
Stability and Catalase-Like Activity of a Mononuclear Non-Heme Oxoiron(IV) Complex in Aqueous Solution

Department of Chemistry, University of Pannonia, 8201 Veszprém, Hungary
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(18), 3236; https://doi.org/10.3390/molecules24183236
Submission received: 27 August 2019
/
Revised: 4 September 2019
/
Accepted: 5 September 2019
/
Published: 5 September 2019
(This article belongs to the Special Issue Biomimetic Radical Chemistry and Applications)
Abstract
:Heme-type catalase is a class of oxidoreductase enzymes responsible for the biological defense against oxidative damage of cellular components caused by hydrogen peroxide, where metal-oxo species are proposed as reactive intermediates. To get more insight into the mechanism of this curious reaction a non-heme structural and functional model was carried out by the use of a mononuclear complex [FeII(N4Py*)(CH3CN)](CF3SO3)2 (N4Py* = N,N-bis(2-pyridylmethyl)- 1,2-di(2-pyridyl)ethylamine) as a catalyst, where the possible reactive intermediates, high-valent FeIV=O and FeIII–OOH are known and spectroscopically well characterized. The kinetics of the dismutation of H2O2 into O2 and H2O was investigated in buffered water, where the reactivity of the catalyst was markedly influenced by the pH, and it revealed Michaelis–Menten behavior with KM = 1.39 M, kcat = 33 s−1 and k2(kcat/KM) = 23.9 M−1s−1 at pH 9.5. A mononuclear [(N4Py)FeIV=O]2+ as a possible intermediate was also prepared, and the pH dependence of its stability and reactivity in aqueous solution against H2O2 was also investigated. Based on detailed kinetic, and mechanistic studies (pH dependence, solvent isotope effect (SIE) of 6.2 and the saturation kinetics for the initial rates versus the H2O2 concentration with KM = 18 mM) lead to the conclusion that the rate-determining step in these reactions above involves hydrogen-atom transfer between the iron-bound substrate and the Fe(IV)-oxo species.
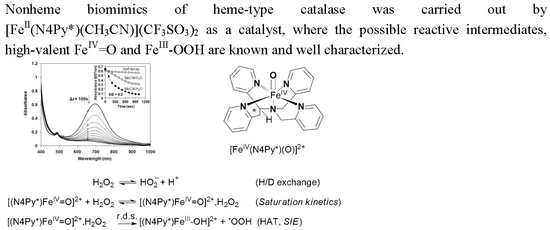
1. Introduction
Superoxide dismutases (SODs), catalase-peroxidases (KatGs) and catalases are specialized oxidoreductase enzymes for the degradation of reactive oxygen species (ROS), e.g., hydrogen peroxide, hydroxyl and superoxide radicals to avoid their accumulation and prevent the oxidative damage of cellular components, that may lead to a number of diseases such as cancer, Alzheimer’s diseases and aging [1,2,3,4]. For example, the hydroxyl and/or hydroperoxyl radicals may cause lipid peroxidation, membrane damage, DNA oxidation and cell death [5,6]. As a fine coupling of SODs and catalases, the former enzymes catalyze the dismutation of superoxide into dioxygen (1-electon oxidation) and H2O2, whilst the latter enzymes eliminate the H2O2 via its decomposition by disproportionation into O2 (2-electron oxidation) and H2O, resulting in the optimal intracellular concentration of a H2O2 molecule [7,8,9], which acts as a second messenger in signal-transduction pathways. Otherwise, it is worth to note, that the therapeutic potential of H2O2 makes this molecule also a valuable target in cancer killing via chemo- and radiotherapy, and in stroke therapy [10,11,12].
Two main classes of catalase enzymes are known, an iron and manganese-containing proteins. Although both types of catalases exhibit high catalytic activities, there are significant differences, including the active sites and the catalytic mechanisms [13]. Monofunctional catalases (EC 1.11.1.6) are heme-containing enzymes, that catalyze the dismutation of hydrogen peroxide (2H2O2 = 2H2O + O2), where the catalytic mechanism is well-characterized with a high-valent oxoiron(IV) porphyrin π-cation radical, compound I, [(P•+)FeIV=O]+ (P = porphyrinate dianion), being responsible for hydrogen peroxide oxidation [14,15,16]. Manganese catalases such as Lactobacillus plantarum [17,18], Thermus thermophilus [19,20], Thermoleophilium album [21] and Pyrobaculum calidifontis VA1 [22] are found in several bacterial organisms, and possess a binuclear manganese center with a cycle between Mn(II)-Mn(II) and Mn(III)-Mn(III) states during turnover.
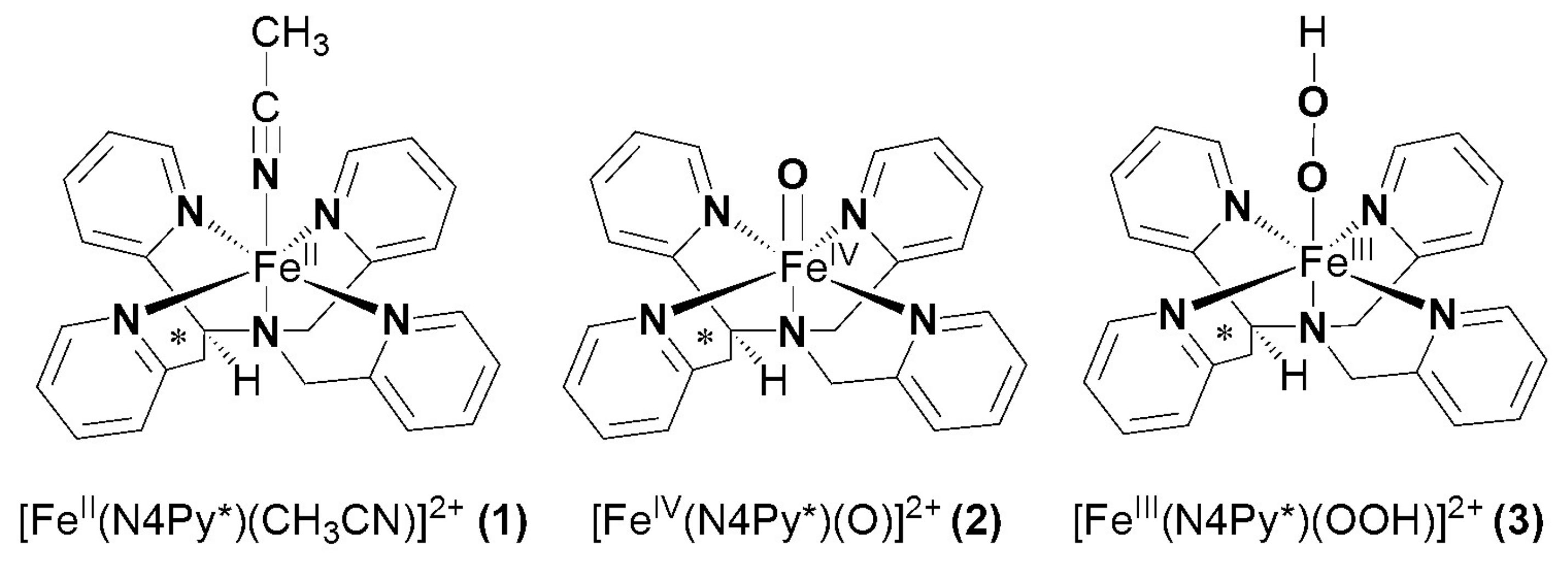
Synthetic compounds as biomimics of catalase enzymes may have potential biomedical application as therapeutic agents against oxidative stress. Besides the heme-type models, a great number of manganese, copper, ruthenium and non-heme iron complexes have been designed and studied as catalase models [23,24,25,26,27,28,29,30,31,32,33,34,35]. However, comparative studies between heme and non-heme models are scarce. The non-heme models are mainly binuclear complexes [27,28,29], only a small number of mononuclear iron compounds have been studied [12,36,37]. The direct dismutation of H2O2 with terminal and bridging oxo ligands has been described for only a few complexes of Fe, Cr, Mn, V and Ru [38,39,40,41,42]. Mononuclear oxoiron(IV) complexes are of interest from a bioinorganic viewpoint, since similar intermediates are frequently invoked as the active species in the active site of numerous proteins and in biomimetic iron-containing catalytic systems. Most of these results were obtained in organic solvent due to the lack of solubility or activity in aqueous solution. Due to the increasing importance of catalase activity, we have focused on the development of such a non-heme iron-containing system that shows catalase-like activity in aqueous solution. To get more insight into the mechanism of H2O2 dismutation the mononuclear complex [FeII(N4Py*)(CH3CN)](CF3SO3)2 (1) (N4Py* = N,N-bis(2-pyridylmethyl)-1,2-di(2-pyridyl)ethylamine) was chosen as a catalyst, where the possible reactive intermediates high-valent FeIV=O (2) and FeIII-OOH (3) are known and spectroscopically well characterized (Scheme 1) [43,44,45,46].
2. Results and Discussion
2.1. Catalase-Like Reactivity of [FeII(N4Py*)(CH3CN)](CF3SO3)2 in Aqueous Solution
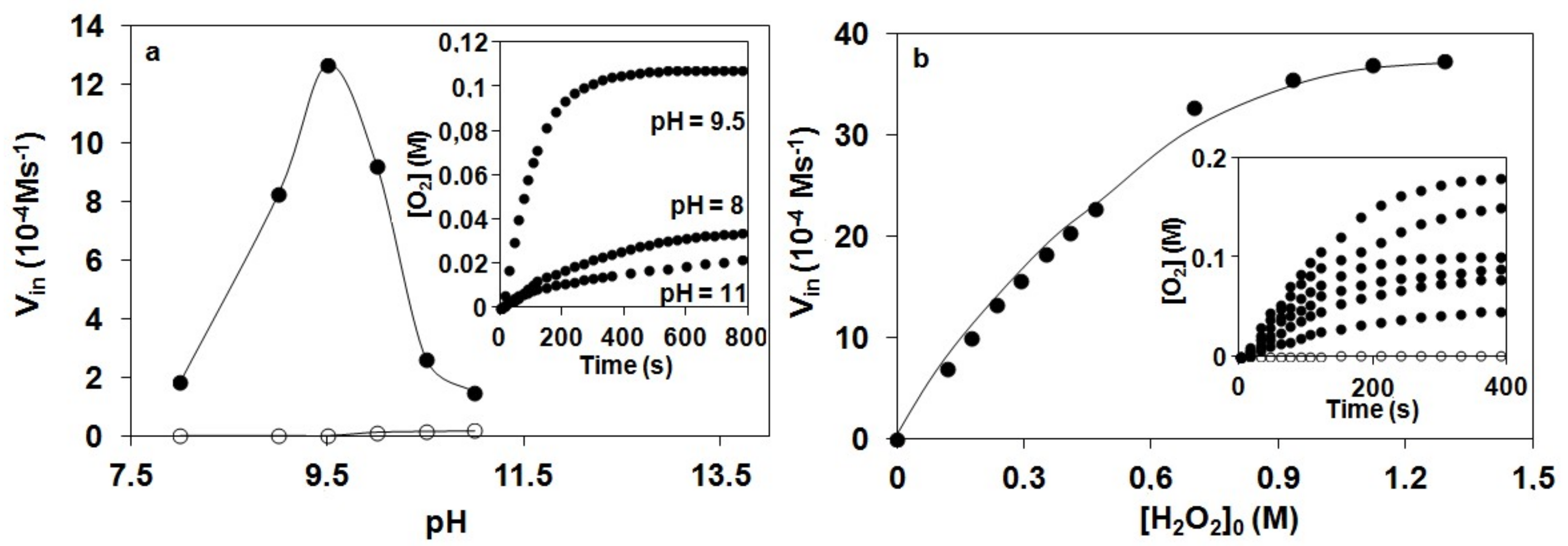
The catalase-like activity of the complex [FeII(N4Py*)(CH3CN)](CF3SO3)2 to disproportionate H2O2 into H2O and O2 was investigated in aqueous solution at 20 °C by gasvolumetric measurements of evolved dioxygen. To gain further information on the mechanism of catalase activity of our iron complex, we first examined pH-dependence of catalase activity. It was reported that the coordination and dissociation of peroxides on metal-porphyrins are pH dependent reactions [47,48]. Moreover, they reported that the coordination is accelerated at a higher pH region and that the subsequent O–O bond cleavage leading to the formation of high-valent oxo-Fe(IV) or oxo-Fe(V) species is pH-independent (only at higher pH region, where the protonation of the distal oxygen in the peroxo-complex can be excluded) irreversible reaction. These results suggest that the coordination of peroxides is a crucial step for the formation of high-valent Fe species, and the mechanism of catalase activity involves the coordination of H2O2, which is considered to be pH-dependent as well. Therefore, we hypothesized that formation of reactive intermediate 2 is accelerated at pH 9.5 and catalase activity is increased as compared at pH 8. As shown in Figure 1, O2 production of 1 in 50 mM borate buffer (pH 9.5) was significantly higher than that in phosphate buffer (pH 8). Vin value under this condition was determined to be Vin = 1.13 × 10−3 Ms−1, which is approximately seven times higher than that at pH 8, and 8.5 times higher than that at pH 11. This indicates that the rate-determining step was faster at pH 9.5 than at pH 8, which may be explained by the higher concentration of the more nucleophilic HO2–.
The pH dependence of H2O2 dismutation was further studied between pH 7 and pH 11. It was found that the initial rate of the disproportionation of H2O2 increases with increasing pH and goes through a maximum. The pH profile of 1 exhibits a sharp optimum at pH ~9.5, whereas catalases in general exhibit a broad pH optimum extending from pH 5.6 to 8.5 [48]. In control experiments, in the absence of the complex, the pH of the solution did not change in the presence of H2O2, and no significant O2 volume was evolved. We believe that the activity is influenced by the protonation state of H2O2. Assuming that hydrogen peroxide is activated by a direct interaction with the FeIV=O group of the complex, decomposition is expected to be favored by a high pH because of the larger concentration of the hydroperoxide anion (HOO− is more nucleophilic than H2O2). On the other hand, at higher pH values, the complex may be destroyed by the formation of the mineral forms of iron or catalytically inactive, insoluble μ-oxo-diiron(III) species.
Detailed kinetic studies on the disproportionation of H2O2 were performed in aqueous solution (pH 9.5; 0.025 M Na2B4O7.10H2O/0.1 M HCl; I = 0.15 M KNO3) at 20 °C by volumetric measurements of evolved dioxygen. To determine the dependence of the rates on the substrate concentration, solutions of the complex [FeII(N4Py*)(CH3CN)](CF3SO3)2 were treated with increasing amounts of H2O2 (1:400–5300). Plots of the amount of dioxygen evolved versus time at [1]0 constant, are shown in Figure 1a. The initial rates values were calculated from the maximum slope of the O2 versus time curves. Under this experimental condition, saturation kinetics was found for the initial rates (Vin = –d[H2O2]/dt) versus the H2O2 concentration (Figure 1b). An analysis of the data based on the Michaelis–Menten model (Vin = kcat[cat][S]0/(KM + [S]0)), originally developed for enzyme kinetics, was applied. A nonlinear least square fit was applied to calculate the Michaelis–Menten parameters, where kcat is the turnover number, KM is the Michaelis constant, S is the substrate initial concentration and [cat] is the catalyst concentration. The results were KM = 1.39 M, kcat = 33 s−1 and k2(kcat/KM) = 23.9 M−1s−1. The data presented illustrate that the catalyst had a relatively high turnover number (kcat) but appeared to bind peroxide very badly. The KM value was greater than the values for the natural enzymes from Thermus thermophilus (KM = 0.083 M) [19,20], Tricholoma album (KM = 0.015 M) [21] and Lactobacillus plantarum (KM = 0.35 M) [17,18] indicating a lower affinity to the substrate. The kcat value equaled 33 s−1, however, was 3–4 times magnitudes lower when compared to the natural enzymes Thermus thermophilus (kcat = 2.6 × 105 s−1), Tricholoma album (kcat = 2.0 × 105 s−1), Lactobacillus plantarum (kcat = 2.6 × 104 s−1) and the heme-containing catalases (kcat = 4 × 107 s−1). Despite this iron complex presents lower values of catalytic efficiency than other models (Table 1) [49,50,51,52], it must be emphasized that this value was obtained in water and in pH close to the natural, representing an advantage of the title complex with respect to most of the published models, whose studies have been conducted in organic solvent due to the lack of solubility or activity in aqueous solution.
2.2. Catalase-Like Reactivity Mediated by [(N4Py*)FeIV=O](ClO4)2 in Aqueous Solution
Rohde and co-workers have shown that the independently prepared [(N4Py)FeIV=O]2+ reacts rapidly with near-stoichiometric H2O2 resulting in dioxygen and [FeII(N4Py)(CH3CN)]2+ in acetonitrile [54]. Later Browne and co-workers have found clear evidence for the reaction of FeIII-OOH with H2O2 in methanol [55]. In their case the oxoiron(IV) intermediate can also be formed by homolytic cleavage of the O–O bond of an FeIII–OOH, but the rate of its formation is much lower than the FeIII–OOH-mediated H2O2 disproportionation observed with high excess H2O2 under catalytic conditions. As a continuity of these studies, we attempted to directly investigate the reactivity of the possible intermediates (FeIV=O, FeIII–OOH) during the catalase reaction in aqueous solution.
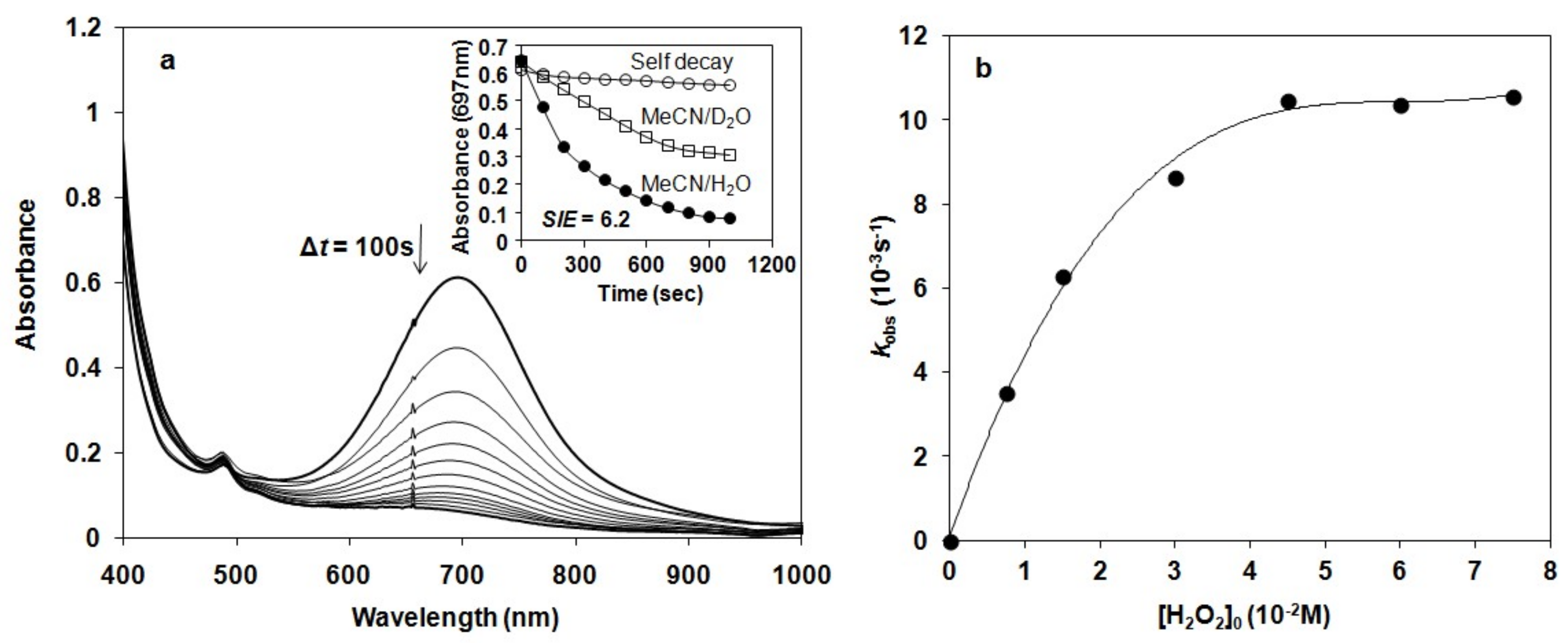
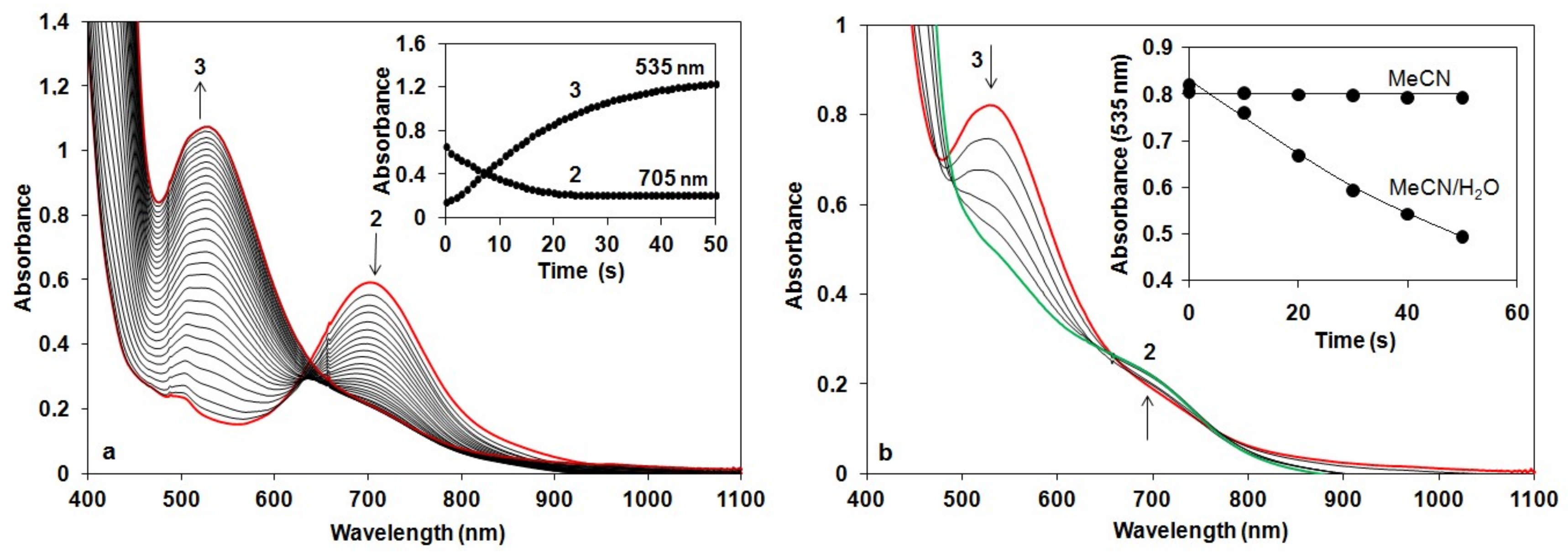
We have shown earlier that complex 1 forms very stable high valent oxoiron(IV) species (2) with PhIO in CH3CN (t1/2 = 233 h at R.T., λmax = 705 nm, ε = 400 M−1cm−1) [43]. As a test of our oxoiron(IV) species we firstly investigated its reaction with excess H2O2 (75 equiv.) in acetonitrile at 10 °C, which resulted in the formation of a relatively stable transient purple species with a characteristic absorbance maximum at λmax 535 nm (ε = 1100 M−1 cm−1; Figure 2a). It had a half-life of about 3 min even at 25 °C, but its decay can be remarkably enhanced by the addition of H2O into the FeIII–OOH-containing solution (CH3CN/H2O = 1:1) with a kobs value of about 12.3 × 10−3 s−1 at 10 °C, resulting in the formation of 2 (Figure 2b). It is worth to note that at higher pH the decay was so fast, that we were not able to follow it. These results might suggest that a high-valent oxoiron(IV) species was one of the possible intermediates that may be responsible for the dismutation of H2O2 in aqueous solution.
In the iron-catalyzed oxidation of H2O2 with terminal oxidants four processes can be proposed as the rate-controlling step, namely the formation of FeIII–OOH or high-valent oxoiron(IV), or their reaction with the substrate (H2O2). To avoid this difficulty, and to get more insight into the mechanism of the H2O2 oxidation process we synthesized the oxoiron(IV) complex 2 by an in situ reaction of 1 with PhIO in acetonitrile, and investigated its stability and reactivity with H2O2 in a buffered H2O–CH3CN mixture (v/v = 1:1). In this way the role of the oxoiron(IV) species could be directly investigated. The UV-vis spectra of 2 in buffered solutions were almost identical to that observed in the acetonitrile. The observed blue shift on the λmax values (from 705 to 697 nm) might be explained by the interaction (H-bridge) of the oxoiron(IV) with the H2O molecule(s).
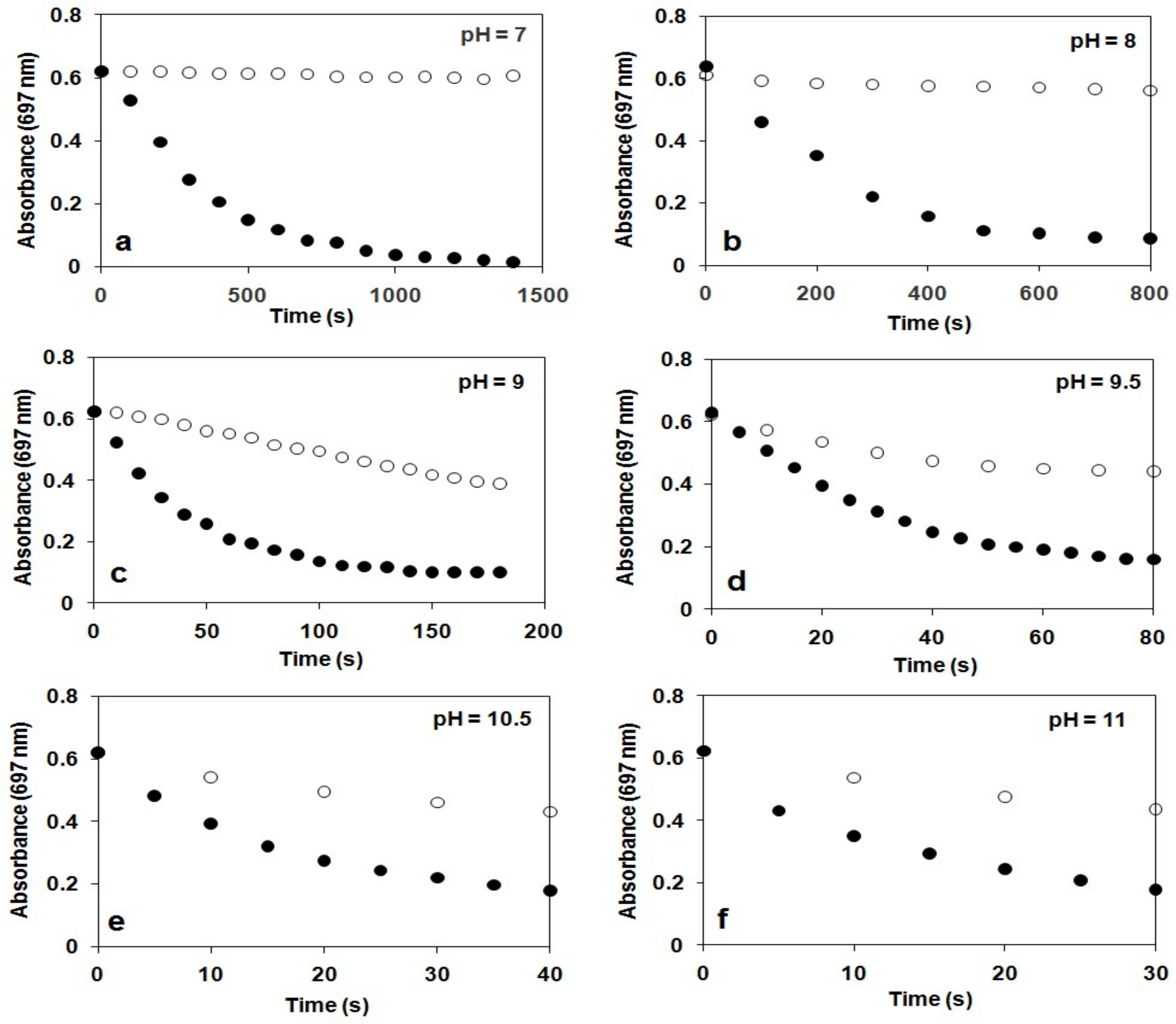
The stability of 2 was found to depend significantly on the pH value of reaction solutions, in which 2 was stable at pH 7–8 (ksd = 0.43 × 10−3 s−1, 0.64 × 10−3 s−1 with t1/2 = 180 and 150 min at pH 7 and 8 at 10 °C, respectively), but decayed at a fast rate with increasing pH at pH 9–11 (ksd = 3.51 × 10−3 s−1, and 7.27 × 10−3 s−1, 23 × 10−3 s−1, 39 × 10−3 s−1 and 46 × 10−3 s−1 with t1/2 = 4, 3, 2, 1.7 and 1 min at pH 9, 9.5, 10, 10.5 and 11 at 10 °C, respectively; Figure 3). This is the second example that the stability of oxoiron(IV) complex is controlled by the pH of reaction solutions [56].
The pH dependence of the reactivity of 2 against H2O2 was also examined in the range pH 7–11 in a buffered H2O–MeCN mixture (v/v = 1:1) at 10 °C (Figure 3). Upon addition of 10 equiv. H2O2 to the solution of 2, the characteristic absorption band of 2 (λmax = 697 nm) disappeared rapidly, and no formation of FeIII–OOH was observed. Pseudo-first-order fitting of the kinetic data allowed us to calculate kobs values to be 2.96 × 10−3 s−1, 6.29 × 10−3 s−1, 37.9 × 10−3 s−1, 41.6 × 10−3 s−1, 60.3 × 10−3 s−1, 75.3 × 10−3 s−1 and 84 × 10−3 s−1 at pH 7, 8, 9, 9.5, 10, 10.5 and 11 at 10 °C, respectively.
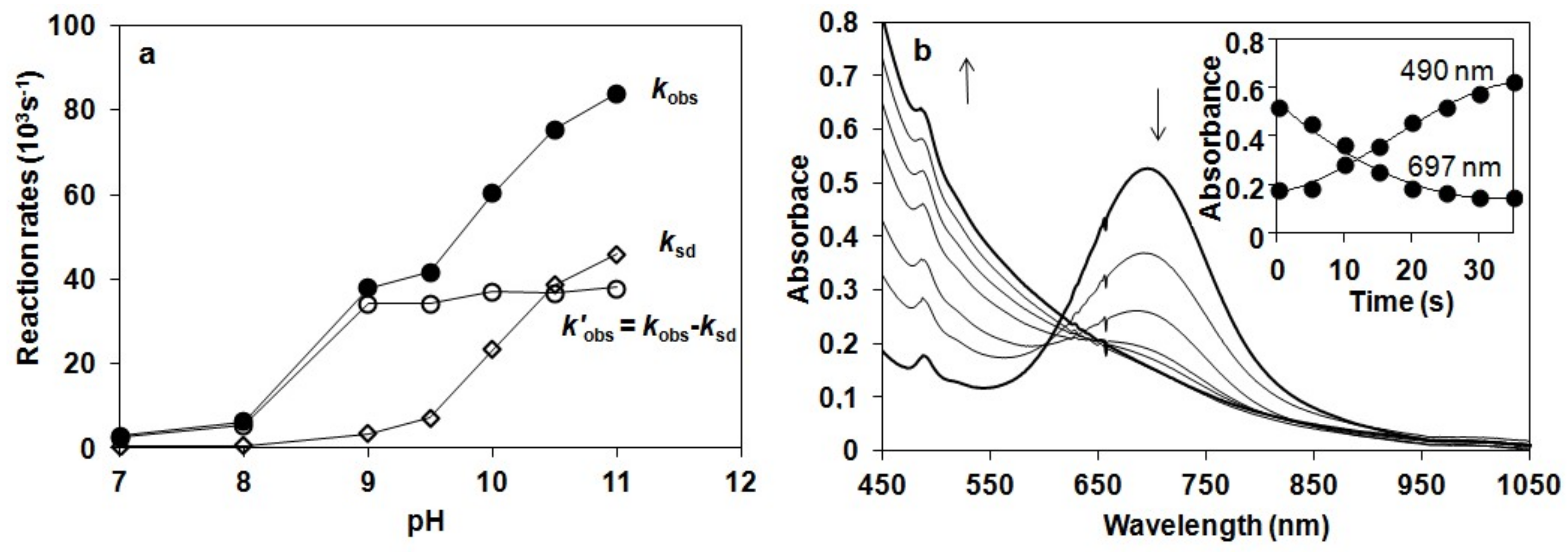
The reactivity of 2 was found to depend significantly on the pH value of reaction solutions. The maximum rate of H2O2 dismutation, k’obs (k’obs = kobs − ksd from the −d[2]/dt = kobs[2] = (ksd + k’obs)[2]) could be observed at pH 9, where the self decay process (ksd) could be neglected (Figure 4a). The increase of the kobs at higher pH could be explained by the self decay of 2. Addition of 10 equiv. H2O2 at pH 10 resulted in a decrease in absorbance at λmax = 697 nm concomitant with an increase at 490 nm within 40 s at 10 °C, and an isosbestic point obtained at approximately λmax = 620 nm. This spectrum including a weak absorption band at 700 nm with a shoulder around 490 nm corresponded to the spectrum of [(N4Py*)FeIII-O-FeIII(N4Py*)]4+ (Figure 4b).
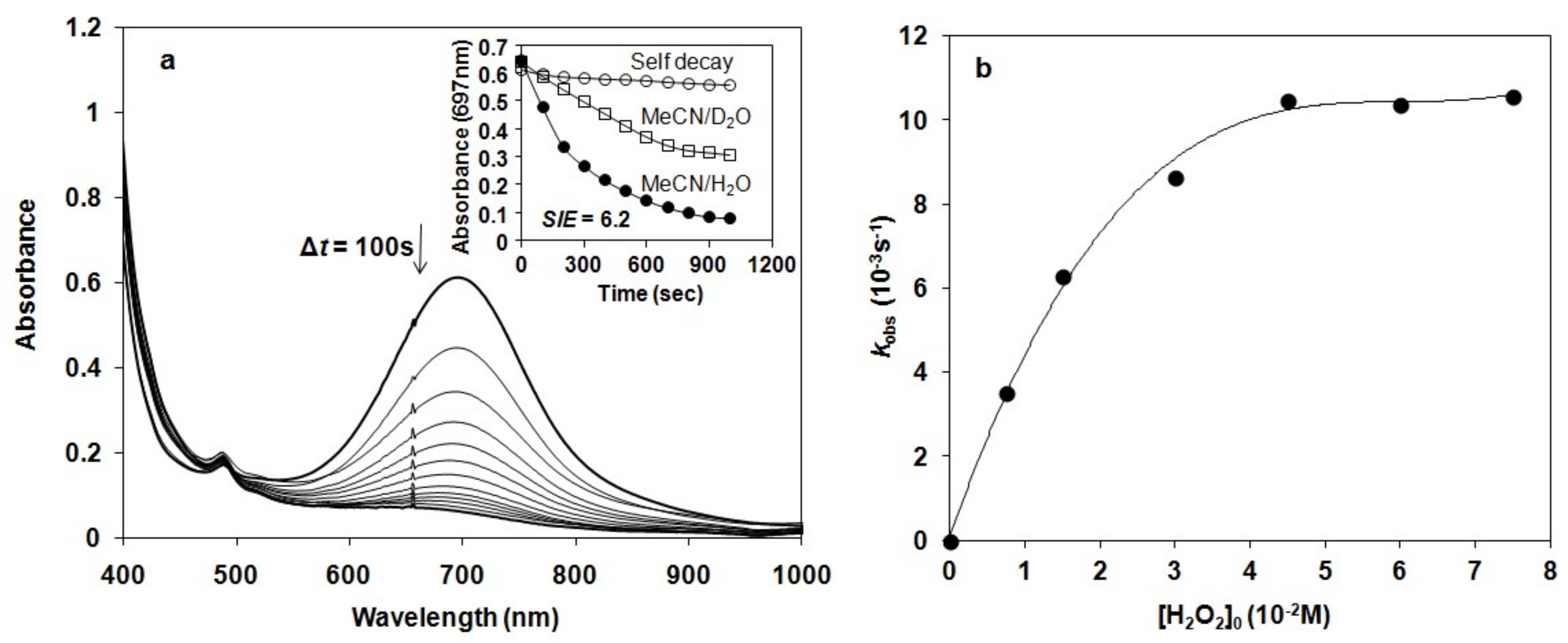
Detailed kinetic and mechanistic studies were carried out in buffered water/acetonitril mixture (v/v = 1:1) in pH 8, close to the natural at 10 °C, where the self decay process can be excluded. The reactivity of 2 was monitored by UV-vis spectroscopy and the rate of its rapid decomposition was measured at 697 nm (Figure 5a). Pseudo-first order fitting of the kinetic data allowed us to determine kobs values. These results indicate a direct reaction between 2 and H2O2. In order to investigate the possible involvement of a hydrogen atom in the rate-determining step we investigated the reactivity of 2 with H2O2 in buffered MeCN/D2O/H2O (v/v = 1:0.75:0.25). Solutions of 2 in the presence of D2O at pH 8 were somewhat less reactive against H2O2, yielding a solvent kinetic isotope effect of 6.2. This value was significantly smaller than that was obtained for the H–D isotope effect for [RuIVO(bpy)2(py)] at pH 2.3 (KIE = 22.1 ± 1.2), but almost identical with that was measured at pH 9.7 (KIE = 8 ± 2.9) at 25 °C [40]. The most straightforward interpretation of the proton dependence was that the pathways involve the acid-base pre-equilibrium of H2O2 (H2O2 = HO2−+ H+) and the concomitant rate-controlling hydrogen-atom-transfer (HAT) between the FeIV=O species and the OH (or OD) group of H2O2 (D2O2) [57] forming a peroxyl radical.
To determine the dependence of the rates on the substrate concentration, solutions of the complex [(N4Py*)FeIV=O](CF3SO3)2 were treated with increasing amounts of H2O2 (1:5–50). Under this experimental condition, saturation kinetics was found for the kobs versus the H2O2 concentration (Figure 5b). At low H2O2 concentration, a k’ value of about 0.47 M−1s−1 was obtained at 10 °C (k’ = kobs/[H2O2] assuming a first order dependence). The reactivity of 2 was lower than that of [(N4Py)FeIV=O]2+ (N4Py = N,N’-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine) in CH3CN (k’ value of 8 M−1s−1 at 25 °C), but significantly higher than that of [(tmc)(CH3CN)FeIV=O]2+ (tmc = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane; k2 value of 0.035 ± 0.002 M−1s−1 at 25 °C) in CH3CN. Furthermore, a k’ value of 12.7 ± 1.3 M−1s−1 had been reported for the oxoruthenium(IV) complex [RuIVO(bpy)2(py)] at 25 °C (H2O, pH 7.92) [40]. Based on literature data, it can be concluded that [(N4Py*)FeIV=O]2+ is more reactive in O–H bond activation (H2O2) than in C–H bond activation (hydrocarbons) [46].
Substrates saturation behaviors implied a rapid equilibrium between the unbound substrate and the iron complex as a result of hydrogen bridge bond. Under conditions of high substrate concentration, the primary species in solution was the FeIVO–H2O2 (FeIVO–HO2−) complex. The rate of the reaction was dependent only on the decomposition of the FeIVO–H2O2 (FeIVO–HO2−) complex (r.d.s.) to the product and free precursor complex (Scheme 2) [40,57]. A nonlinear least square fit was applied to calculate the Michaelis–Menten parameters. The results were KM = 0.018 M, kcat = 0.014 s−1 and k2(kcat/KM) = 0.754 M−1s−1. An apparent KM value for bovine liver catalase (BLC) was determined to be 0.093 M. By contrast, the KM values of KatGs (catalase-peroxidase) were much lower (0.0042 M for SynKatG, 0.0025 M for MtbKatG and 0.0059 M for BpKatG, all at pH 7) [48], but was almost identical with the value for the natural enzyme from Tricholoma album (KM = 0.015 M) indicating a high affinity to the substrate, appearing to bind to peroxide very strongly [21].
3. Materials and Methods
The N4Py* ligand, and its [FeII(N4Py*)(CH3CN)](CF3SO3)2 (1) complex were prepared according to published procedures [31]. UV/Vis spectra were recorded with an Agilent 8453 diode-array spectrophotometer (Agilent Technologies, Hewlett-Packard-Strasse 8, Waldbronn, Germany) with quartz cells.
Catalytic reactions were carried out at 20 °C in a 30 cm3 reactor containing a stirring bar under air. In a typical experiment the appropriate aqueous solution (19 cm3 0.1 M KH2PO4/0.1 M NaOH pH 7, 8; 0.025 M Na2B4O7.10H2O/0.1 M HCl pH 9, 9.5, 10; or 0.05 M NaHCO3/0.1 M KOH pH 10.5, 11 buffer and I = 0.15 M KNO3) was added to the complex dissolved in 1 cm3 DMF, and the flask was closed with a rubber septum. H2O2 was injected by syringe through the septum. The reactor was connected to a graduated burette filled with oil, and the evolved dioxygen was measured volumetrically at time intervals of 15 s. Initial rates were expressed as Ms−1 by taking the volume of the solution into account, and calculated from the maximum slope of the evolved dioxygen versus time.
Stoichiometric reactions were carried out under thermostated conditions at 10 °C in 1 cm quartz cuvettes. In a typical experiment [FeII(N4Py*)(CH3CN)](CF3SO3)2 (1) (3 × 10−3 M) was dissolved in acetonitrile (1.0 cm3), then iodosobenzene (4.5 × 10−3 M) was added to the solution. The mixture was stirred for 50 min then excess iodosobenzene was removed by filtration. The acetonitril solution was than diluted with the appropriate buffered aqueous solution (1.0 cm3), and the decay of 2 was followed by monitoring the decrease in absorbance at 697 nm (ε = 400 M−1 cm−1) in the absence or in the presence of H2O2 under a pseudo-first order condition of excess H2O2.
4. Conclusions
It was found earlier that non-heme oxoiron(IV) complexes were able to carry out electrophilic transformations including O–H activation of H2O2 via homolytic O–H bond cleavage in acetonitrile as a functional catalase model. As a continuity of this study, efforts were made to work out a functional model in aqueous solution, close to the natural, where the postulated oxoiron(IV) intermediate behaved as an electrophilic oxidant. In summary, we reported one of the first examples of catalytic and stoichiometric H2O2 dismutation into O2 and H2O in aqueous solution mediated by electrophilic oxoiron(IV) intermediate, where the reactivity of 2 was markedly influenced by the pH. Based on detailed mechanistic studies on H2O2 oxidation that were investigated with in situ generated oxoiron(IV) species, plausible mechanisms were proposed, in which the H2O2 oxidation occurred by the HAT mechanism. To put together the stoichiometric and catalytic results it could be said that the highest catalytic activity of the H2O2 dismutation could be observed at pH 9.5, where the concentration of the more nucleophilic hydroperoxide anion (HOO−) was high, and the self-decay of the oxoiron(IV) intermediate could be neglected. These results were in good agreement with the electrophilic reactivity of oxoiron(IV) intermediates proposed for heme-type monoiron catalases, and might help us to understand the mechanism of the detoxification of H2O2 in biological systems.
Author Contributions
Individual contribution of authors were as follows: B.K., Organic synthesis; B.S., Reaction kinetics; G.S., Senior supervisor and advisor; and J.K., Project leader, writer of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Financial support of the Hungarian National Research Fund (OTKA K108489), and GINOP-2.3.2-15-2016-00049 are gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zamocky, M.; Furtmuller, P.G.; Obinger, C. Evolution of catalases from bacteria to humans. Antioxid. Redox Signal. 2008, 10, 1527–1548. [Google Scholar] [CrossRef]
- Kunsch, C.; Medford, R.M. Oxidative Stress as a Regulator of Gene Expression in the Vasculature. Circ. Res. 1999, 85, 753–766. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 2005, 115, 500–508. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals, antioxidants, and human disease: Curiosity, cause, or consequence? Lancet 1994, 344, 721–724. [Google Scholar] [CrossRef]
- Choua, S.; Pacheco, P.; Coquelet, C.; Bienvenüe, E. Catalase-like activity of a water-soluble complex of Ru(II). J. Inorg. Biochem. 1997, 65, 79–85. [Google Scholar] [CrossRef]
- Hempel, N.; Carrico, P.M.; Melendez, J.A. Manganese superoxide dismutase (Sod2) and redoxcontrol of signaling events that drive metastasis. Anticancer Agents Med. Chem. 2011, 11, 191–201. [Google Scholar] [CrossRef]
- Sampson, N.; Koziel, R.; Zenzmaier, C.; Bubendorf, L.; Plas, E.; Jansen-Durr, P.; Berger, P. ROS Signaling by NOX4 Drives Fibroblast-to-Myofibroblast Differentation in the Diseased Prostatic Stroma. Mol. Endocrinol. 2011, 25, 503–515. [Google Scholar] [CrossRef]
- Gao, M.C.; Jia, X.D.; Wu, Q.F.; Cheng, Y.; Chen, F.R.; Zhang, J. Silencing Prx1 and/or Prx5 sensitizes human esophageal cancer cells to ionizing radiation and increases apoptosis via intracellular ROS accumulation. Acta Pharmacol. Sin. 2011, 32, 528–536. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, Y.; Su, Y. Peroxiredoxins, a novel target in cancer radiotherapy. Cancer Lett. 2009, 286, 154–160. [Google Scholar] [CrossRef]
- Holley, A.K.; Miao, L.; St Clair, D.K.; St Clair, W.H. Redox-Modulated Phenomena and Radiation Therapy: The Central Role of Superoxide Dismutases. Antioxid. Redox Signal. 2014, 20, 1567–1589. [Google Scholar] [CrossRef] [Green Version]
- Armogida, M.; Nistico, R.; Mercuri, N.B. Therapeutic potential of targeting hydrogen peroxide metabolism in the treatment of brain ischaemia. Br. J. Pharmacol. 2012, 166, 1211–1224. [Google Scholar] [CrossRef] [Green Version]
- Beyer, W.F.; Fridovich, I. Catalases-with and without heme. Basic Life Sci. 1988, 49, 651–661. [Google Scholar]
- Nicholls, P.; Fita, I.; Loewen, P.C. Enzymology and structure of catalases. Adv. Inorg. Chem. 2001, 51, 51–106. [Google Scholar]
- Ko, T.P.; Day, J.; Malkin, A.J.; McPherson, A. Structure of orthorhombic crystals of beef liver catalase. Acta Crystallogr. 1999, 55, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Ivancich, A.; Jouve, H.M.; Sartor, B.; Gaillard, J. EPR investigation of compound I in Proteus mirabilis and bovin liver catalases: Formation of porphyrin and tyrosyl radical intermediates. Biochemistry 1997, 36, 9356–9364. [Google Scholar] [CrossRef]
- Kono, Y.; Fridovich, I. Isolation and characterization of the pseudocatalase of Lactobacillus plantarum. J. Biol. Chem. 1983, 258, 6015–6019. [Google Scholar]
- Barynin, V.V.; Whittaker, M.M.; Antonyuk, S.V.; Lamzin, V.S.; Harrison, P.M.; Artymiuk, P.J.; Whittaker, J.W. Crystal Structure of Manganese Catalase from Lactobacillus plantarum. Structure 2001, 9, 725–738. [Google Scholar] [CrossRef]
- Antonyuk, S.V.; Melik-Adman, V.R.; Popov, A.N.; Lamzin, V.S.; Hempstead, P.D.; Harrison, P.M.; Artymyuk, P.J.; Barynin, V.V. Three-dimensional structure of the enzyme dimanganese catalase from Thermus thermophilus at 1 Å resolution. Crystallogr. Rep. 2000, 45, 105–113. [Google Scholar] [CrossRef]
- Barynin, V.V.; Grebenko, A.I. T-catalase is nonheme catalase of the extremely thermophilic bacterium Thermus thermophilus HB8. Dokl. Akad. Nauk. USSR 1986, 286, 461–464. [Google Scholar]
- Allgood, G.S.; Perry, J.J. Characterization of a manganese-containing catalase from the obligate thermophile Thermoleophilum album. J. Bacteriol. 1986, 168, 563–567. [Google Scholar] [CrossRef]
- Amo, T.; Atomi, H.; Imanaka, T. Unique Presence of a Manganase Catalase in a Hyperthermophilic Archaeon, Pyrobaculum calidifontis VA1. J. Bacteriol. 2002, 184, 3305–3312. [Google Scholar] [CrossRef]
- Gao, J.; Martell, A.E.; Reibenspies, J.H. Novel dicopper(II) catalase-like model complexes: Synthesis, crystal structure, properties and kinetic studies. Inorg. Chim. Acta 2003, 346, 32–42. [Google Scholar] [CrossRef]
- Boelrijk, A.E.M.; Dismukes, G.C. Mechanism of Hydrogen Peroxide Dismutation by a Dimanganese Catalase Mimic: Dominant Role of an Intramolecular Base on Substrate Binding Affinity and Rate Acceleration. Inorg. Chem. 2000, 39, 3020. [Google Scholar] [CrossRef]
- Paschke, J.; Kirsch, M.; Korth, H.G.; Groot, H.; Sustmann, R. Catalase-Like Activity of a Non-Heme Dibenzotetraaza[14]annulene–Fe(III) Complex under Physiological Conditions. J. Am. Chem. Soc. 2001, 123, 11099–11100. [Google Scholar] [CrossRef]
- Okuno, T.; Ito, S.; Ohba, S.; Nishida, Y. µ-Oxo bridged diiron(III) complexes and hydrogen peroxide: Oxygenation and catalase-like activities. J. Chem. Soc. Dalton. Trans. 1997, 24, 3547–3551. [Google Scholar] [CrossRef]
- Mauerer, B.; Crane, J.; Schuler, J.; Wieghardt, K.; Nuber, B. A Hemerythrin Model Complex with Catalase Activity. Angew. Chem. Int. Ed. Engl. 1993, 32, 289–291. [Google Scholar] [CrossRef]
- Ménage, S.; Vincent, J.M.; Lambeaux, C.; Fontecave, M. µ-Oxo-bridged diiron(III) complexes and H2O2: Monooxygenase and catalase-like activities. J. Chem. Soc. Dalton Trans. 1994, 21, 2081–2084. [Google Scholar] [CrossRef]
- Sigel, H.; Wiss, K.; Fischer, B.E.; Prijs, B. Metal ions and hydrogen peroxide. Catalase-like activity of copper(2+) ion in aqueous solution and its promotion by the coordination of 2,2′-bipyridyl. Inorg. Chem. 1979, 18, 1354–1358. [Google Scholar] [CrossRef]
- Kaizer, J.; Csonka, R.; Speier, G.; Giorgi, M.; Réglier, M. Synthesis, structure and catalase-like activity of new dicopper(II) complexes with phenylglyoxylate and benzoate ligands. J. Mol. Catal. A Chem. 2005, 236, 12–17. [Google Scholar] [CrossRef]
- Kaizer, J.; Csay, T.; Speier, G.; Réglier, M.; Giorgi, M. Synthesis, structure and catalase-like activity of Cu(N-baa)(2)(phen) (phen=1, 10-phenanthroline, N-baaH = N-benzoylanthranilic acid). Inorg. Chem. Commun. 2006, 9, 1037–1039. [Google Scholar] [CrossRef]
- Pap, J.S.; Horvath, B.; Speier, G.; Kaizer, J. Synthesis and catalase-like activity of dimanganese complexes with phthalazine-based ligands. Transit. Met. Chem. 2011, 36, 603–609. [Google Scholar] [CrossRef]
- Pap, J.S.; Kripli, B.; Bors, I.; Bogáth, D.; Giorgi, M.; Kaizer, J.; Speier, G. Transition metal complexes bearing flexible N-3 or N3O donor ligands: Reactivity toward superoxide radical anion and hydrogen peroxide. J. Inorg. Biochem. 2012, 117, 60–70. [Google Scholar] [CrossRef]
- Kaizer, J.; Csay, T.; Kovari, P.; Speier, G.; Parkanyi, L. Catalase mimics of a manganese(II) complex: The effect of axial ligands and pH. J. Mol. Catal. A Chem. 2008, 280, 203–209. [Google Scholar] [CrossRef]
- Kaizer, J.; Kripli, B.; Speier, G.; Parkanyi, L. Synthesis, structure, and catalase-like activity of a novel manganese(II) complex: Dichloro[1,3-bis(2 ‘-benzimidazolylimino) isoindoline] manganese(II). Polyhedron 2009, 28, 933–936. [Google Scholar] [CrossRef]
- Horn, A., Jr.; Parrilha, G.I.; Melo, K.V.; Fernandes, C.; Horner, M.; Visentin, I.C.; Santos, J.A.S.; Santos, M.S.; Eleutherio, E.C.A.; Pereira, M.D. An iron-based cytosolic catalase and superoxide dismutase mimic complex. Inorg. Chem. 2010, 49, 1274–1276. [Google Scholar] [CrossRef]
- Carvalho, N.M.F.; Horn, A., Jr.; Faria, R.B.; Bortoluzzi, A.J.; Drago, V.; Antunes, O.A.C. Synthesis, characterization, X-ray molecular structure and catalase-like activity of a non-heme iron complex: Dichloro[N-propanoate-N,N-bis-(2-pyridylmethyl)amine] iron(III). Inorg. Chim. Acta 2006, 359, 4250–4258. [Google Scholar] [CrossRef]
- Dickman, M.H.; Pope, M.T. Peroxo and Superoxo Complexes of Chromium, Molybdenum, and Tungsten. Chem. Rev. 1994, 94, 569–584. [Google Scholar] [CrossRef]
- Wu, A.J.; Penner-Hahn, J.E.; Pecoraro, V.L. Structural, Spectroscopic, and Reactivity Models for the Manganese Catalases. Chem. Rev. 2004, 104, 903–938. [Google Scholar] [CrossRef]
- Gilbert, J.; Roecker, L.; Meyer, T.J. Hydrogen Atom Transfer in the Oxidation of Hydrogen Peroxide by [(bpy) 2(py)Ru IV=O] 2+ and by [(bpy) 2(py)Ru III-OH] 2+. Inorg. Chem. 1987, 26, 1126. [Google Scholar] [CrossRef]
- Crans, D.C.; Smee, J.J.; Gaidamauskas, E.; Yang, L. The Chemistry and Biochemistry of Vanadium and the Biological Activities Exerted by Vanadium Compounds. Chem. Rev. 2004, 104, 849–902. [Google Scholar] [CrossRef]
- Pires, B.M.; Silva, D.M.; Visentin, L.C.; Drago, V.; Carvalho, N.M.F.; Faria, R.B.; Antunes, O.A.C. Synthesis, characterization and catalase-like activity of the tetranuclear iron(III) complex involving a (μ-oxo)(μ-hydroxo)bis(μ-alkoxo)tetra(μ-carboxylato)tetrairon core. Inorg. Chim. Acta 2013, 407, 69–81. [Google Scholar] [CrossRef]
- Lakk-Bogáth, D.; Csonka, R.; Speier, G.; Reglier, M.; Simaan, A.J.; Naubron, J.V.; Giorgi, M.; Lazar, K.; Kaizer, J. Formation, Characterization, and Reactivity of Nonheme Iron(IV)-Oxo Complex Derived from the Chiral Pentadentate Ligand asN4Py. Inorg. Chem. 2016, 55, 10090. [Google Scholar] [CrossRef]
- Turcas, R.; Lakk-Bogáth, D.; Speier, G.; Kaizer, J. Steric Control and Mechanism of Benzaldehyde Oxidation by Polypyridyl Oxoiron(IV) Complexes: Aromatic versus Benzylic Hydroxylation of Aromatic Aldehydes. Dalton. Trans. 2018, 47, 3248. [Google Scholar] [CrossRef]
- Lakk-Bogáth, D.; Kripli, B.; Meena, B.I.; Speier, G.; Kaizer, J. Catalytic and stoichiometric oxidation of N,N-dimethylanilines mediated by nonheme oxoiron(IV) complex with tetrapyridyl ligand. Polyhedron 2019, 169, 169–175. [Google Scholar] [CrossRef]
- Lakk-Bogáth, D.; Kripli, B.; Meena, B.I.; Speier, G.; Kaizer, J. Catalytic and stoichiometric C-H oxidation of benzylalcohols and hydrocarbons mediated by nonheme oxoiron(IV) complex with chiral tetrapyridyl ligand. Inorg. Chem. Commun. 2019, 104, 165–170. [Google Scholar] [CrossRef]
- Kubota, R.; Imamura, S.; Shimizu, T.; Asayama, S.; Kawakami, H. Synthesis of Water-Soluble Dinuclear Mn-Porphyrin with Multiple Antioxidative Activities. ACS Med. Chem. Lett. 2014, 5, 639–643. [Google Scholar] [CrossRef] [Green Version]
- Jakopitsch, C.; Vlasits, J.; Wiseman, B.; Loewen, P.C.; Obinger, C. Redox Intermediates in the Catalase Cycle of Catalase-Peroxidases from Synechocystis PCC 6803, Burkholderia pseudomallei, and Mycobacterium tuberculosis. Biochemistry 2007, 46, 1183–1193. [Google Scholar] [CrossRef]
- Gelasco, A.; Bensiek, S.; Pecoraro, V.L. The [Mn2(2-OHsalpn)2]2-,1-,0 System: An Efficient Functional Model for the Reactivity and Inactivation of the Manganese Catalases. Inorg. Chem. 1998, 37, 3301–3309. [Google Scholar] [CrossRef]
- Larson, E.J.; Pecoraro, V.L. [Mn(III)(2-OHsalpn)]2 is an efficient functional model for the manganese catalases. J. Am. Chem. Soc. 1993, 115, 7928–7929. [Google Scholar]
- Kaizer, J.; Baráth, G.; Speier, G.; Réglier, M.; Giorgi, M. Synthesis, structure and catalase mimics of novel homoleptic manganese(II) complexes of 1,3-bis(2’-pyridylimino) isoindoline, Mn(4R-ind)2 (R= H., Me). Inorg. Chem. Commun. 2007, 10, 292–294. [Google Scholar] [CrossRef]
- Signorella, S.; Palopoli, C.; Ledesma, G. Rationally designed mimics of antioxidant manganoenzymes: Role of structural features in the quest for catalysts with catalaseand superoxide dismutase activity. Coord. Chem. Rev. 2018, 365, 75–102. [Google Scholar] [CrossRef]
- Chance, B.; Greenstein, D.S.; Roughton, F.J. The mechanism of catalase action. I. Steady-state analysis. Arch. Biochem. Biophys. 1952, 37, 301–321. [Google Scholar] [CrossRef]
- Braymer, J.J.; O’Neill, K.P.; Rohde, J.U.; Lim, M.H. The Reaction of a High-Valent Nonheme Oxoiron(IV) Intermediate with Hydrogen Peroxide. Angew. Chem. Int. Ed. 2012, 51, 1–6. [Google Scholar] [CrossRef]
- Chen, J.; Draksharapu, A.; Angelone, D.; Unjaroen, D.; Padamati, S.K.; Hage, R.; Swart, M.; Duboc, C.; Browne, W.R. H2O2 Oxidation by FeIII-OOH Intermediates and Its Effect on Catalytic Efficiency. ACS Catal. 2018, 8, 9665–9674. [Google Scholar] [CrossRef]
- Sastri, C.V.; Seo, M.S.; Park, M.J.; Kim, K.M.; Nam, W. Formation, stability, and reactivity of a mononuclear nonheme oxoiron(IV) complex in aqueous solution. Chem. Commun. 2005, 1405–1407. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Gersten, S.W.; Meyer, T.J. H-D Kinetic Isotope Effects of 16 and 22 in the Oxidation of H2O2. J. Am. Chem. Soc. 1982, 104, 6872–6873. [Google Scholar] [CrossRef]
Scheme 1.
Structures of (1), (2) and (3).
Figure 1.
Kinetics of hydrogen peroxide degradation catalyzed by 1 in water: (a) pH dependence of hydrogen peroxide degradation determined by volumetrically measuring the evolved dioxygen in the presence (●) and in the absence (○) of 1. The inset shows the time traces for the reaction of 0.275 mM 1 with 0.35 M H2O2 at pH 8, 9.5 and 11 at 20 °C. (b) Vin versus [H2O2]0 at [1] = 2.75 × 10−4 M, pH 9.5 (borate buffer) and 20 °C. The inset shows the time traces for the reaction of 0.275 mM 1 with H2O2 (0.11–1.29 M).
Figure 1.
Kinetics of hydrogen peroxide degradation catalyzed by 1 in water: (a) pH dependence of hydrogen peroxide degradation determined by volumetrically measuring the evolved dioxygen in the presence (●) and in the absence (○) of 1. The inset shows the time traces for the reaction of 0.275 mM 1 with 0.35 M H2O2 at pH 8, 9.5 and 11 at 20 °C. (b) Vin versus [H2O2]0 at [1] = 2.75 × 10−4 M, pH 9.5 (borate buffer) and 20 °C. The inset shows the time traces for the reaction of 0.275 mM 1 with H2O2 (0.11–1.29 M).

Figure 2.
Reaction of 2 with H2O2 in acetonitrile: (a) UV-Vis spectra of the reaction of 1.5 mM 2 in CH3CN with 75 equiv of H2O2 at 10 °C (path length, 1 cm). Inset: Time course of the reaction monitored at 705 nm (2) and 535 (3). (b) UV/Vis spectra of the decay of 3 generated based on (a). Inset: Time course of the decay of 3 in CH3CN and CH3CN/H2O (v/v = 1:1) solution at 10 °C.
Figure 2.
Reaction of 2 with H2O2 in acetonitrile: (a) UV-Vis spectra of the reaction of 1.5 mM 2 in CH3CN with 75 equiv of H2O2 at 10 °C (path length, 1 cm). Inset: Time course of the reaction monitored at 705 nm (2) and 535 (3). (b) UV/Vis spectra of the decay of 3 generated based on (a). Inset: Time course of the decay of 3 in CH3CN and CH3CN/H2O (v/v = 1:1) solution at 10 °C.
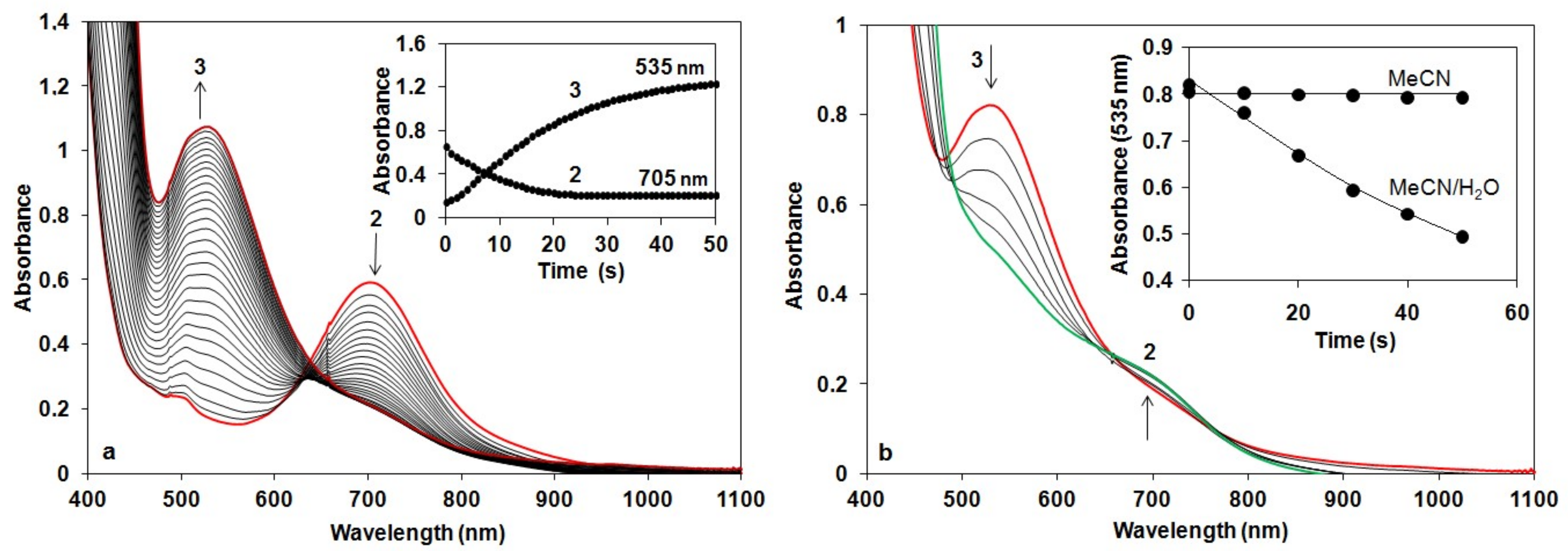
Figure 3.
Time course of the decay of 2 monitored at 697 nm at different pH in the presence (●) and in the absence (○) of H2O2 at 10 °C. Conditions: [2] = 1.5 mM; [H2O2]0 = 15 mM in MeCN/H2O (2 cm3, v/v = 1:1, path = 1 cm). (a) pH 7: 0.1 M KH2PO4/0.1 M NaOH. (b) pH 8: 0.025 M Na2B4O7.10H2O/0.1 M HCl. (c) pH 9: 0.05 M NaHCO3/0.1 M KOH. (d) pH 9.5: 0.05 M NaHCO3/0.1 M KOH. (e) pH 10.5: 0.05 M NaHCO3/0.1 M KOH. (f) pH 11: 0.05 M NaHCO3/0.1 M KOH. I = 0.15 M KNO3.
Figure 3.
Time course of the decay of 2 monitored at 697 nm at different pH in the presence (●) and in the absence (○) of H2O2 at 10 °C. Conditions: [2] = 1.5 mM; [H2O2]0 = 15 mM in MeCN/H2O (2 cm3, v/v = 1:1, path = 1 cm). (a) pH 7: 0.1 M KH2PO4/0.1 M NaOH. (b) pH 8: 0.025 M Na2B4O7.10H2O/0.1 M HCl. (c) pH 9: 0.05 M NaHCO3/0.1 M KOH. (d) pH 9.5: 0.05 M NaHCO3/0.1 M KOH. (e) pH 10.5: 0.05 M NaHCO3/0.1 M KOH. (f) pH 11: 0.05 M NaHCO3/0.1 M KOH. I = 0.15 M KNO3.
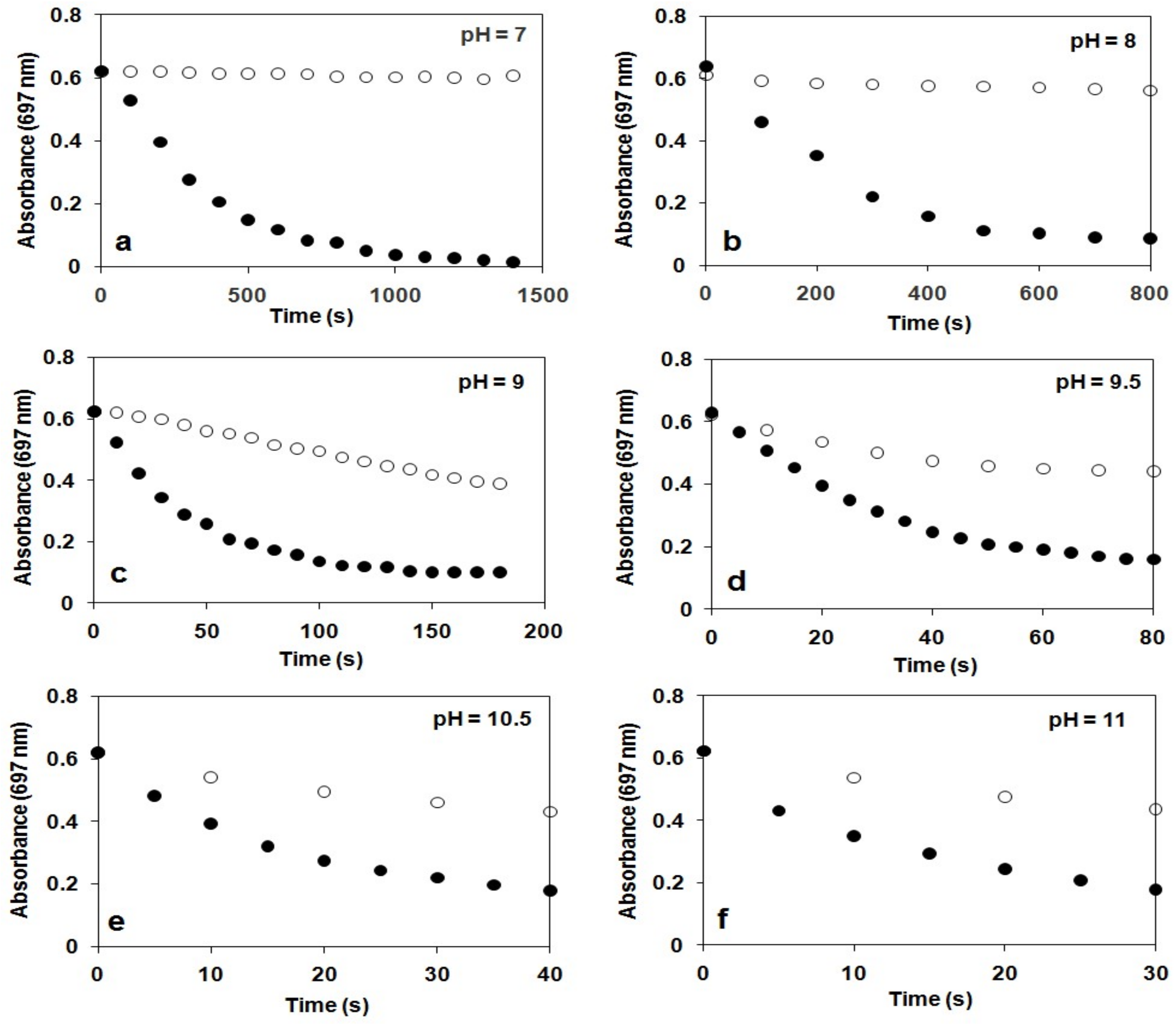
Figure 4.
(a) Reaction rates of the decay of 2 monitored at 697 nm at different pH values in the presence (●) and in the absence (○) of H2O2 and their normalized values (◊) in buffered CH3CN/H2O (v/v = 1:1) solution (pH 7–11) at 10 °C. (b) Reaction of 2 with H2O2 in buffered CH3CN/H2O: UV-Vis spectra of the reaction of 1.5 mM 2 in buffered CH3CN/H2O (pH 10, v/v = 1:1) with 10 equiv of H2O2 at 10 °C (path length, 1 cm). Inset: Time course of the reaction monitored at 697 (2) and 490 nm in buffered CH3CN/H2O (v/v = 1:1) solution (pH 10) at 10 °C.
Figure 4.
(a) Reaction rates of the decay of 2 monitored at 697 nm at different pH values in the presence (●) and in the absence (○) of H2O2 and their normalized values (◊) in buffered CH3CN/H2O (v/v = 1:1) solution (pH 7–11) at 10 °C. (b) Reaction of 2 with H2O2 in buffered CH3CN/H2O: UV-Vis spectra of the reaction of 1.5 mM 2 in buffered CH3CN/H2O (pH 10, v/v = 1:1) with 10 equiv of H2O2 at 10 °C (path length, 1 cm). Inset: Time course of the reaction monitored at 697 (2) and 490 nm in buffered CH3CN/H2O (v/v = 1:1) solution (pH 10) at 10 °C.

Figure 5.
Kinetic studies on the reaction of 2 with H2O2 in buffered MeCN/H2O solution at pH 8 and 10 °C. (a) UV-vis spectral change of 1.5 mM 2 upon addition of 10 equiv of H2O2. Inset shows time course of the decay of in the absence (○) and in the presence of H2O2 in MeCN/D2O (□) and MeCN/H2O (●) solution, respectively. (b) Plot of kobs versus [H2O2]0 at [2] = 1.5 mM, pH 8 and 10 °C.
Figure 5.
Kinetic studies on the reaction of 2 with H2O2 in buffered MeCN/H2O solution at pH 8 and 10 °C. (a) UV-vis spectral change of 1.5 mM 2 upon addition of 10 equiv of H2O2. Inset shows time course of the decay of in the absence (○) and in the presence of H2O2 in MeCN/D2O (□) and MeCN/H2O (●) solution, respectively. (b) Plot of kobs versus [H2O2]0 at [2] = 1.5 mM, pH 8 and 10 °C.
Scheme 2.
Proposed mechanism for the oxoiron(IV)-mediated H2O2 oxidation.
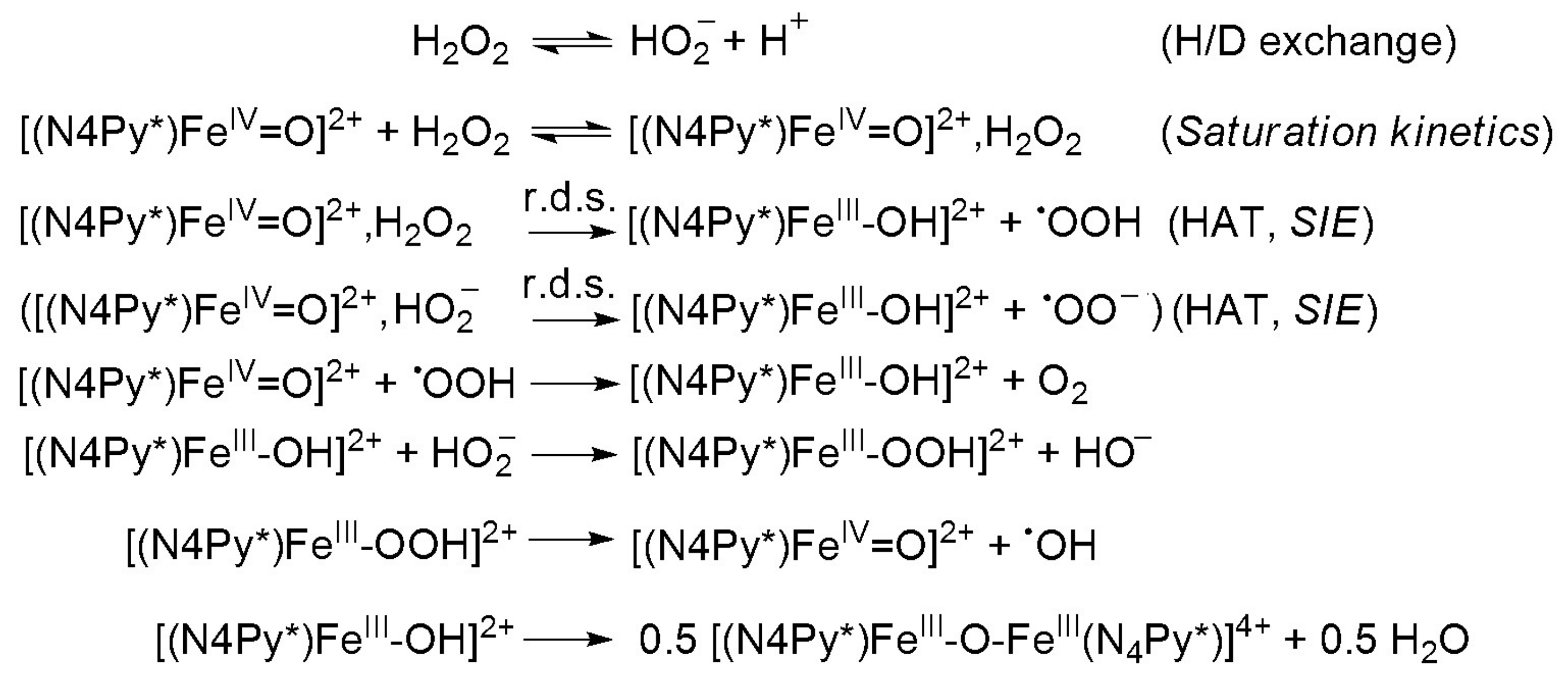

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
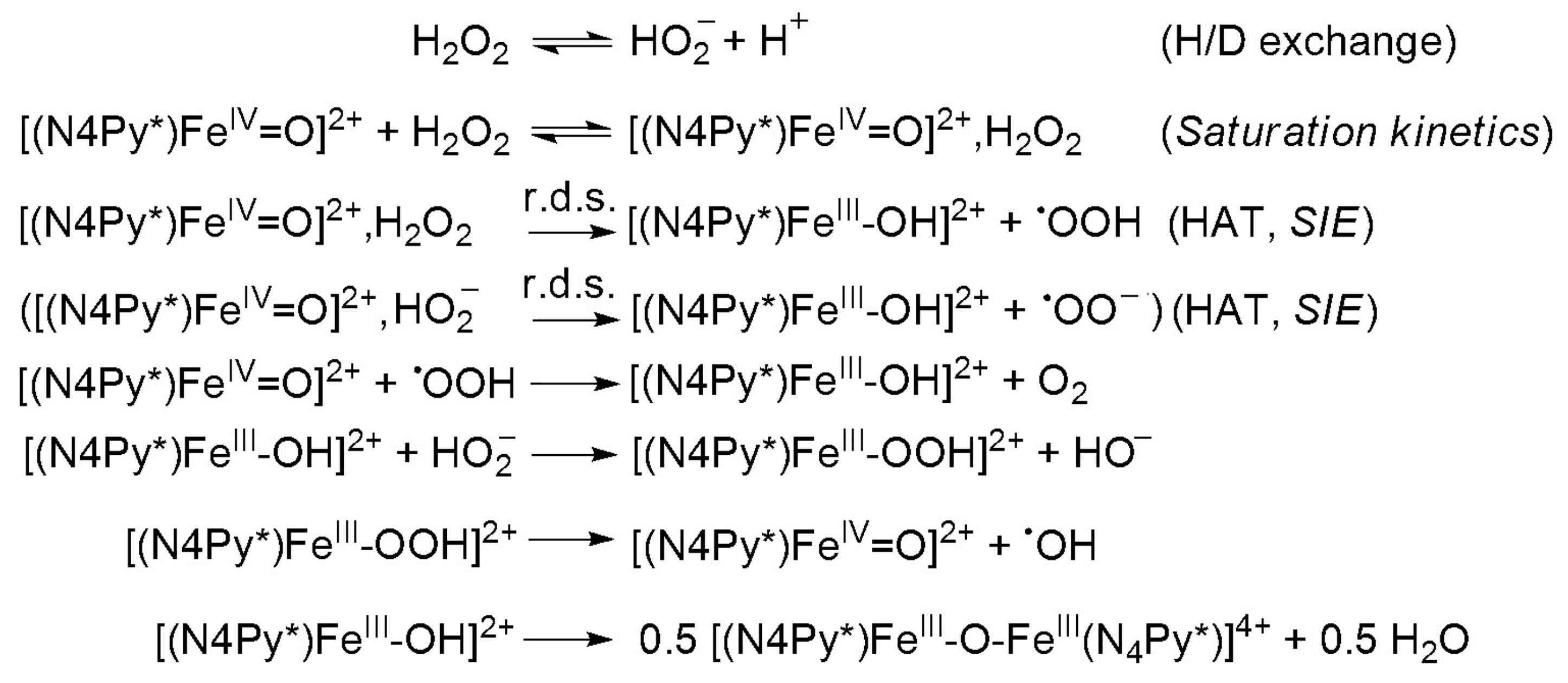{kind=link}
Table 1.
Kinetic parameters of reported catalase, catalase-peroxidase and their synthetic models.
| Entry | Complex/Enzyme | KM (M) | kcat (s−1) | kcat/KM (s−1M−1) | Solvent | Refs. |
|---|---|---|---|---|---|---|
| 1 | SynKatG 1 | 0.0042 | H2O, pH 7 | [48] | ||
| 2 | BpKatG 2 | 0.0059 | H2O, pH 7 | [48] | ||
| 3 | MtbKatG 3 | 0.0025 | 1.2 × 103 | 5 × 108 | H2O, pH 7 | [48] |
| 4 | BLC 4 | 0.093 | 4.0 × 107 | H2O, pH 7 | [53] | |
| 5 | [FeII(N4Py*)(CH3CN)](ClO4)2 | 1.39 | 33.2 | 23.9 | H2O, pH 9.5 | this work. |
| 6 | [(N4Py*)FeIV=O](ClO4)2 | 0.018 | 0.014 | 0.754 | CH3CN/H2O, pH 8 | this work |
| 7 | [Fe4(μ -O) μ-OH)(μ-OAc)4(L2)]3+, 5 | 1.010 | 1.41 × 10−4 | 1.40 × 10−4 | H2O | [42] |
| 8 | [Fe4(μ-O) μ-OH)(μ-OAc)4(L2)]3+, 5 | 2.882 | 3.50 × 10−3 | 1.21 × 10−3 | H2O, pH 7.2 | [42] |
| 9 | [Fe4(μ-O) μ-OH)(μ-OAc)4(L2)]3+, 5 | 0.749 | 5.37 × 10−2 | 7.17 × 10−2 | CH3CN | [42] |
| 10 | T. thermophilus | 0.083 | 2.6 × 105 | 3.13 × 106 | H2O | [19,20] |
| 11 | T. album | 0.015 | 2.6 × 104 | 1.73 × 106 | H2O | [21] |
| 12 | L. plantarum | 0.35 | 2.0 × 105 | 0.57 × 106 | H2O | [17,18] |
| 13 | [Mn(indH)Cl2] 6 | 0.49 | 38.9 | 79.2 | H2O, pH 9.5 | [30] |
| 14 | [Mn(ind)2] 6 | 0.019 | 0.06 | 3.2 | DMF | [51] |
| 15 | [Mn(X-salpn)O]2 7 | 10–102 | 4.2–21.9 | 305–990 | CH3CN | [49,50] |
1 Catalase-peroxidase from Synechocystis PCC6803. 2 Catalase-peroxidase from Burkholderia pseudomallei. 3 Catalase peroxidase from Mycobacterium tuberculosis. 4 Bovine liver catalase. 5 HL = 1,3-bis[2-aminoethyl)amino]-2-propanol. 6 IndH = 1,3-bis(2′-pyridylimino)-isoindoline. 7 H2salpn = N,N’-bis(salicylidene)-1,3-diaminopropane.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kripli, B.; Sólyom, B.; Speier, G.; Kaizer, J. Stability and Catalase-Like Activity of a Mononuclear Non-Heme Oxoiron(IV) Complex in Aqueous Solution. Molecules 2019, 24, 3236. https://doi.org/10.3390/molecules24183236
AMA Style
Kripli B, Sólyom B, Speier G, Kaizer J. Stability and Catalase-Like Activity of a Mononuclear Non-Heme Oxoiron(IV) Complex in Aqueous Solution. Molecules. 2019; 24(18):3236. https://doi.org/10.3390/molecules24183236
Chicago/Turabian StyleKripli, Balázs, Bernadett Sólyom, Gábor Speier, and József Kaizer. 2019. "Stability and Catalase-Like Activity of a Mononuclear Non-Heme Oxoiron(IV) Complex in Aqueous Solution" Molecules 24, no. 18: 3236. https://doi.org/10.3390/molecules24183236