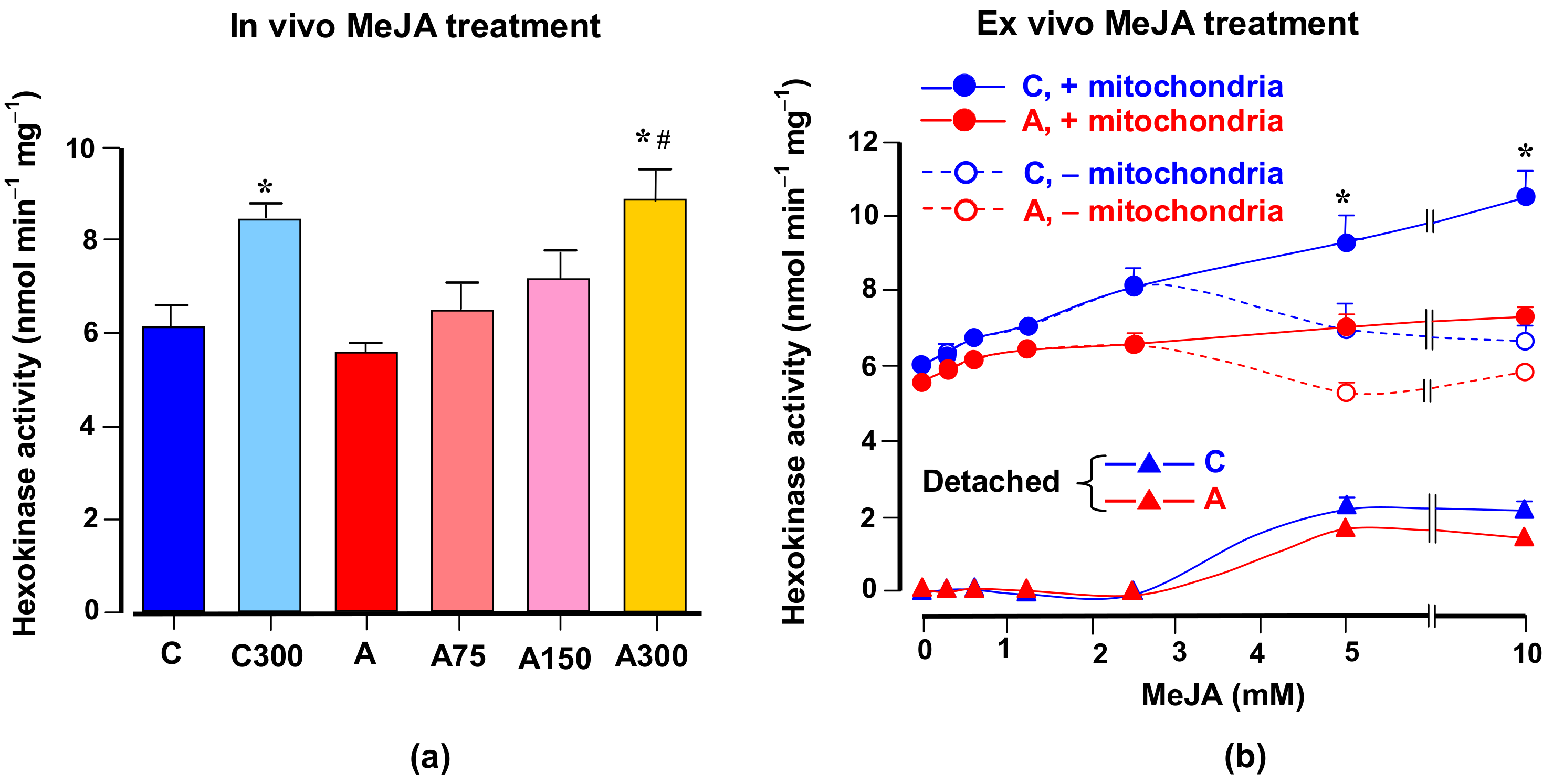
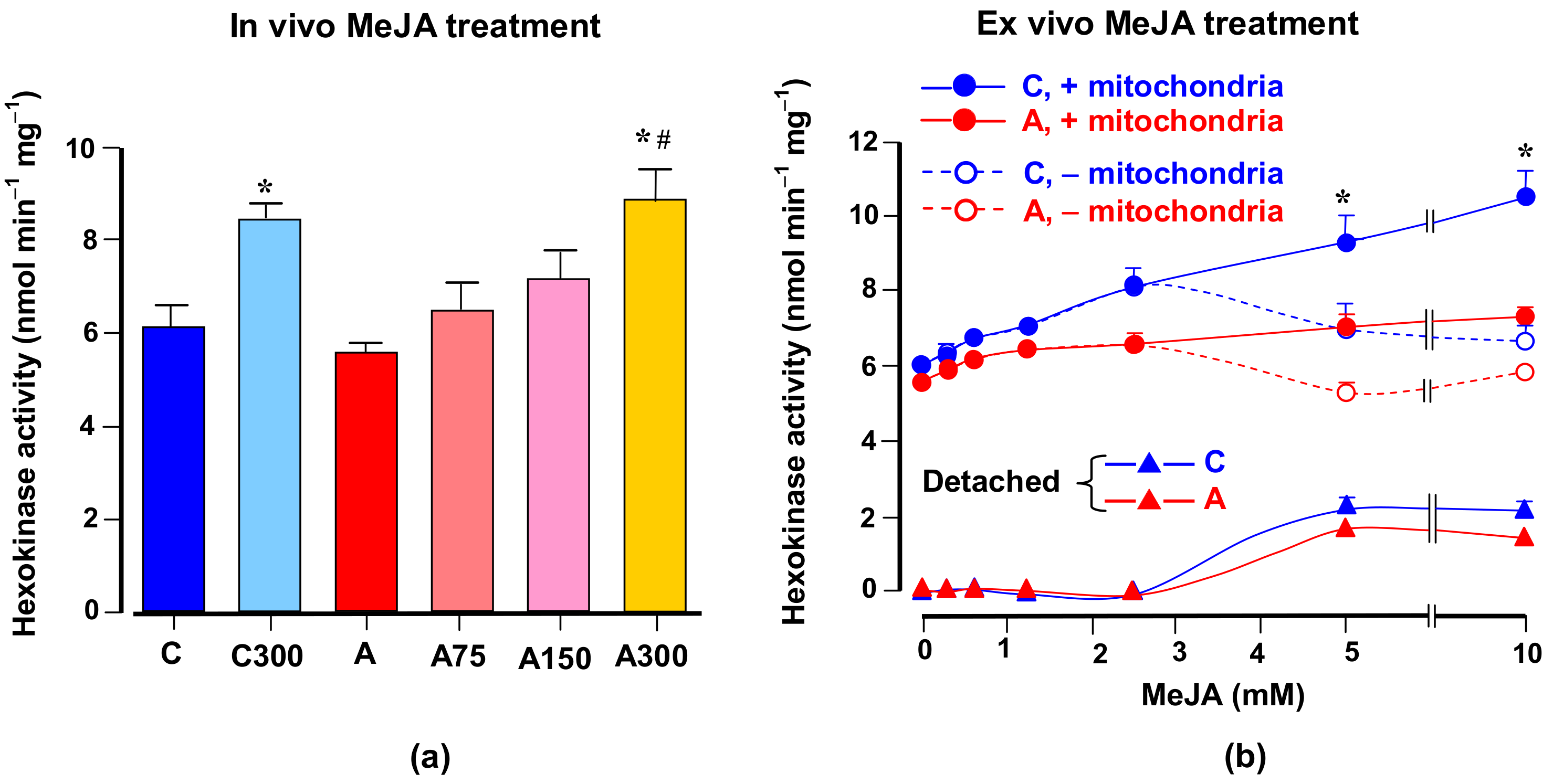
3.6. Hexokinase Activity
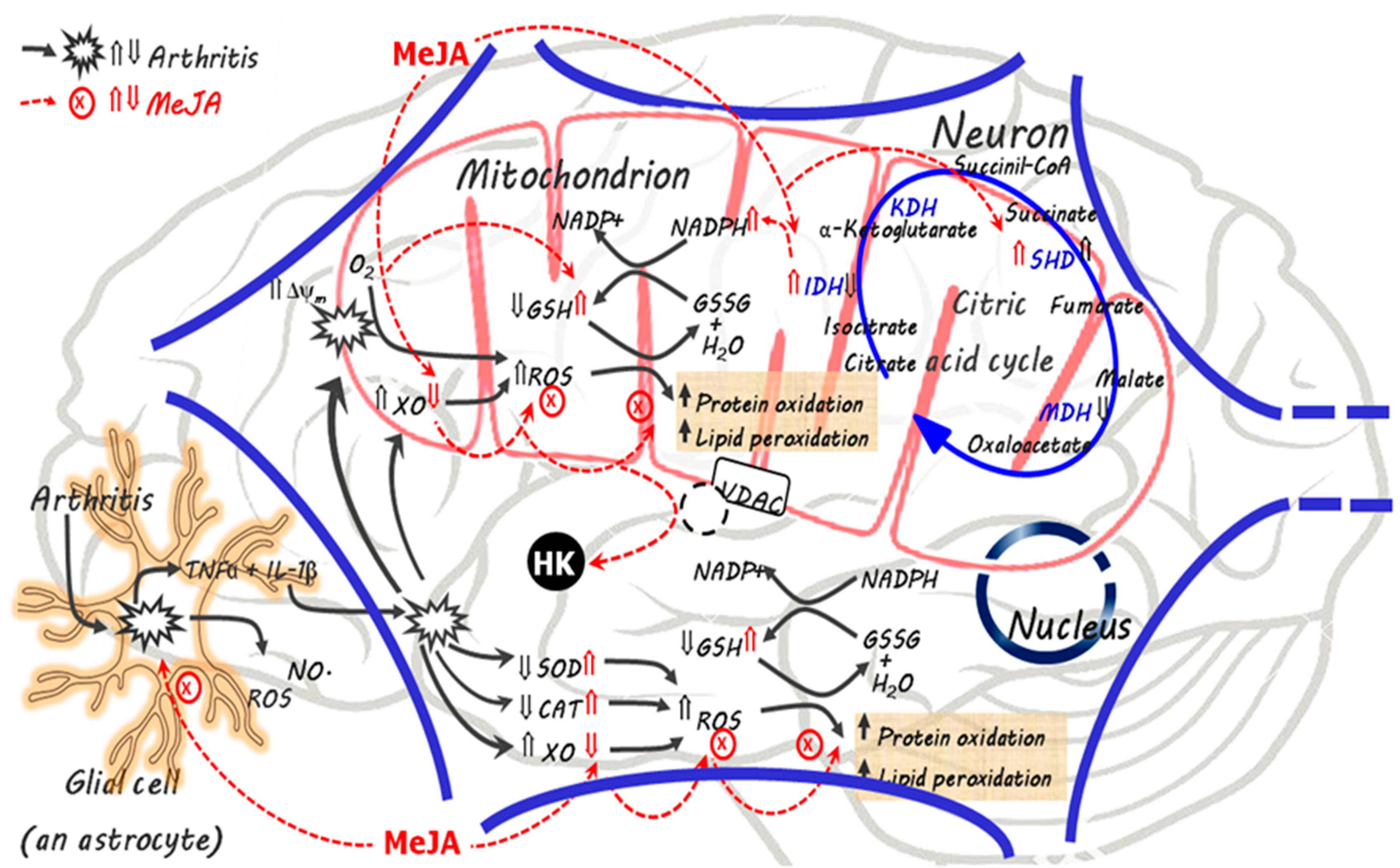
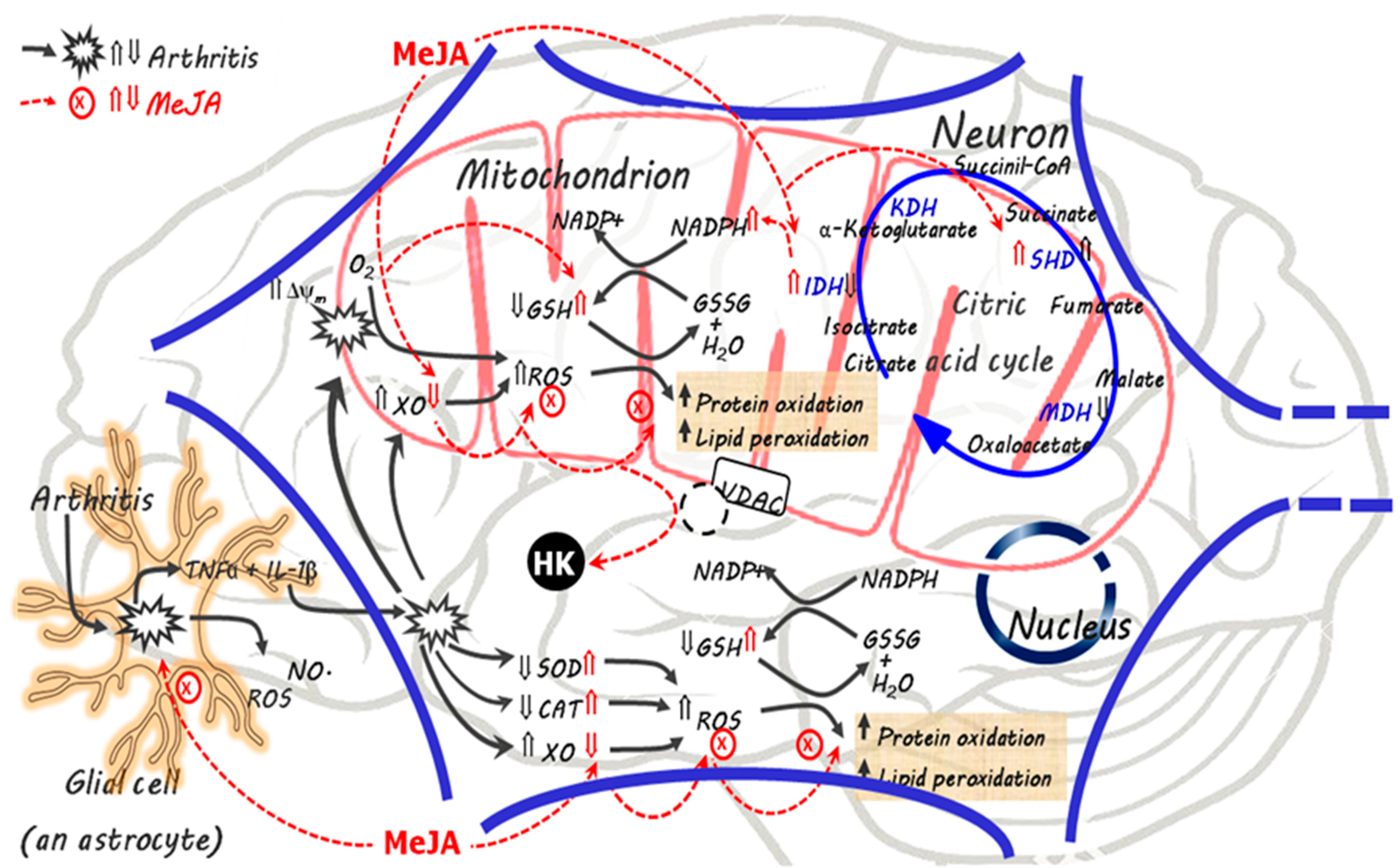
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raviv, Z.; Cohen, S.; Reischer-Pelech, D. The anti-cancer activities of jasmonates. Cancer Chemother. Pharmacol. 2013, 71, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Inoue, N.; Ito, Y.; Kubota, K.; Sugimoto, A.; Kakuda, T.; Fushiki, T. Sedative effects of the jasmine tea odor and (R)-(−)-linalool, one of its major odor components on autonomic nerve activity and mood states. Eur. J. Appl. Physiol. 2005, 95, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Cesari, I.M.; Carvalho, E.; Rodrigues, M.F.; Mendonça, B.S.; Amôedo, N.D.; Rumjanek, F.D. Methyl jasmonate: Putative mechanisms of action on cancer cells cycle, metabolism and apoptosis. Int. J. Cell Biol. 2014, 2014, ID572097. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, M.W.; Zhang, L.; Zhang, L. Methyl jasmonate and its potential in cancer therapy. Plant Signal Behav. 2015, 10, e106219. [Google Scholar] [CrossRef] [PubMed]
- Goldin, N.; Arzoine, L.; Heyfets, A.; Israelson, A.; Zaslavsky, Z.; Bravman, T.; Bronner, V.; Notcovich, A.; Shoshan-Barmatz, V.; Flescher, E. Methyl jasmonate binds to and detaches mitochondria bound hexokinase. Oncogene 2008, 27, 4636–4643. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.T.; Lee, H.J.; Yoo, E.S.; Hong, J.; Bao, B.; Choi, J.S.; Jung, J.H. New jasmonate analogues as potential anti-inflammatory agents. Bioorg. Med. Chem. 2008, 16, 10228–10235. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, S.S.; Park, K.J.; An, H.J.; Choi, Y.H.; Lee, N.H.; Hyun, C.G. Methyl jasmonate inhibits lipopolysaccharide-induced inflammatory cytokine production via mitogen-activated protein kinase and nuclear factor-κB pathways in RAW 264.7 cells. Die Pharm. 2016, 71, 540–543. [Google Scholar] [CrossRef]
- Lee, H.J.; Maeng, K.; Dang, H.T.; Kang, G.J.; Ryou, C.; Jung, J.H.; Kang, H.K.; Prchal, J.T.; Yoo, E.S.; Yoon, D. Anti-inflammatory effect of methyl dehydrojasmonate (J2) is mediated by the NF-kappaB pathway. J. Mol. Med. 2011, 89, 83–90. [Google Scholar] [CrossRef]
- Sá-Nakanishi, A.B.; Soni-Neto, J.; Moreira, L.S.; Gonçalves, G.A.; Silva-Comar, F.M.S.; Bracht, L.; Bersani-Amado, C.A.; Peralta, R.M.; Bracht, A.; Comar, J.F. Anti-inflammatory and antioxidant actions of methyl jasmonate are associated with metabolic modifications in the liver of arthritic rats. Oxid. Med. Cell. Longev. 2018, 2018, 2056250. [Google Scholar] [CrossRef]
- Uhlig, T.; Moe, R.H.; Kvien, T.K. The burden of disease in rheumatoid arthritis. Pharmacoeconomics 2014, 32, 841–851. [Google Scholar] [CrossRef]
- Kitas, G.D.; Gabriel, S.E. Cardiovascular disease in rheumatoid arthritis: State of the art and future perspectives. Ann. Rheum. Dis. 2011, 70, 8–14. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2209. [Google Scholar] [CrossRef] [PubMed]
- Misko, T.P.; Radabaugh, M.R.; Highkin, M.; Abrams, M.; Friese, O.; Gallavan, R.; Bramson, C.; Le Graverand, M.P.H.; Lohmander, L.S.; Roman, D. Characterization of nitrotyrosine as a biomarker for arthritis and joint injury. Osteoarthr. Cart. 2013, 21, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Stamp, L.K.; Khalilova, I.; Tarr, J.M.; Senthilmhan, R.; Turner, R.; Haigh, R.C.; Winyard, P.G.; Kettle, A.J. Myeloperoxidase and oxidative stress in rheumatoid arthritis. Rheumatology 2012, 51, 1796–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemarechal, H.; Allanore, Y.; Chenevier-Gobeaux, C.; Kahan, A.; Ekindjian, O.G.; Borderie, D. Serum protein oxidation in patients with rheumatoid arthritis and effects of infliximab therapy. Clin. Chim. Acta 2006, 372, 147–153. [Google Scholar] [CrossRef]
- Haruna, Y.; Morita, Y.; Yada, T.; Satoh, M.; Fox, D.A.; Kashihara, N. Fluvastatin reverses endothelial dysfunction and increased vascular oxidative stress in rat adjuvant-induced arthritis. Arthritis Rheum. 2007, 56, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Comar, J.F.; Sá-Nakanishi, A.B.; Oliveira, A.L.; Wendt, M.M.N.; Bersani-Amado, C.A.; Ishii-Iwamoto, E.L.; Peralta, R.M.; Bracht, A. Oxidative state of the liver of rats with adjuvant-induced arthritis. Free Rad. Biol. Med. 2013, 58, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Schubert, A.C.; Wendt, M.M.N.; Sá-Nakanishi, A.B.; Bersani-Amado, C.A.; Peralta, R.M.; Comar, J.F.; Bracht., A. Oxidative status and oxidative metabolism of the heart from rats with adjuvant-induced arthritis. Exp. Mol. Pathol. 2016, 100, 393–401. [Google Scholar] [CrossRef]
- Bracht, A.; Silveira, S.S.; Castro-Ghizoni, C.V.; Sá-Nakanishi, A.B.; Oliveira, M.R.N.; Bersani-Amado, C.A.; Peralta, R.M.; Comar, J.F. Oxidative changes in the blood and serum albumin differentiate rats with monoarthritis and polyarthritis. Springer Plus 2016, 5, 36–50. [Google Scholar] [CrossRef]
- Roubenoff, R. Rheumatoid cachexia: A complication of rheumatoid arthritis moves into the 21st century. Arthritis Res. Ther. 2009, 11, 108–109. [Google Scholar] [CrossRef]
- Wendt, M.N.M.; de Oliveira, M.C.; Castro, L.S.; Franco-Salla, G.B.; Parizotto, A.V.; Silva, F.M.S.; Natali, M.R.M.; Bersani-Amado, C.A.; Bracht, A.; Comar, J.F. Fatty acids uptake and oxidation are increased in the liver of rats with adjuvant-induced arthritis. Biochim. Biophys. Acta (BBA) Mol. Basis. Dis. 2019, 1865, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Ames-Sibin, A.P.; Barizão, C.L.; Castro-Ghizoni, C.V.; Silva, F.M.S.; Sá-Nakanishi, A.B.; Bracht, L.; Bersani-Amado, C.A.; Natali, M.R.M.; Bracht, A.; Comar, J.F. β-Caryophyllene, the major constituent of copaiba oil, reduces systemic inflammation and oxidative stress in arthritic rats. J. Cell. Biochem. 2018, 119, 10262–10277. [Google Scholar] [CrossRef] [PubMed]
- Castro-Ghizoni, C.V.; Ames, A.P.A.; Lameira, O.A.; Bersani-Amado, C.A.; Sá-Nakanishi, A.B.; Bracht, L.; Natali, M.R.M.; Peralta, R.M.; Bracht, A.; Comar, J.F. Anti-inflammatory and antioxidant actions of copaiba oil (Copaifera reticulata) are associated with histological modifications in the liver of arthritic rats. J. Cell. Biochem. 2017, 118, 3409–3423. [Google Scholar] [CrossRef] [PubMed]
- Wartolowska, K.; Hough, M.G.; Jenkinson, M.; Andersson, J.; Wordsworth, B.P.; Tracey, I. Structural changes of the brain in rheumatoid arthritis. Arthritis Rheum. 2012, 64, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Bekkelund, S.I.; Pierre-Jerome, C.; Husby, G.; Mellgren, S.I. Quantitative cerebral MR in rheumatoid arthritis. Am. J. Neuroradiol. 1995, 16, 767–772. [Google Scholar] [PubMed]
- Wendt, M.M.N.; Sá-Nakanishi, A.B.; Ghizoni, C.V.C.; Bersani-Amado, C.A.; Peralta, R.M.; Bracht., A.; Comar, J.F. Oxidative state and oxidative metabolism in the brain of rats with adjuvant-induced arthritis. Exp. Mol. Pathol. 2015, 98, 549–557. [Google Scholar] [CrossRef]
- Solomon, U.; Taghogho, E.A. Methyl jasmonate attenuates memory dysfunction and decreases brain levels of biomarkers of neuroinflammation induced by lipopolysaccharide in mice. Brain Res. Bull. 2017, 131, 133–141. [Google Scholar] [CrossRef]
- Eduviere, A.T.; Umukoro, S.; Aderibigbe, A.O.; Ajayi, A.M.; Adewole, F.A. Methyl jasmonate enhances memory performance through inhibition of oxidative stress and acetylcholinesterase activity in mice. Life Sci. 2015, 132, 20–26. [Google Scholar] [CrossRef]
- Umukoro, S.; Aluko, O.M.; Eduviere, A.T.; Owoeye, O. Evaluation of adaptogenic-like property of methyl jasmonate in mice exposed to unpredictable chronic mild stress. Brain Res. Bull. 2016, 121, 105–114. [Google Scholar] [CrossRef]
- Fedatto, Z., Jr.; Ishii-Iwamoto, E.L.; Bersani-Amado, C.; Maciel, E.R.M.; Bracht, A.; Kelmer-Bracht, A.M. Glucose phosphorylation capacity and glycolysis in the liver of arthrityic rats. Inflamm. J. 2000, 49, 128–132. [Google Scholar]
- Pearson, C.M.; Wood, F.D. Studies of arthritis and other lesions induced in rats by the injection of mycobacterial adjuvant. Am. J. Pathol. Phila. 1963, 42, 93–95. [Google Scholar]
- Umukoro, S.; Olugbemide, A.S. Antinociceptive effects of methyl jasmonate in experimental animals. J. Nat. Med. 2011, 65, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Katyare, S.S.; Rajan., R.R. Influence of thyroid hormone treatment on the respiratory activity of cerebral mitochondria from hypothyroid rats. A critical re-assessment. Exp. Neurol. 2005, 195, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Garland, D.; Oliver, C.N.; Amici, A.; Climent, I.; Lenz, A.G.; Ahn, B.W.; Shaltiel, S.; Stadtman, E.R. Determination of carbonyl content in oxidatively modified proteins. Meth. Enzymol. 1990, 186, 464–478. [Google Scholar] [CrossRef]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Meth. Enzymol. 1978, 52, 302–310. [Google Scholar] [CrossRef]
- Siqueira, I.R.; Fochesatto, C.; Torres, I.L.S.; Dalmaz, C.; Netto, C.A. Aging affects oxidative state in hippocampus, hypothalamus and adrenal glands of Wistar rats. Life Sci. 2005, 78, 271–278. [Google Scholar] [CrossRef]
- Biazon, A.C.B.; Wendt, M.M.N.; Moreira, J.R.; Castro-Ghizoni, C.V.; Soares, A.A.; Silveira, S.S.; Sá-Nakanishi, A.B.; Bersani-Amado, C.A.; Peralta, R.M.; Bracht, A.; et al. The in vitro antioxidant capacities of hydroalcoholic extracts from roots and leaves of Smallanthus sonchifolius (yacon) do not correlate with their in vivo antioxidant action in diabetic rats. J. Biosci. Med. 2016, 4, 15–27. [Google Scholar] [CrossRef]
- Bryan, N.S.; Grisham, M.B. Methods to detect nitric oxide and its metabolites in biological samples. Free Rad. Biol. Med. 2007, 43, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Bergmeyer, H.U. Methods of Enzymatic Analysis; Verlag Chemie/Academic Press: London, UK; Weinheim, Germany, 1974; ISBN 352725370X. [Google Scholar]
- Marklund, S.; Marklund, G. Involvement of the superoxide anion radical in the oxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef]
- Bradley, P.P.; Christensen, R.D.; Rothstein, G. Cellular and extracellular myeloperoxidase in pyogenic inflammation. Blood 1982, 60, 618–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galilea, J.; Canela, E.I.; Bozal, J. The course analysis of guanine and hypoxanthine transformation to uric acid by bovine liver guanine aminohydrolase and xanthine oxidase. J. Mol. Catal. 1981, 12, 27–36. [Google Scholar] [CrossRef]
- Maciel, E.N.; Vercesi, A.E.; Castilho, R.F. Oxidative stress in (Ca2+)-induced membrane permeability transition in brain mitochondria. J. Neurochem. 2001, 79, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Sá-Nakanishi, A.B.; Soares, A.A.; de Oliveira, A.L.; Comar, J.F.; Peralta, R.M.; Bracht, A. Effects of treating old rats with an aqueous Agaricus blazei extract on oxidative and functional parameters of the brain tissue and brain mitochondria. Oxid. Med. Cell. Longev. 2014, 2014, 563179. [Google Scholar] [CrossRef] [PubMed]
- Saling, S.C.; Comar, J.F.; Mito, M.S.; Peralta, R.M.; Bracht, A. Actions of juglone on energy metabolism in the rat liver. Toxicol. Appl. Pharmacol. 2011, 257, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Acharya, M.M.; Katyare, S.S. Structural and functional alterations in mitochondrial membrane in picrotoxin-induced epileptic rat brain. Exp. Neurol. 2005, 192, 79–88. [Google Scholar] [CrossRef]
- Maeng, O.; Kim, Y.C.; Shin, H.J.; Li, J.O.; Huh, T.L.; Kang, K.I.; Kim, Y.S.; Paik, S.G.; Lee, H. Cytosolic NADP+-dependent isocitrate dehydrogenase protects macrophages from LPS-induced nitric oxide and reactive oxygen species. Biochem. Biophys. Res. Comm. 2004, 317, 558–564. [Google Scholar] [CrossRef]
- Hussain, M.M.; Zannis, V.I.; Plaitakis, A. Characterization of glutamate dehydrogenase isoproteins purified from the cerebellum of normal subjects and patients with degenerative neurological disorders, and from human neoplastic cell lines. J. Biol. Chem. 1989, 264, 20730–20735. [Google Scholar]
- Vilela, V.R.; Oliveira., A.L.; Comar, J.F.; Peralta, R.M.; Bracht., A. Effects of tadalafil on the cAMP stimulated glucose output in the rat liver. Chem. Biol. Interact. 2014, 220, 1–11. [Google Scholar] [CrossRef]
- Nicholls, D.G. Mitochondrial membrane potential and aging. Aging Cell 2004, 3, 35–40. [Google Scholar] [CrossRef]
- Hossain, S.J.; Aoshima, H.; Koda, H.; Kiso, Y. Fragrances in oolong tea that enhance the response of GABAA receptors. Biosci. Biotechnol. Biochem. 2004, 68, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, G.A.; Sá-Nakanishi, A.B.; Wendt, M.M.N.; Comar, J.F.; Bersani-Amado, C.A.; Bracht, A.; Peralta, R.M. Green tea extract improves the oxidative state of the liver and brain in rats with adjuvant-induced arthritis. Food Funct. 2015, 6, 2701–2711. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Alsahli, M.; Rahmani, A. Myeloperoxidase as an active disease biomarker: Recent biochemical and pathological perspectives. Med. Sci. 2018, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Ohigashi, H. Targeting NOX, INOS and COX-2 in inflammatory cells: Chemoprevention using food phytochemicals. Int. J. Cancer 2007, 121, 2357–2363. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Shintani, F.; Yagi, G.; Asai, M.; Nozfawa, S. Combination of inflammatory cytokines increases nitrite and nitrate levels in the paraventricular nucleus of conscious rats. Brain Res. 2001, 905, 12–20. [Google Scholar] [CrossRef]
- Alabi, A.O.; Ajayi, A.M.; Ben-Azu, B.; Bakre, A.G.; Umukoro, S. Methyl jasmonate abrogates rotenone- induced parkinsonian-like symptoms through inhibition of oxidative stress, release of pro-inflammatory cytokines, and down-regulation of immnopositive cells of NF-κB and α-synuclein expressions in mice. Neurotoxicology 2019, 74, 172–183. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2005, 417, 1–13. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: London, UK, 2007. [Google Scholar]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Rad. Biol. Med. 2002, 33, 774. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Rad. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Zhang, Y. Methyl jasmonate induces the apoptosis of human colorectal cancer cells via downregulation of EZH2 expression by microRNA-101. Mol. Med. Rep. 2016, 15, 957–962. [Google Scholar] [CrossRef]
- Wang, Y.; Xiang, W.; Wang, M.; Huang, T.; Xiau, X.; Wang, L.; Tao, D.; Dong, L.; Zheng, F.; Jiang, G. Methyl jasmonate sensitizes human bladder cancer cells to gambogic acid-induced apoptosis through down-regulation of EZH2 expression by miR-101. Br. J. Pharmacol. 2014, 171, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Xu, M.; Xo, R.; Mates, A.; Wilson, G.; Pearsall, A.T.; Grishko, V. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthr. Cart. 2010, 18, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Armada, M.; Caramés, B.; Martín, M.; Cillero-Pastor, B.; Lires-Dean, M.; Fuentes-Boquete, I.; Arenas, J.; Blanco, F. Mitochondrial activity is modulated by TNFalpha and IL-1beta in normal human chondrocyte cells. Osteoarthr. Cart. 2006, 14, 1011–1022. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of α-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 2000, 20, 8972–8979. [Google Scholar] [CrossRef] [PubMed]
- Levrat, C.; Larrick, J.W.; Wright, S.C. Tumor necrosis factor induces activation of mitochondrial succinate dehydrogenase. Life Sci. 1991, 49, 1731–1737. [Google Scholar] [CrossRef]
- Li, J.; Chen, K.; Wang, F.; Dai, W.; Li, S.; Feng, J.; Wu, L.; Liu, T.; Xu, S.; Xia, Y.; et al. Methyl jasmonate leads to necrosis and apoptosis in hepatocelular carcinoma via inhibition of glycolysis and represses tumor growth in mice. Oncotarget 2017, 8, 45965–45980. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, H.; Lu, W.; Huang, P. Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1787, 553–570. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira-Maróstica, H.V.; Castro, L.S.; Gonçalves, G.A.; Silva, F.M.S.; Bracht, L.; Bersani-Amado, C.A.; Peralta, R.M.; Comar, J.F.; Bracht, A.; Sá-Nakanishi, A.B. Methyl Jasmonate Reduces Inflammation and Oxidative Stress in the Brain of Arthritic Rats. Antioxidants 2019, 8, 485. https://doi.org/10.3390/antiox8100485
Pereira-Maróstica HV, Castro LS, Gonçalves GA, Silva FMS, Bracht L, Bersani-Amado CA, Peralta RM, Comar JF, Bracht A, Sá-Nakanishi AB. Methyl Jasmonate Reduces Inflammation and Oxidative Stress in the Brain of Arthritic Rats. Antioxidants. 2019; 8(10):485. https://doi.org/10.3390/antiox8100485
Chicago/Turabian StylePereira-Maróstica, Heloisa V., Lorena S. Castro, Geferson A. Gonçalves, Francielli M.S. Silva, Lívia Bracht, Ciomar A. Bersani-Amado, Rosane M. Peralta, Jurandir F. Comar, Adelar Bracht, and Anacharis B. Sá-Nakanishi. 2019. "Methyl Jasmonate Reduces Inflammation and Oxidative Stress in the Brain of Arthritic Rats" Antioxidants 8, no. 10: 485. https://doi.org/10.3390/antiox8100485