Cigarette Smoke Induces the Risk of Metabolic Bone Diseases: Transforming Growth Factor Beta Signaling Impairment via Dysfunctional Primary Cilia Affects Migration, Proliferation, and Differentiation of Human Mesenchymal Stem Cells

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
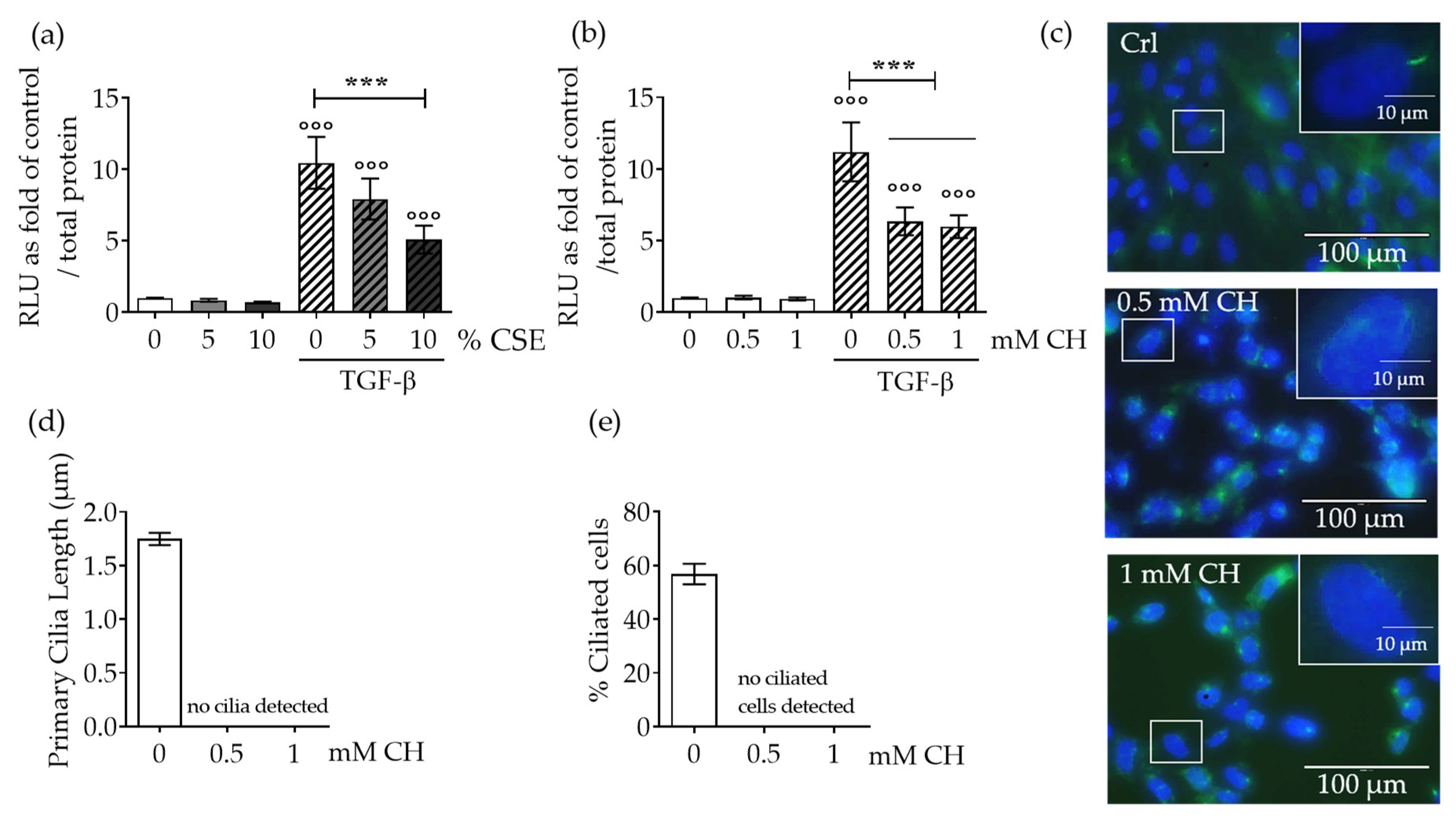
2.1. CSE Downregulated TGF-β Signaling Through Disruption of Primary Cilia on SCP-1 Cells
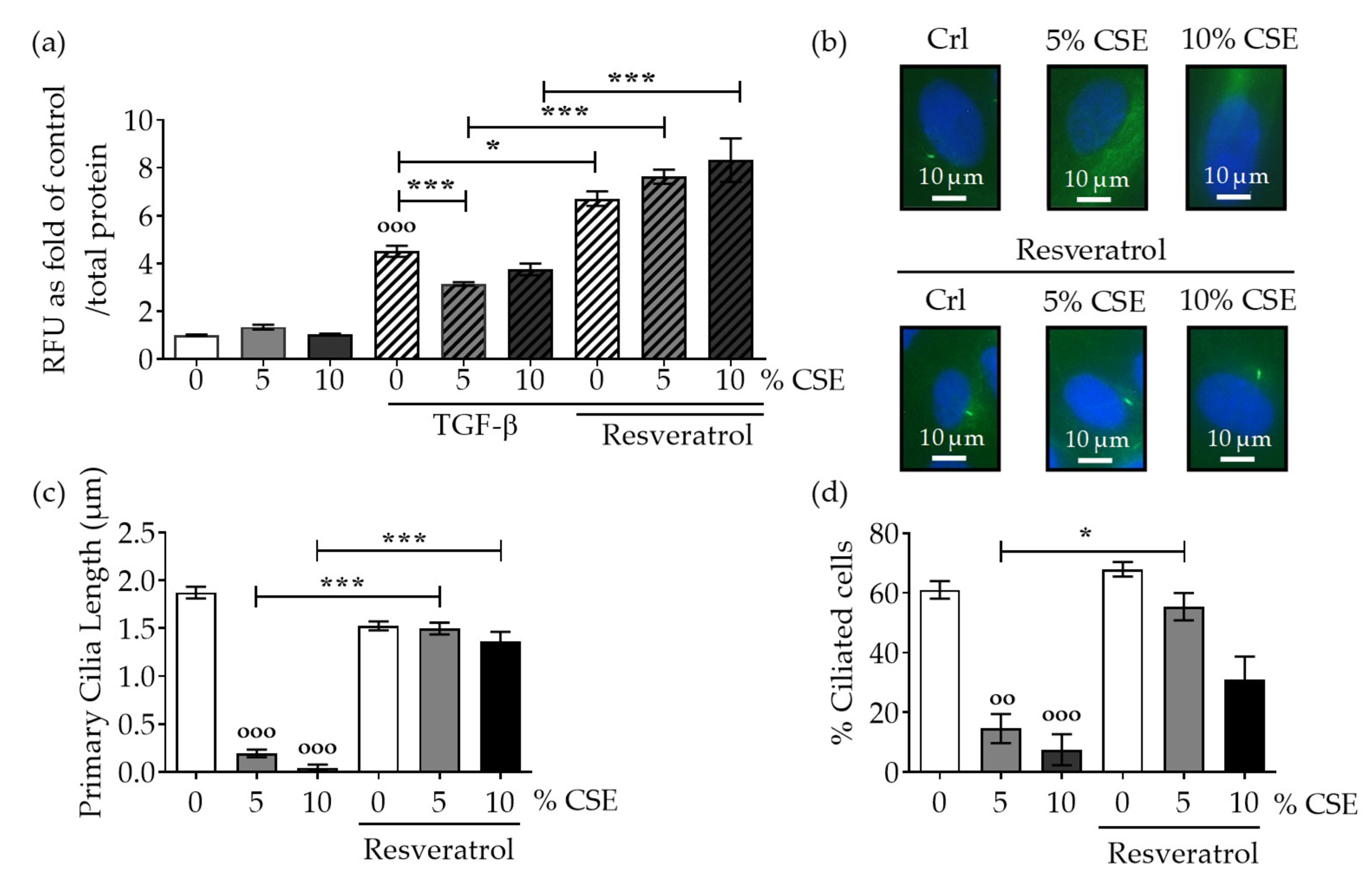
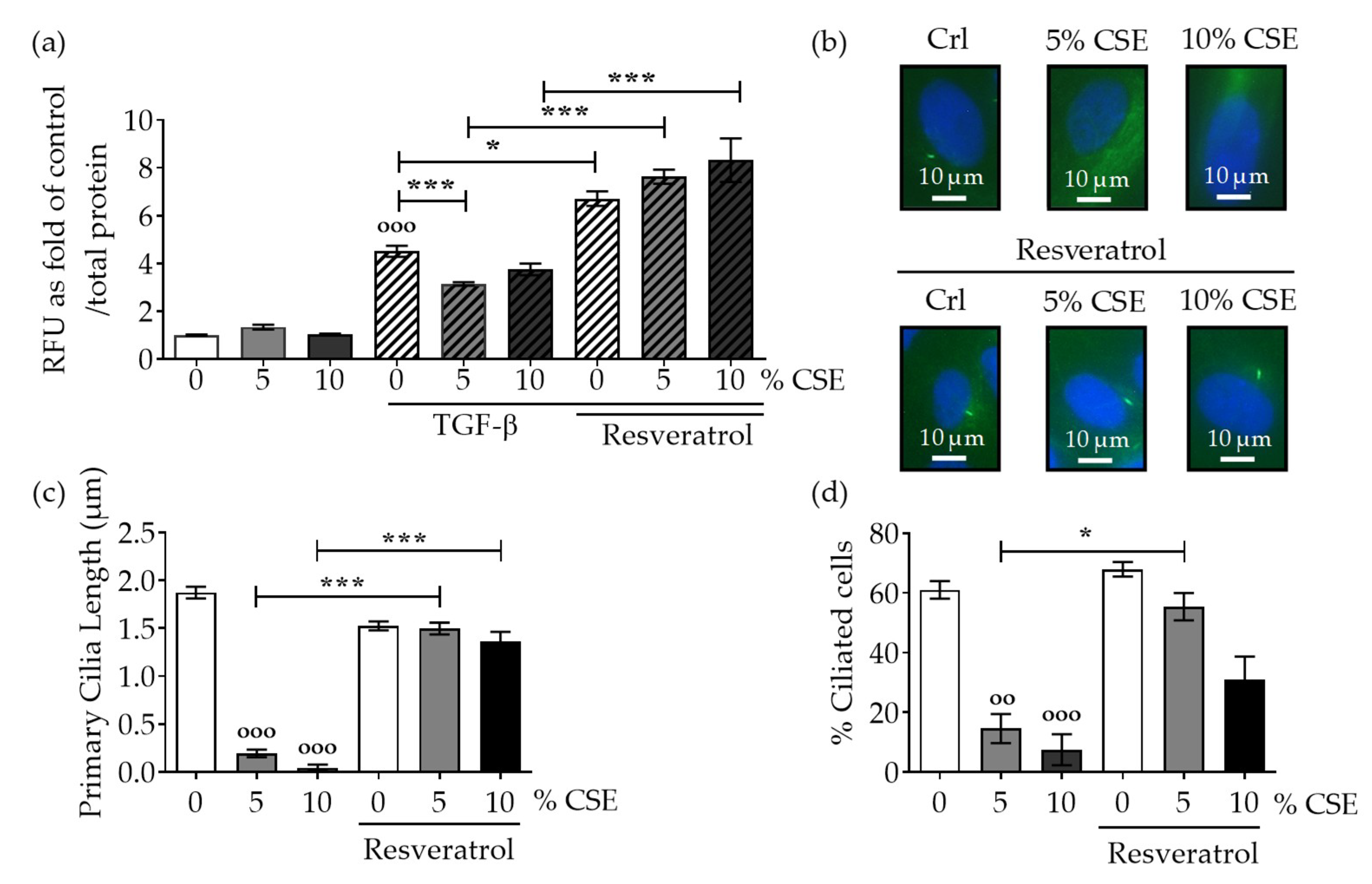
2.2. Protection of Primary Cilia Structure with Resveratrol rescues TGF-β Signaling Suppressed by CSE
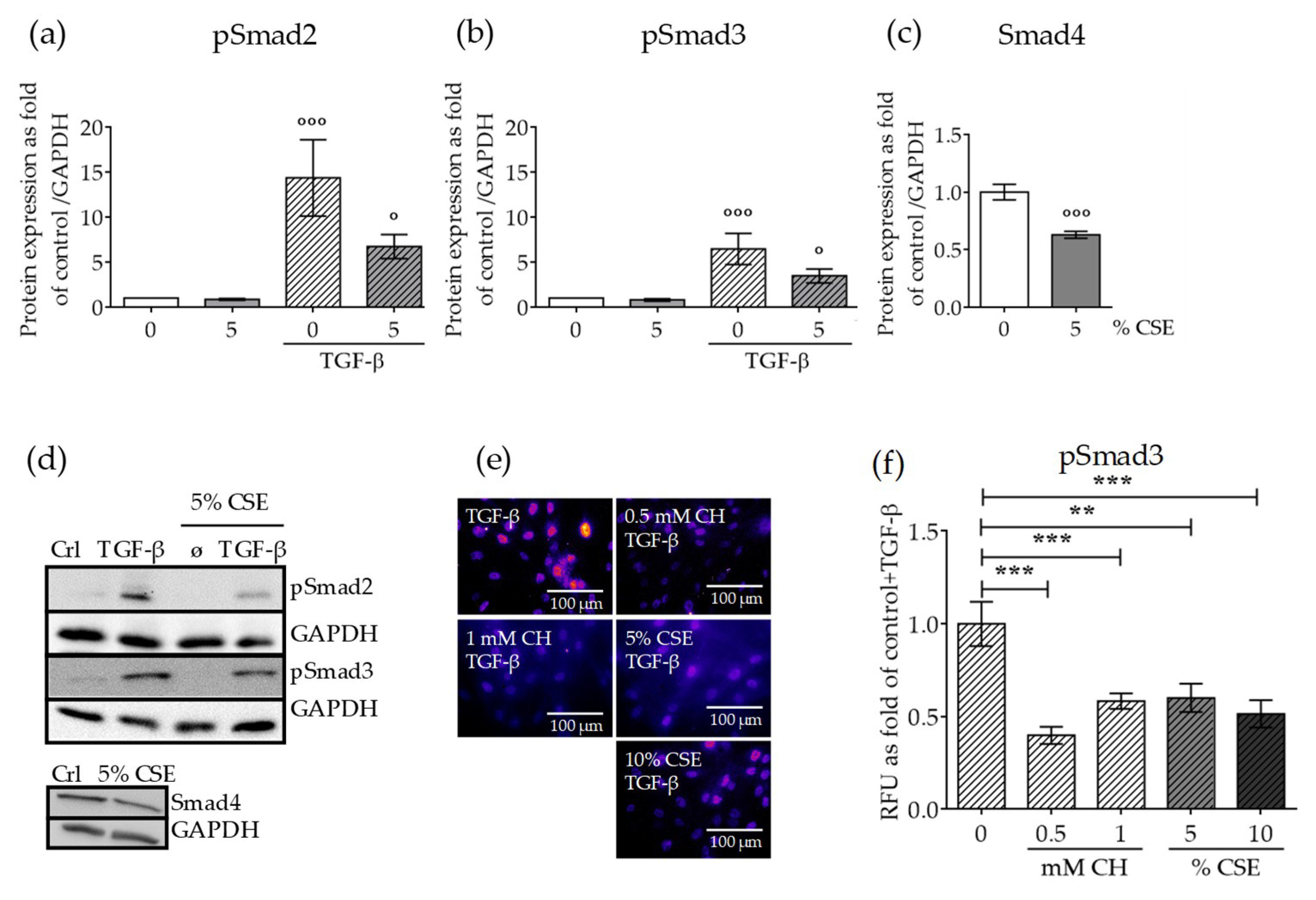
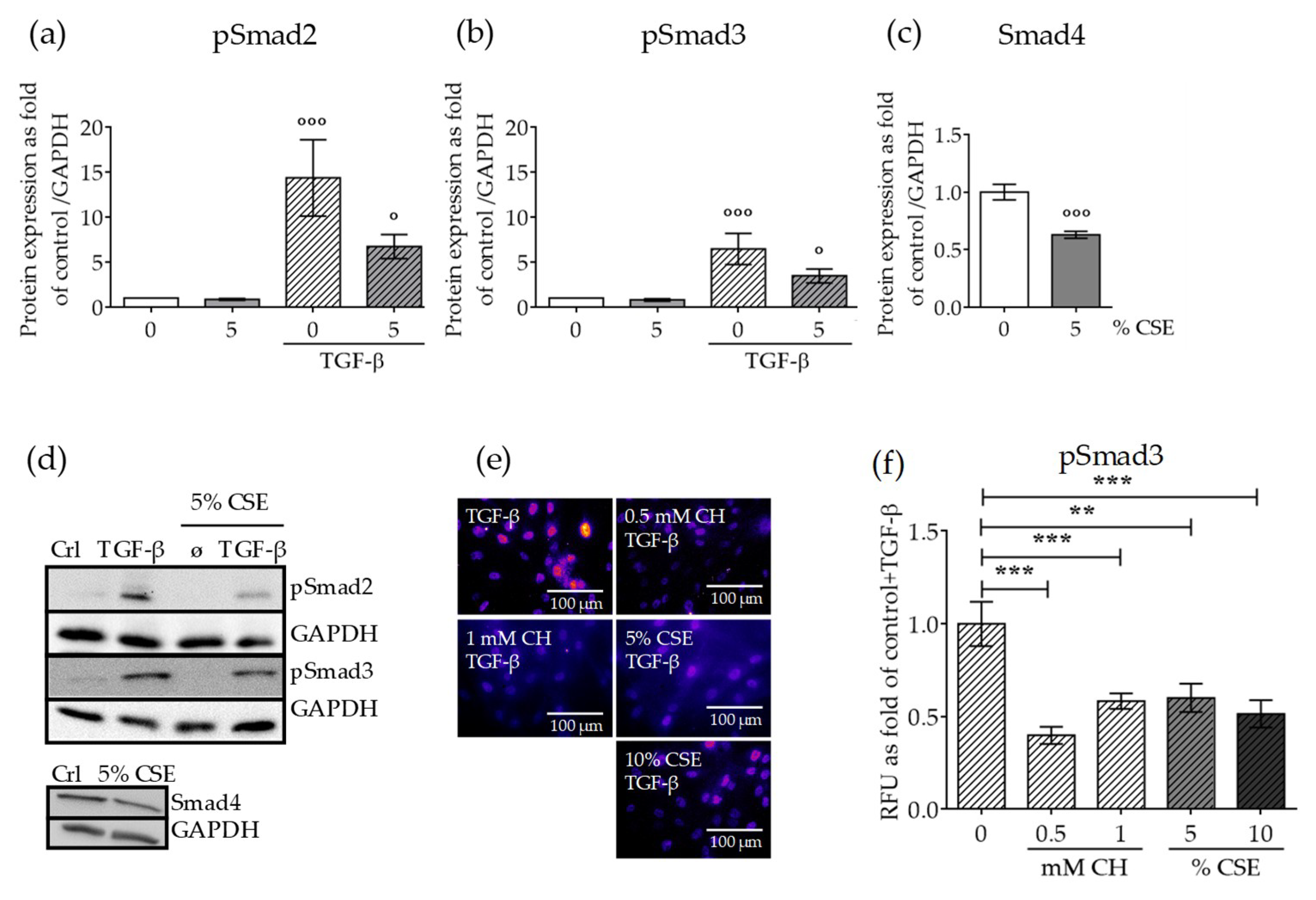
2.3. CSE Reduced the Levels of Downstream TGF-β Pathway Mediators and the Nuclear Translocation of the Active Complex
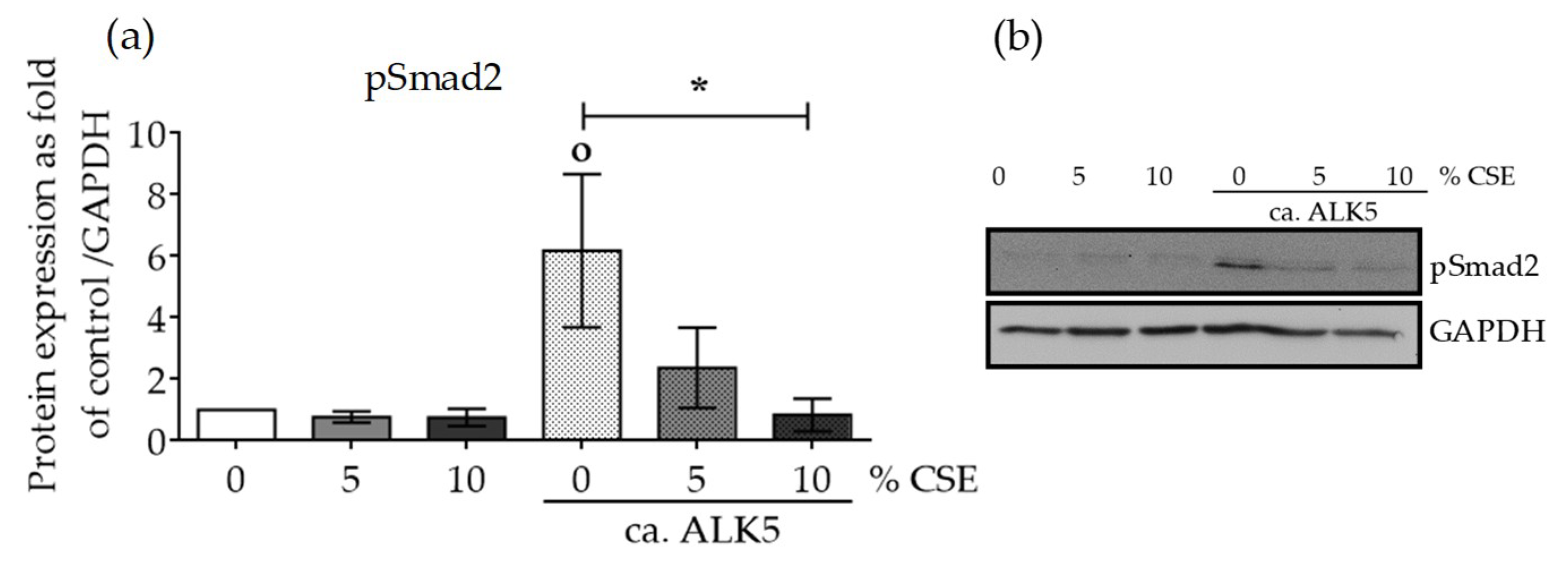
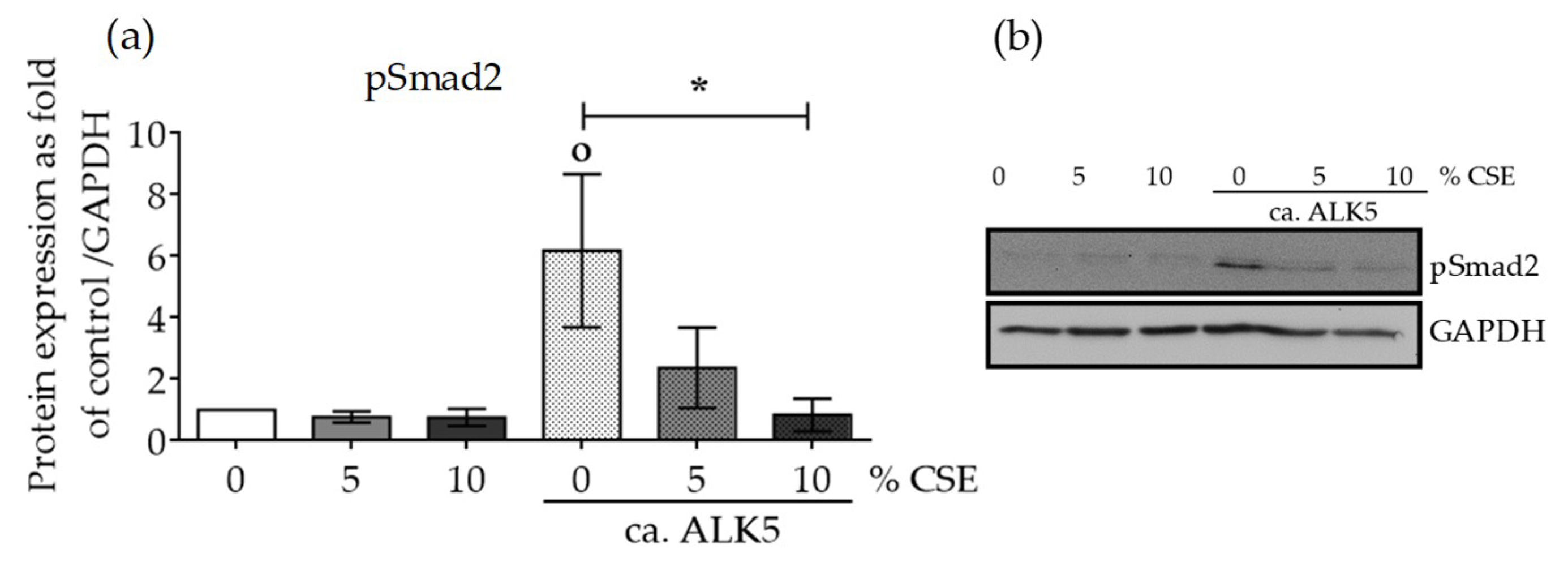
2.4. CSE Perturbed Normal TGF-β Receptor Type I Function
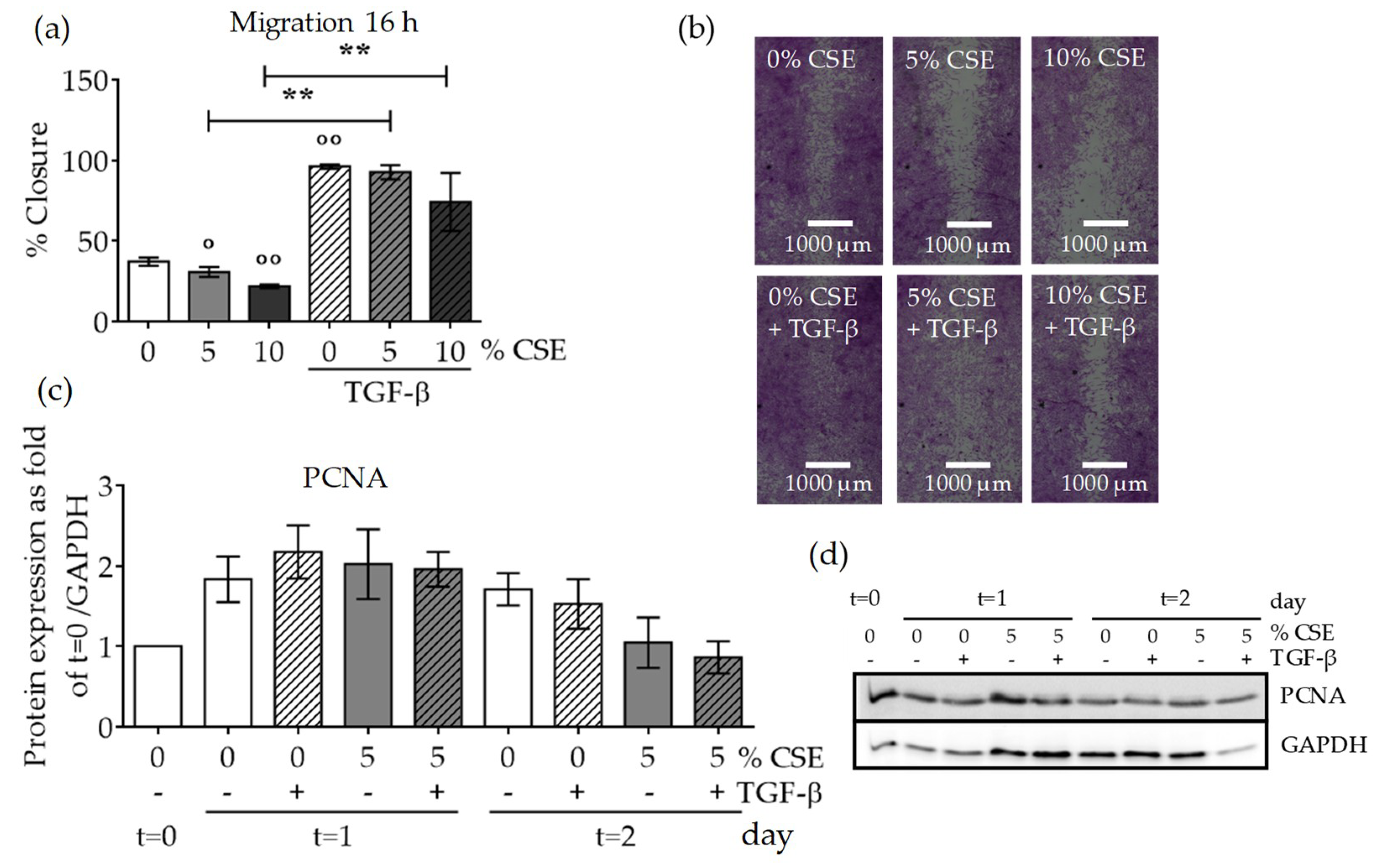
2.5. CSE Affected SCP-1 Cell Migration and Proliferation
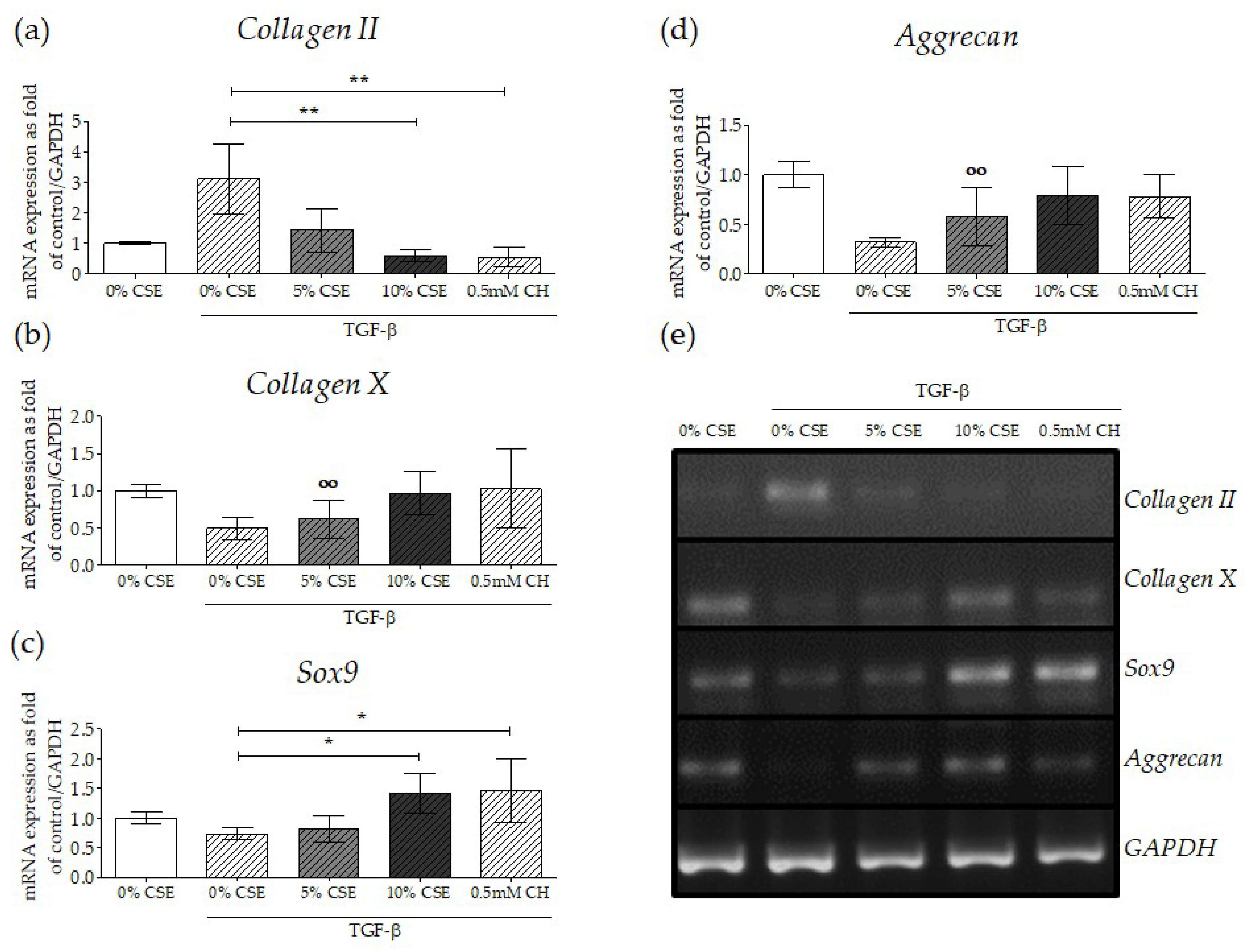
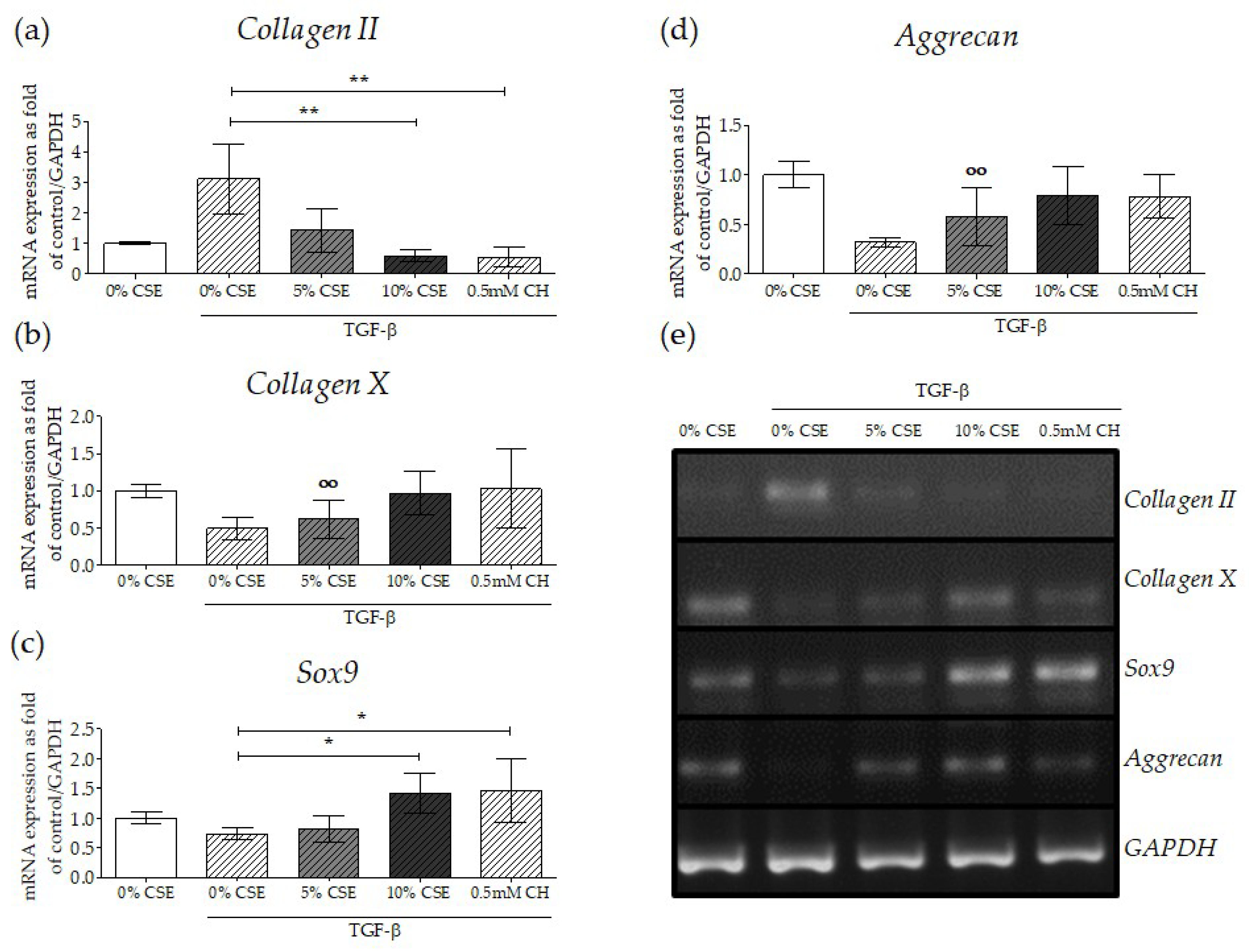
2.6. Impaired TGF-β Signaling by CSE Negatively Affected Osteochondral Progenitor Cell Differentiation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. SCP-1 Cells Culture and Chondrogenic Differentiation
4.3. Cigarette Smoke Extract Generation
4.4. Transient SCP-1 Cells Infections and Reporter Assay
4.5. Inmunofluorescence Staining
4.6. Western Blot Analysis
4.7. SCP-1 Cells Migration Assay—Scratch Assay
4.8. Semi-Quantitative Reverse-Transcription Polymerase Chain Reaction RT-PCR
4.9. Statistic Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALK5 | TGF-β type I receptor kinase |
| BSA | Bovine serum albumin |
| CAGA | Promotor region of TGF-β target genes |
| CH | Chloral Hydrate |
| CS | Cigarette smoke |
| CSE | Cigarette smoke extract |
| ECL | Electrogenerated chemiluminescence |
| FBS | Fetal bovine serum |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GFP | Green fluorescent protein |
| IFT | Intraflagellar transport system |
| MSCs | Mesenchymal stem cells |
| NUP | Nucleoporin protein |
| PBS | Phosphate-buffered saline |
| PCNA | Proliferating cell nuclear antigen |
| RIPA | Radioimmunoprecipitation assay buffer |
| RLU | Relative fluorescent units |
| SARA | Smad anchor for receptor activation |
| SCP-1 | Single-cell-derived human mesenchymal stem cell line |
| SDS page | Sodium dodecyl sulfate–polyacrylamide gel |
| SRB | Sulforhodamine B |
| TBS-T | Tris-buffered Saline-Tween |
| TGF-β1 | Transforming growth factor β1 |
Appendix A
References
- Abate, M.; Vanni, D.; Pantalone, A.; Salini, V. Cigarette smoking and musculoskeletal disorders. Muscles Ligaments Tendons J. 2013, 3, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.M. Tobacco-related diseases. Semin. Oncol. Nurs. 2003, 19, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Kanis, J.A.; Johnell, O.; Oden, A.; Johansson, H.; De Laet, C.; Eisman, J.A.; Fujiwara, S.; Kroger, H.; McCloskey, E.V.; Mellstrom, D.; et al. Smoking and fracture risk: A meta-analysis. Osteoporos. Int. 2005, 16, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Yoon, V.; Maalouf, N.M.; Sakhaee, K. The effects of smoking on bone metabolism. Osteoporos. Int. 2012, 23, 2081–2092. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.D.; Klesges, R.C. A meta-analysis of the effects of cigarette smoking on bone mineral density. Calcif. Tissue Int. 2001, 68, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.H.; Chan, R.L.; Siu, W.S.; Shum, W.T.; Leung, P.C.; Zhang, L.; Cho, C.H. Deteriorating effect on bone metabolism and microstructure by passive cigarette smoking through dual actions on osteoblast and osteoclast. Calcif. Tissue Int. 2015, 96, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Mäkitie, R.E.; Costantini, A.; Kämpe, A.; Alm, J.J.; Mäkitie, O. New insights into monogenic causes of osteoporosis. Front. Endocrinol. 2019, 10, 70. [Google Scholar] [CrossRef]
- Even Dar, R.; Mazor, Y.; Karban, A.; Ish-Shalom, S.; Segal, E. Risk factors for low bone density in inflammatory bowel disease: Use of glucocorticoids, low body mass index, and smoking. Dig. Dis. 2019, 37, 284–290. [Google Scholar] [CrossRef]
- Feng, X.; McDonald, J.M. Disorders of bone remodeling. Annu. Rev. Pathol. 2011, 6, 121–145. [Google Scholar] [CrossRef]
- Greenblatt, M.B.; Tsai, J.N.; Wein, M.N. Bone turnover markers in the diagnosis and monitoring of metabolic bone disease. Clin. Chem. 2017, 63, 464–474. [Google Scholar] [CrossRef]
- Centrella, M.; McCarthy, T.L.; Canalis, E. Skeletal tissue and transforming growth factor beta. FASEB J. 1988, 2, 3066–3073. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F.; Mundy, G.R. Role of transforming growth factor-beta in bone remodeling. Clin. Orthop. Relat. Res. 1990, 261–276. [Google Scholar] [CrossRef]
- Poniatowski, L.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming growth factor beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Med. Inflam. 2015, 2015, 137823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-smad pathways in tgf-β signaling. Cell Res. 2008, 19, 128. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.; Henle, P.; Kusswetter, M.; Moghaddam, A.; Wentzensen, A.; Richter, W.; Weiss, S. Tgf-beta1 as a marker of delayed fracture healing. Bone 2005, 36, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Li, R.D.; Deng, Z.L.; Hu, N.; Liang, X.; Liu, B.; Luo, J.; Chen, L.; Yin, L.; Luo, X.; Shui, W.; et al. Biphasic effects of tgfbeta1 on bmp9-induced osteogenic differentiation of mesenchymal stem cells. BMB Rep. 2012, 45, 509–514. [Google Scholar] [CrossRef]
- Li, X.L.; Liu, Y.B.; Ma, E.G.; Shen, W.X.; Li, H.; Zhang, Y.N. Synergistic effect of bmp9 and tgf-beta in the proliferation and differentiation of osteoblasts. Genet. Mol. Res. GMR 2015, 14, 7605–7615. [Google Scholar] [CrossRef]
- Lindbæk, L.; Warzecha, C.B.; Koefoed, K.; Mogensen, J.B.; Schmid, F.; Pedersen, L.B.; Larsen, L.A.; Christensen, S. Coordination of tgfβ/bmp signaling is associated with the primary cilium. Cilia 2015, 4, 17. [Google Scholar] [CrossRef]
- Malone, A.M.; Anderson, C.T.; Tummala, P.; Kwon, R.Y.; Johnston, T.R.; Stearns, T.; Jacobs, C.R. Primary cilia mediate mechanosensing in bone cells by a calcium-independent mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 13325–13330. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Cao, X.; Yang, S. Ift80 is required for stem cell proliferation, differentiation, and odontoblast polarization during tooth development. Cell Death Dis. 2019, 10, 63. [Google Scholar] [CrossRef]
- Reilly, M.L.; Benmerah, A. Ciliary kinesins beyond ift: Cilium length, disassembly, cargo transport and signalling. Biol. Cell 2019, 111, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.T.; Morthorst, S.K.; Mogensen, J.B.; Pedersen, L.B. Primary cilia and coordination of receptor tyrosine kinase (rtk) and transforming growth factor beta (tgf-beta) signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028167. [Google Scholar] [CrossRef] [PubMed]
- Labour, M.-N.; Riffault, M.; Christensen, S.T.; Hoey, D.A. Tgfβ1-induced recruitment of human bone mesenchymal stem cells is mediated by the primary cilium in a smad3-dependent manner. Sci. Rep. 2016, 6, 35542. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, V.; Aspera-Werz, R.; Ehnert, S.; Strobel, J.; Tendulkar, G.; Heid, D.; Schreiner, A.; Arnscheidt, C.; Nuessler, A. Resveratrol protects primary cilia integrity of human mesenchymal stem cells from cigarette smoke to improve osteogenic differentiation in vitro. Arch. Toxicol. 2017, 92, 1525–1538. [Google Scholar] [CrossRef] [PubMed]
- Aspera-Werz, R.H.; Ehnert, S.; Heid, D.; Zhu, S.; Chen, T.; Braun, B.; Sreekumar, V.; Arnscheidt, C.; Nussler, A.K. Nicotine and cotinine inhibit catalase and glutathione reductase activity contributing to the impaired osteogenesis of scp-1 cells exposed to cigarette smoke. Oxid. Med. Cell. Longev. 2018, 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.F.; Ehnert, S.; Freude, T.; Egana, J.T.; Schenck, T.L.; Buchholz, A.; Schmitt, A.; Siebenlist, S.; Schyschka, L.; Neumaier, M.; et al. Quercetin protects primary human osteoblasts exposed to cigarette smoke through activation of the antioxidative enzymes ho-1 and sod-1. Sci. World J. 2011, 11, 2348–2357. [Google Scholar] [CrossRef]
- Ehnert, S.; Stefan, D.; Friedrich, B.K.; Britta, B.; Valeska, H.; Mario, H.; Tomas, E.J.; Ulrich, S.; Thomas, F.; Klaus, N.A. N-acetylcyteine and flavonoid rich diet: The protective effect of 15 different antioxidants on cigarette smoke-damaged primary human osteoblasts. Adv. Biosci. Biotechnol. 2012, 3, 1129–1139. [Google Scholar] [CrossRef] [Green Version]
- Holzer, N.; Braun, K.F.; Ehnert, S.; Egana, J.T.; Schenck, T.L.; Buchholz, A.; Schyschka, L.; Neumaier, M.; Benzing, S.; Stockle, U.; et al. Green tea protects human osteoblasts from cigarette smoke-induced injury: Possible clinical implication. Langenbeck’s Arch. Surg. Deutsch. Ges. Fur Chir. 2012, 397, 467–474. [Google Scholar] [CrossRef]
- Moghaddam, A.; Weiss, S.; Wolfl, C.G.; Schmeckenbecher, K.; Wentzensen, A.; Grutzner, P.A.; Zimmermann, G. Cigarette smoking decreases tgf-b1 serum concentrations after long bone fracture. Injury 2010, 41, 1020–1025. [Google Scholar] [CrossRef]
- Martin, A.R.; Villegas, I.; La Casa, C.; de la Lastra, C.A. Resveratrol, a polyphenol found in grapes, suppresses oxidative damage and stimulates apoptosis during early colonic inflammation in rats. Biochem. Pharmacol. 2004, 67, 1399–1410. [Google Scholar]
- Kurus, M.; Firat, Y.; Cetin, A.; Kelles, M.; Otlu, A. The effect of resveratrol in tracheal tissue of rats exposed to cigarette smoke. Inhal. Toxicol. 2009, 21, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Ehnert, S.; Linnemann, C.; Aspera-Werz, R.H.; Bykova, D.; Biermann, S.; Fecht, L.; De Zwart, P.M.; Nussler, A.K.; Stuby, F. Immune cell induced migration of osteoprogenitor cells is mediated by tgf-β dependent upregulation of nox4 and activation of focal adhesion kinase. Int. J. Mol. Sci. 2018, 19, 2239. [Google Scholar] [CrossRef] [PubMed]
- Bahney, C.S.; Hu, D.P.; Miclau, T.; Marcucio, R.S. The multifaceted role of the vasculature in endochondral fracture repair. Front. Endocrinol. 2015, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Wang, X.; Qu, Y.; Lin, Q.; Cheng, K.; Gao, M.; Ren, S.; Zhang, T.; Li, X. Tgf-β1-induced chondrogenesis of bone marrow mesenchymal stem cells is promoted by low-intensity pulsed ultrasound through the integrin-mtor signaling pathway. Stem Cell Res. Ther. 2017, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Tuli, R.; Tuli, S.; Nandi, S.; Huang, X.; Manner, P.A.; Hozack, W.J.; Danielson, K.G.; Hall, D.J.; Tuan, R.S. Transforming growth factor-beta-mediated chondrogenesis of human mesenchymal progenitor cells involves n-cadherin and mitogen-activated protein kinase and wnt signaling cross-talk. J. Biol. Chem. 2003, 278, 41227–41236. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kupcsik, L.; Yao, S.-J.; Alini, M.; Stoddart, M. Mechanical load modulates chondrogenesis of human mesenchymal stem cells through the TGF-β pathway. J. Cell. Mol. Med. 2009, 14, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, A.; Sato, K.; Makino, H.; Ikegami, H.; Takayama, S.; Toyama, Y.; Umezawa, A. Nicotine acts on growth plate chondrocytes to delay skeletal growth through the alpha7 neuronal nicotinic acetylcholine receptor. PLoS ONE 2008, 3, e3945. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.A.; Wilson, R.F.; Patel, P.A.; Palmer, R.M. The effect of smoking on bone healing: A systematic review. Bone Jt. Res. 2013, 2, 102–111. [Google Scholar] [CrossRef]
- Erlebacher, A.; Filvaroff, E.H.; Ye, J.Q.; Derynck, R. Osteoblastic responses to tgf-beta during bone remodeling. Mol. Biol. Cell 1998, 9, 1903–1918. [Google Scholar] [CrossRef]
- Robey, P.G.; Young, M.F.; Flanders, K.C.; Roche, N.S.; Kondaiah, P.; Reddi, A.H.; Termine, J.D.; Sporn, M.B.; Roberts, A.B. Osteoblasts synthesize and respond to transforming growth factor-type beta (tgf-beta) in vitro. J. Cell Biol. 1987, 105, 457–463. [Google Scholar] [CrossRef]
- Harris, S.E.; Bonewald, L.F.; Harris, M.A.; Sabatini, M.; Dallas, S.; Feng, J.Q.; Ghosh-Choudhury, N.; Wozney, J.; Mundy, G.R. Effects of transforming growth factor beta on bone nodule formation and expression of bone morphogenetic protein 2, osteocalcin, osteopontin, alkaline phosphatase, and type i collagen mrna in long-term cultures of fetal rat calvarial osteoblasts. J. Bone Miner. Res. 1994, 9, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Blumenfeld, I.; Srouji, S.; Lanir, Y.; Laufer, D.; Livne, E. Enhancement of bone defect healing in old rats by tgf-β and igf-1. Exp. Gerontol. 2002, 37, 553–565. [Google Scholar] [CrossRef]
- Veland, I.R.; Awan, A.; Pedersen, L.B.; Yoder, B.K.; Christensen, S.T. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 2009, 111, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Pietenpol, J.A.; Moses, H.L. A tale of two proteins: Differential roles and regulation of smad2 and smad3 in tgf-beta signaling. J. Cell. Biochem. 2007, 101, 9–33. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Weng, T.; Zhang, J.; Wang, J.; Li, W.; Wan, H.; Lan, Y.; Cheng, X.; Hou, N.; Liu, H.; et al. Smad4 is required for maintaining normal murine postnatal bone homeostasis. J. Cell Sci. 2007, 120, 2162–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.F.; Cheng, S.L. Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-beta in normal human osteoblastic cells. J. Biol. Chem. 2002, 277, 15514–15522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tan, X.; Li, W.; Wang, Y.; Wang, J.; Cheng, X.; Yang, X. Smad4 is required for the normal organization of the cartilage growth plate. Dev. Biol. 2005, 284, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Kurisaki, A.; Kose, S.; Yoneda, Y.; Heldin, C.H.; Moustakas, A. Transforming growth factor-beta induces nuclear import of smad3 in an importin-beta1 and ran-dependent manner. Mol. Biol. Cell 2001, 12, 1079–1091. [Google Scholar] [CrossRef]
- Xiao, Z.; Liu, X.; Lodish, H.F. Importin beta mediates nuclear translocation of smad 3. J. Biol. Chem. 2000, 275, 23425–23428. [Google Scholar] [CrossRef]
- Xu, L.; Alarcon, C.; Col, S.; Massague, J. Distinct domain utilization by smad3 and smad4 for nucleoporin interaction and nuclear import. J. Biol. Chem. 2003, 278, 42569–42577. [Google Scholar] [CrossRef]
- Xu, L.; Kang, Y.; Col, S.; Massague, J. Smad2 nucleocytoplasmic shuttling by nucleoporins can/nup214 and nup153 feeds tgfbeta signaling complexes in the cytoplasm and nucleus. Mol. Cell 2002, 10, 271–282. [Google Scholar] [CrossRef]
- Clement, C.A.; Ajbro, K.D.; Koefoed, K.; Vestergaard, M.L.; Veland, I.R.; Henriques de Jesus, M.P.; Pedersen, L.B.; Benmerah, A.; Andersen, C.Y.; Larsen, L.A.; et al. Tgf-beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 2013, 3, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Nucleocytoplasmic shuttling of smad proteins. Cell Res. 2009, 19, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Sivakumar, P.; Jones, C.J.; Chen, Q.; Peters, D.M.; Mosher, D.F.; Humphries, M.J.; Kielty, C.M. Fibronectin regulates latent transforming growth factor-beta (tgf beta) by controlling matrix assembly of latent tgf beta-binding protein-1. J. Biol. Chem. 2005, 280, 18871–18880. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massague, J. Cell adhesion protein receptors as targets for transforming growth factor-beta action. Cell 1987, 51, 189–197. [Google Scholar] [CrossRef]
- Noda, M.; Camilliere, J.J. In vivo stimulation of bone formation by transforming growth factor-beta. Endocrinology 1989, 124, 2991–2994. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. Tgf-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef]
- Ehnert, S.; Sreekumar, V.; Aspera-Werz, R.H.; Sajadian, S.O.; Wintermeyer, E.; Sandmann, G.H.; Bahrs, C.; Hengstler, J.G.; Godoy, P.; Nussler, A.K. Tgf-beta1 impairs mechanosensation of human osteoblasts via hdac6-mediated shortening and distortion of primary cilia. J. Mol. Med. 2017, 95, 653–663. [Google Scholar] [CrossRef]
- Veland, I.R.; Lindbaek, L.; Christensen, S.T. Linking the primary cilium to cell migration in tissue repair and brain development. Bioscience 2014, 64, 1115–1125. [Google Scholar] [CrossRef]
- Wahl, E.A.; Schenck, T.L.; Machens, H.G.; Egana, J.T. Acute stimulation of mesenchymal stem cells with cigarette smoke extract affects their migration, differentiation, and paracrine potential. Sci. Rep. 2016, 6, 22957. [Google Scholar] [CrossRef]
- Dangelo, M.; Sarment, D.P.; Billings, P.C.; Pacifici, M. Activation of transforming growth factor beta in chondrocytes undergoing endochondral ossification. J. Bone Miner. Res. 2001, 16, 2339–2347. [Google Scholar] [CrossRef]
- Einhorn, T.A.; Gerstenfeld, L.C. Fracture healing: Mechanisms and interventions. Nat. Rev. Rheumatol. 2015, 11, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.K.; Huang, L.; Cao, D.; Yip, Y.W.; Tsang, W.M.; Yam, G.H.; Pang, C.P.; Cheung, H.S. Cigarette smoking hinders human periodontal ligament-derived stem cell proliferation, migration and differentiation potentials. Sci. Rep. 2015, 5, 7828. [Google Scholar] [CrossRef] [PubMed]
- Mwale, F.; Stachura, D.; Roughley, P.; Antoniou, J. Limitations of using aggrecan and type x collagen as markers of chondrogenesis in mesenchymal stem cell differentiation. J. Orthop. Res. 2006, 24, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.; Li, J.; Gorski, D.J.; Bartels, A.K.; Shewman, E.F.; Wysocki, R.W.; Cole, B.J.; Bach, B.R., Jr.; Mikecz, K.; Sandy, J.D.; et al. Controlled treadmill exercise eliminates chondroid deposits and restores tensile properties in a new murine tendinopathy model. J. Biomech. 2013, 46, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Deren, M.E.; Yang, X.; Guan, Y.; Chen, Q. Biological and chemical removal of primary cilia affects mechanical activation of chondrogenesis markers in chondroprogenitors and hypertrophic chondrocytes. Int. J. Mol. Sci. 2016, 17, 188. [Google Scholar] [CrossRef]
- Abushahba, W.; Olabisi, O.O.; Jeong, B.-S.; Boregowda, R.K.; Wen, Y.; Liu, F.; Goydos, J.S.; Lasfar, A.; Cohen-Solal, K.A. Non-canonical smads phosphorylation induced by the glutamate release inhibitor, riluzole, through gsk3 activation in melanoma. PLoS ONE 2012, 7, e47312. [Google Scholar] [CrossRef]
- Bocker, W.; Yin, Z.; Drosse, I.; Haasters, F.; Rossmann, O.; Wierer, M.; Popov, C.; Locher, M.; Mutschler, W.; Docheva, D.; et al. Introducing a single-cell-derived human mesenchymal stem cell line expressing htert after lentiviral gene transfer. J. Cell. Mol. Med. 2008, 12, 1347–1359. [Google Scholar] [CrossRef]
- Ehnert, S.; Freude, T.; Ihle, C.; Mayer, L.; Braun, B.; Graeser, J.; Flesch, I.; Stockle, U.; Nussler, A.K.; Pscherer, S. Factors circulating in the blood of type 2 diabetes mellitus patients affect osteoblast maturation-description of a novel in vitro model. Exp. Cell Res. 2015, 332, 247–258. [Google Scholar] [CrossRef]
- Su, Y.; Han, W.; Giraldo, C.; De Li, Y.; Block, E.R. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am. J. Respir. Cell Mol. Biol. 1998, 19, 819–825. [Google Scholar] [CrossRef]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of smad3 and smad4 to critical tgf beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Suh, N.; Roberts, A.B.; Birkey Reffey, S.; Miyazono, K.; Itoh, S.; ten Dijke, P.; Heiss, E.H.; Place, A.E.; Risingsong, R.; Williams, C.R.; et al. Synthetic triterpenoids enhance transforming growth factor beta/smad signaling. Cancer Res. 2003, 63, 1371–1376. [Google Scholar] [PubMed]
- Ehnert, S.; Baur, J.; Schmitt, A.; Neumaier, M.; Lucke, M.; Dooley, S.; Vester, H.; Wildemann, B.; Stockle, U.; Nussler, A.K. Tgf-beta1 as possible link between loss of bone mineral density and chronic inflammation. PLoS ONE 2010, 5, e14073. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct tgf-beta type i receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Catalog No. | Company | Dilution |
|---|---|---|---|
| phospho-Smad2 | 3108 | Cell Signaling | 1:1000 |
| phospho-Smad3 | 9520 | Cell Signaling | 1:1000 |
| Smad4 | 9515 | Cell Signaling | 1:1000 |
| PCNA | ab92522 | Abcam | 1:1000 |
| HRP antirabbit IgG | sc-2004 | Santa Cruz | 1:10000 |
| Smad3 | 9523 | Cell Signaling | 1:50 |
| Alexa 488 antirabbit IgG | A21206 | Invitrogen | 1:1000 |
| Acetylated αTubulin (6-11b-1) | sc-23950 | Santa Cruz | 1:100 |
| Alexa 488 antimouse IgG | A10667 | Invitrogen | 1:1000 |
| Gene | Accession Number | Forward Primer (5′–3′) | Reverse Primer (3’–5’) | Product Length (bp) | Annealing Temperature (°C) | Cycles. (N°) |
|---|---|---|---|---|---|---|
| Aggrecan | NM_001135.3 | CTTGGACTTGGGCAAACTGC | CACTAAAGTCAGGCAGGCCA | 143 | 60 | 35 |
| Collagen II | NM_001844.4 | TGGATGCCACACTCAAGTCC | GCTGCTCCACCAGTTCTTCT | 254 | 60 | 35 |
| Collagen X | NM_000493.3 | AAACCTGGACAACAGGGACC | CGACCAGGAGCACCATATCC | 124 | 60 | 35 |
| SOX9 | NM_000346.3 | GAAGGACCACCCGGATTACA | GCCTTGAAGATGGCGTTGG | 120 | 60 | 35 |
| GAPDH | NM_002046.4 | GTCAGTGGTGGACCTGACCT | AGGGGTCTACATGGCAACTG | 420 | 56 | 30 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aspera-Werz, R.H.; Chen, T.; Ehnert, S.; Zhu, S.; Fröhlich, T.; Nussler, A.K. Cigarette Smoke Induces the Risk of Metabolic Bone Diseases: Transforming Growth Factor Beta Signaling Impairment via Dysfunctional Primary Cilia Affects Migration, Proliferation, and Differentiation of Human Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 2915. https://doi.org/10.3390/ijms20122915
Aspera-Werz RH, Chen T, Ehnert S, Zhu S, Fröhlich T, Nussler AK. Cigarette Smoke Induces the Risk of Metabolic Bone Diseases: Transforming Growth Factor Beta Signaling Impairment via Dysfunctional Primary Cilia Affects Migration, Proliferation, and Differentiation of Human Mesenchymal Stem Cells. International Journal of Molecular Sciences. 2019; 20(12):2915. https://doi.org/10.3390/ijms20122915
Chicago/Turabian StyleAspera-Werz, Romina H., Tao Chen, Sabrina Ehnert, Sheng Zhu, Theresa Fröhlich, and Andreas K. Nussler. 2019. "Cigarette Smoke Induces the Risk of Metabolic Bone Diseases: Transforming Growth Factor Beta Signaling Impairment via Dysfunctional Primary Cilia Affects Migration, Proliferation, and Differentiation of Human Mesenchymal Stem Cells" International Journal of Molecular Sciences 20, no. 12: 2915. https://doi.org/10.3390/ijms20122915
APA StyleAspera-Werz, R. H., Chen, T., Ehnert, S., Zhu, S., Fröhlich, T., & Nussler, A. K. (2019). Cigarette Smoke Induces the Risk of Metabolic Bone Diseases: Transforming Growth Factor Beta Signaling Impairment via Dysfunctional Primary Cilia Affects Migration, Proliferation, and Differentiation of Human Mesenchymal Stem Cells. International Journal of Molecular Sciences, 20(12), 2915. https://doi.org/10.3390/ijms20122915