o-Vanillin Derived Schiff Bases and Their Organotin(IV) Compounds: Synthesis, Structural Characterisation, In-Silico Studies and Cytotoxicity

 ,
,  ,
, 
 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
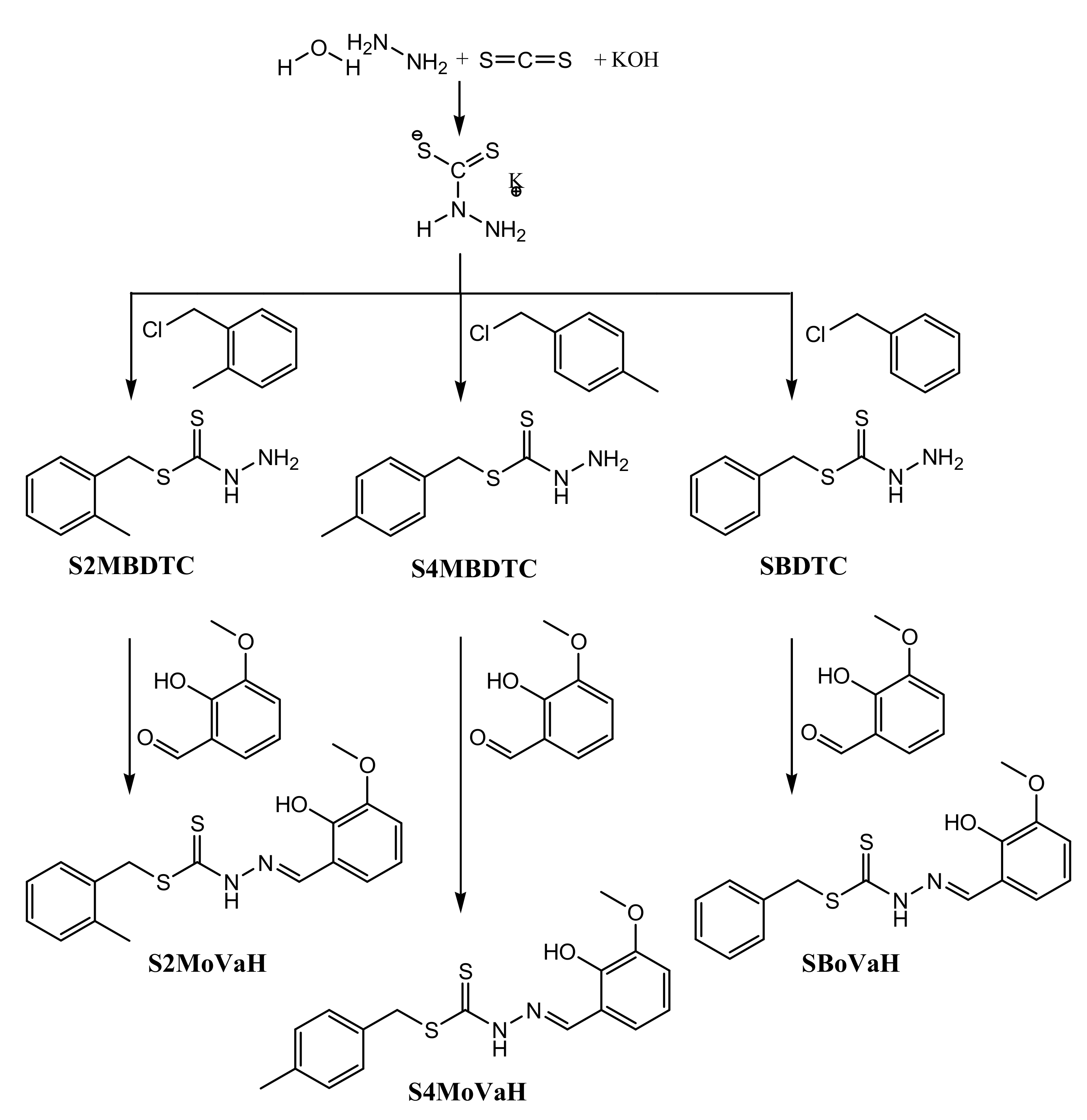
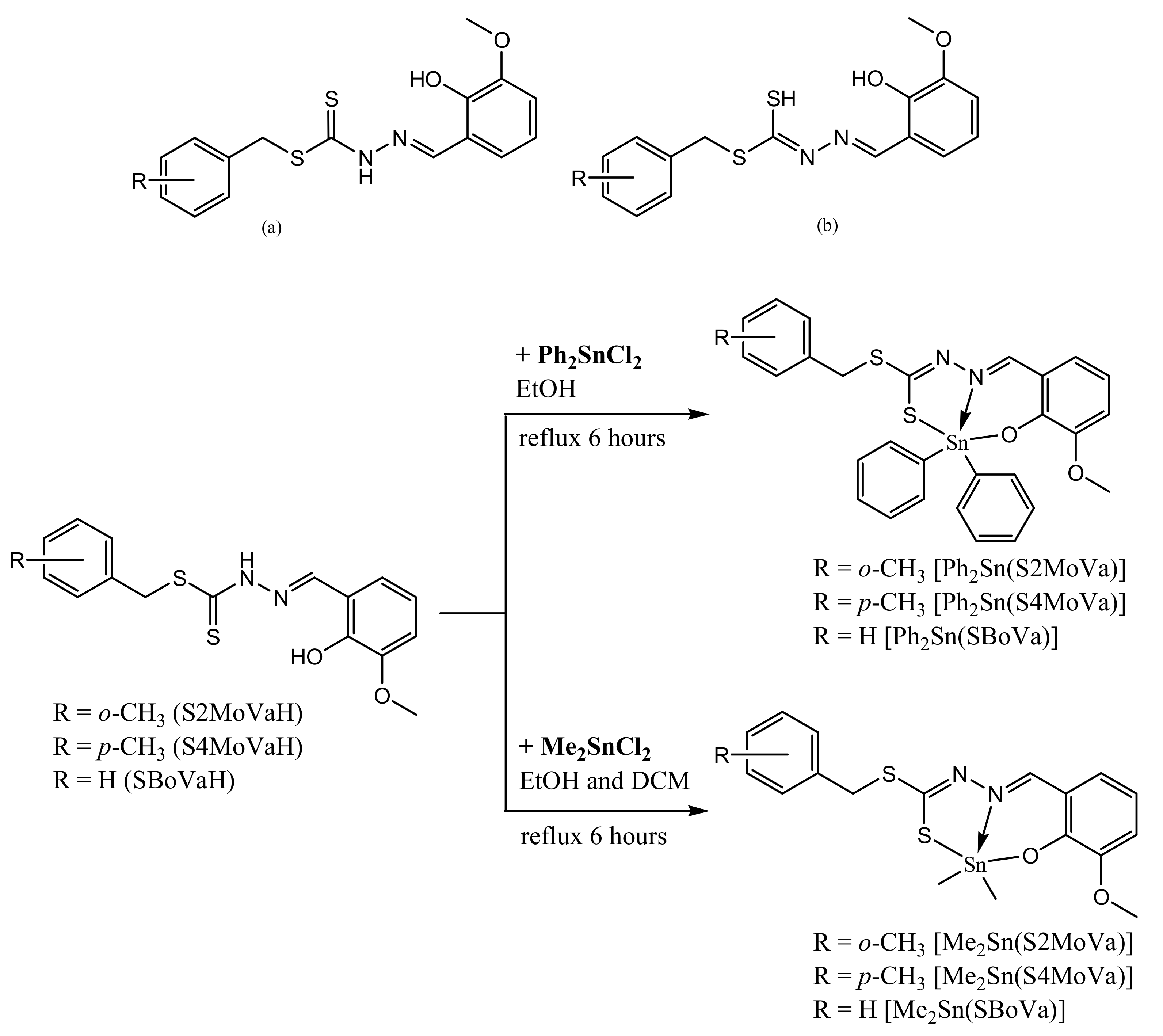
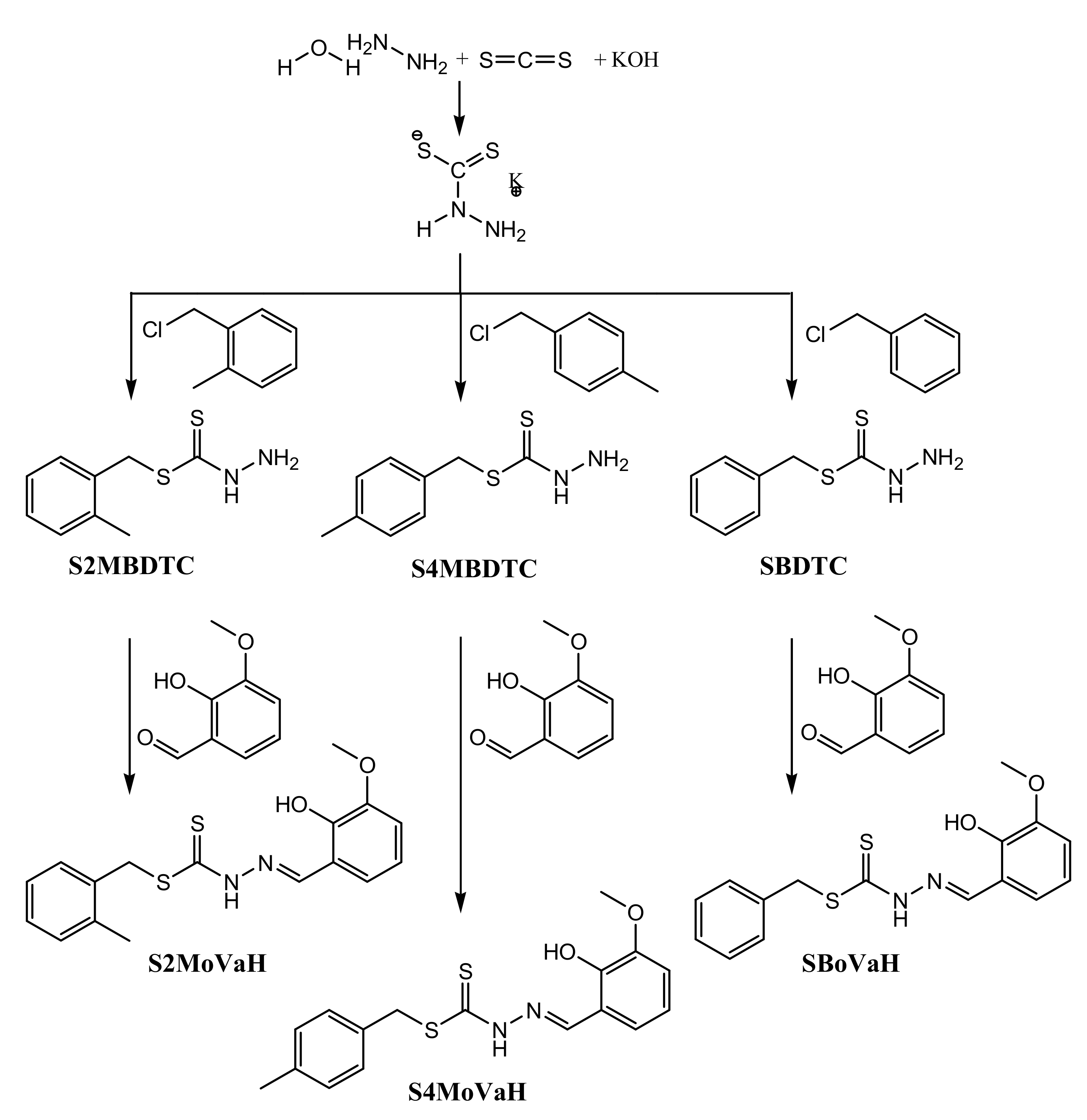
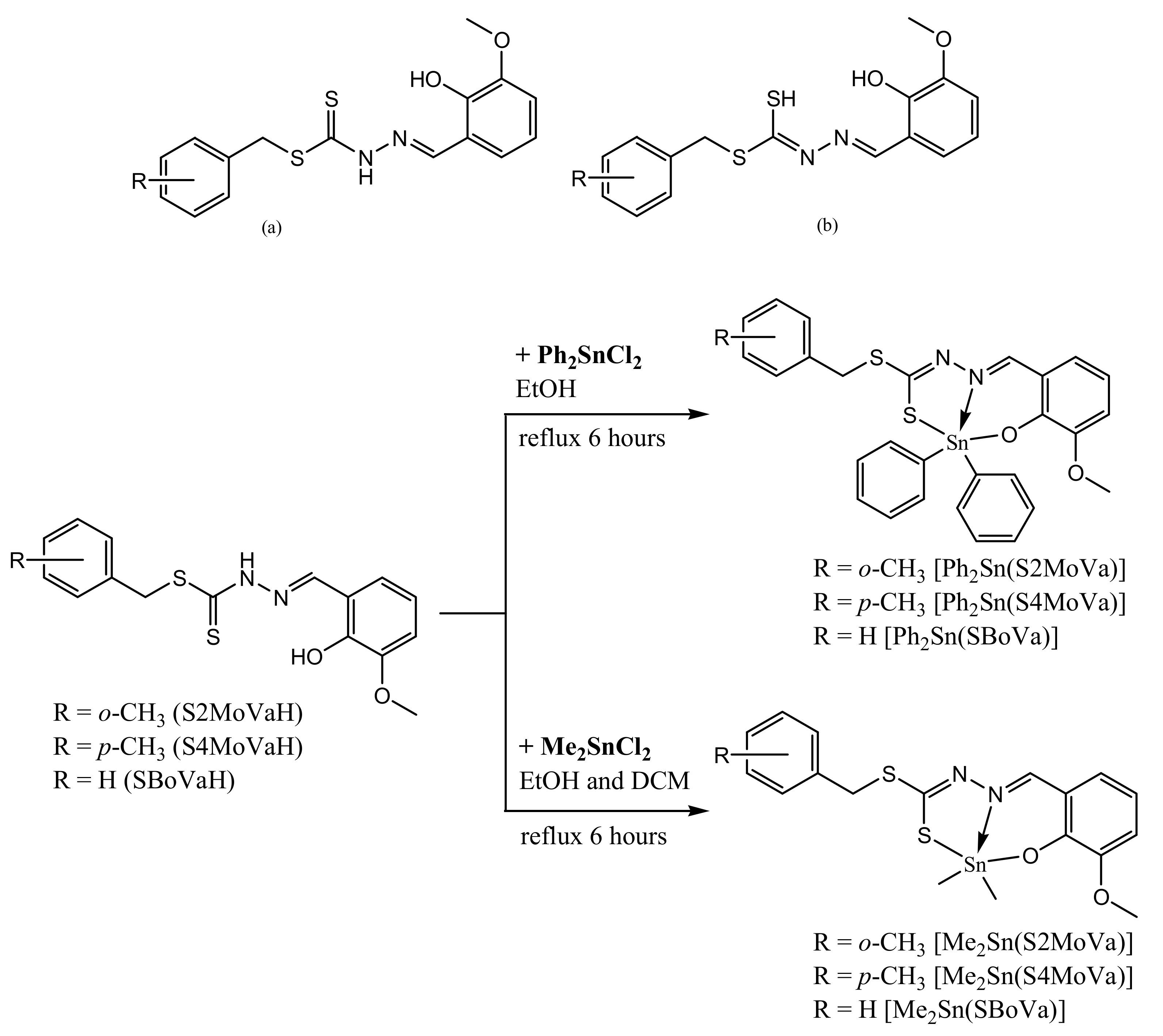
2.1. Synthesis
2.2. IR Spectral Analysis
2.3. NMR Spectroscopic Analysis
2.4. Mass Spectral Analysis
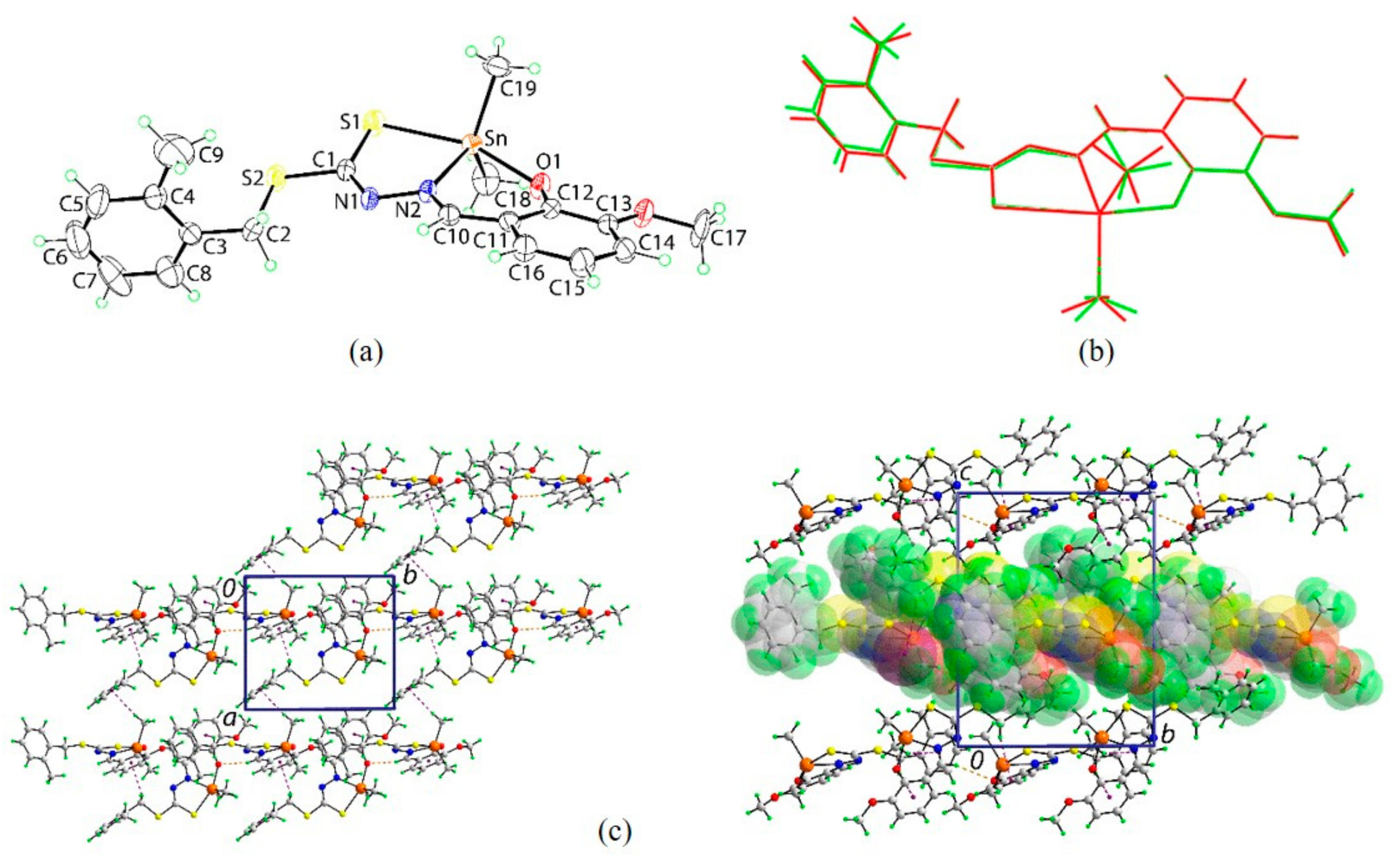
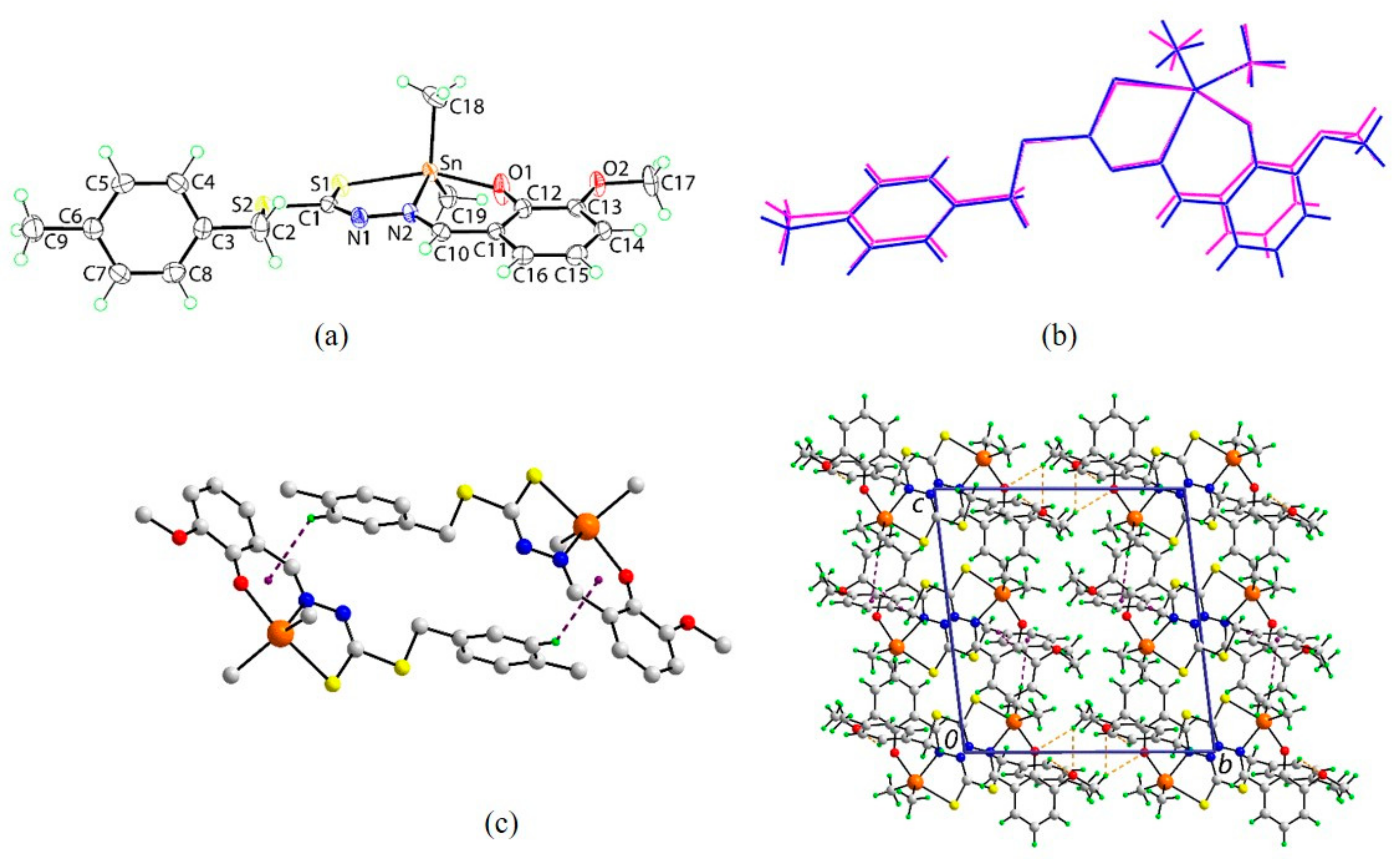
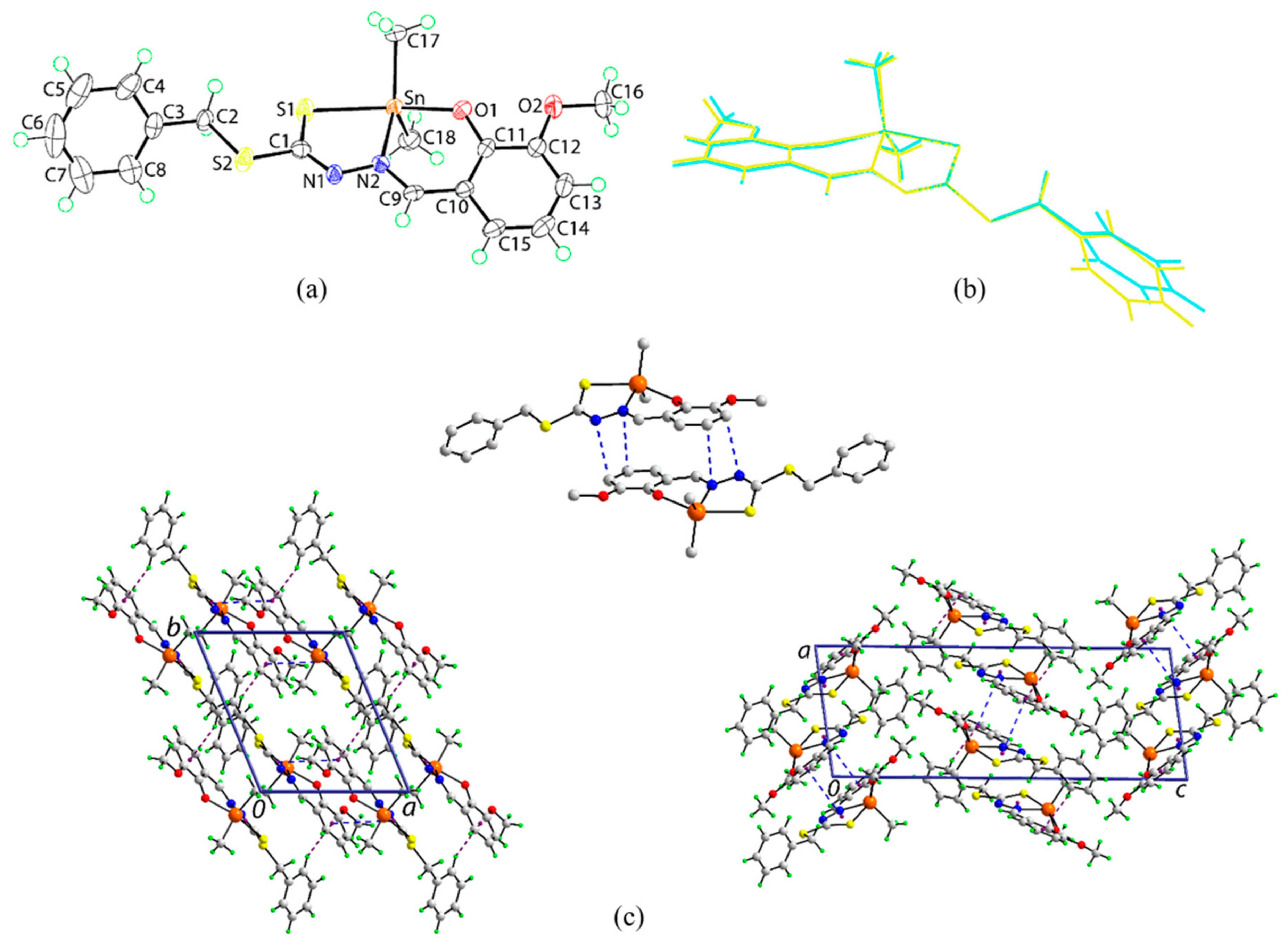
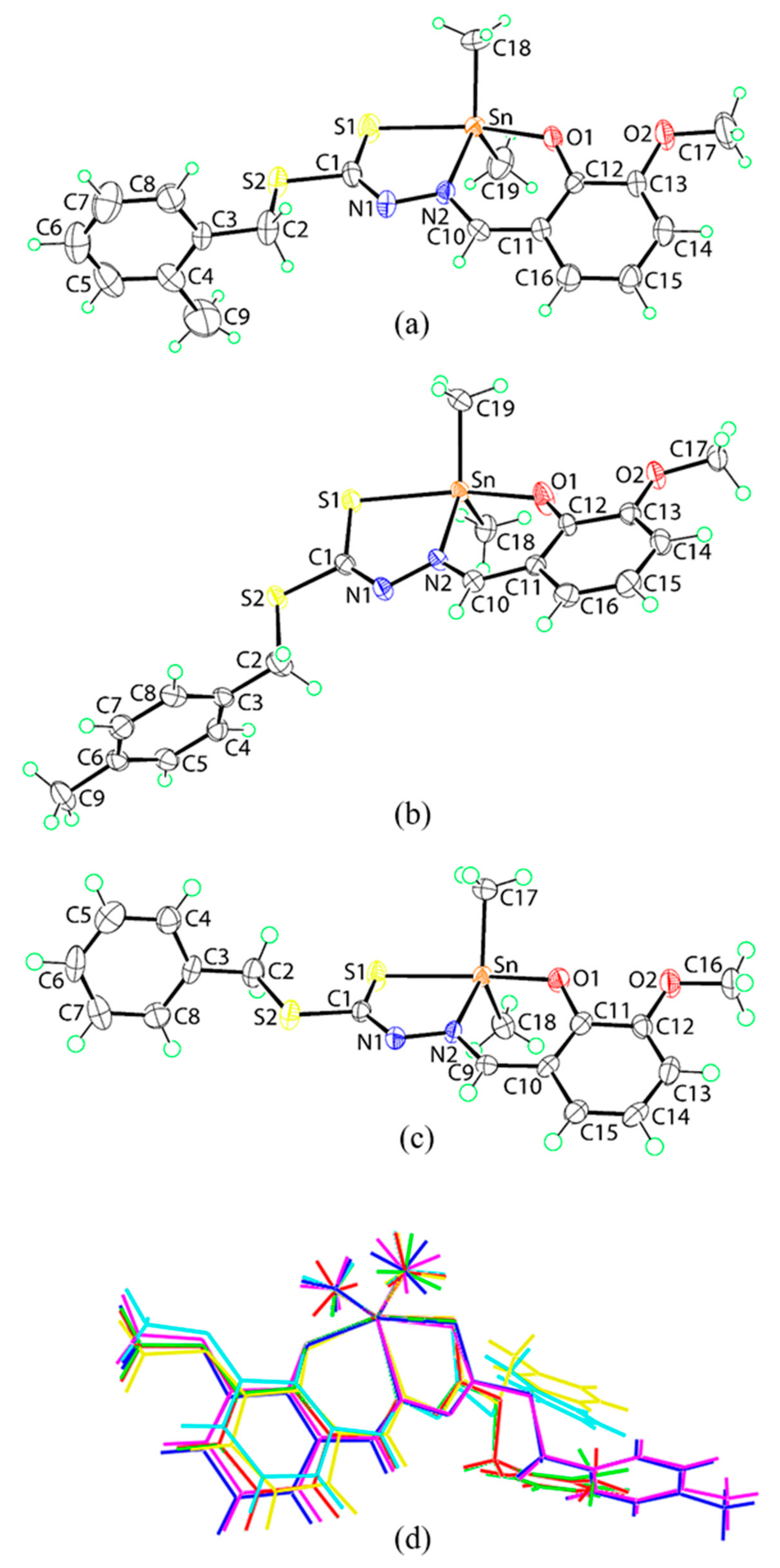
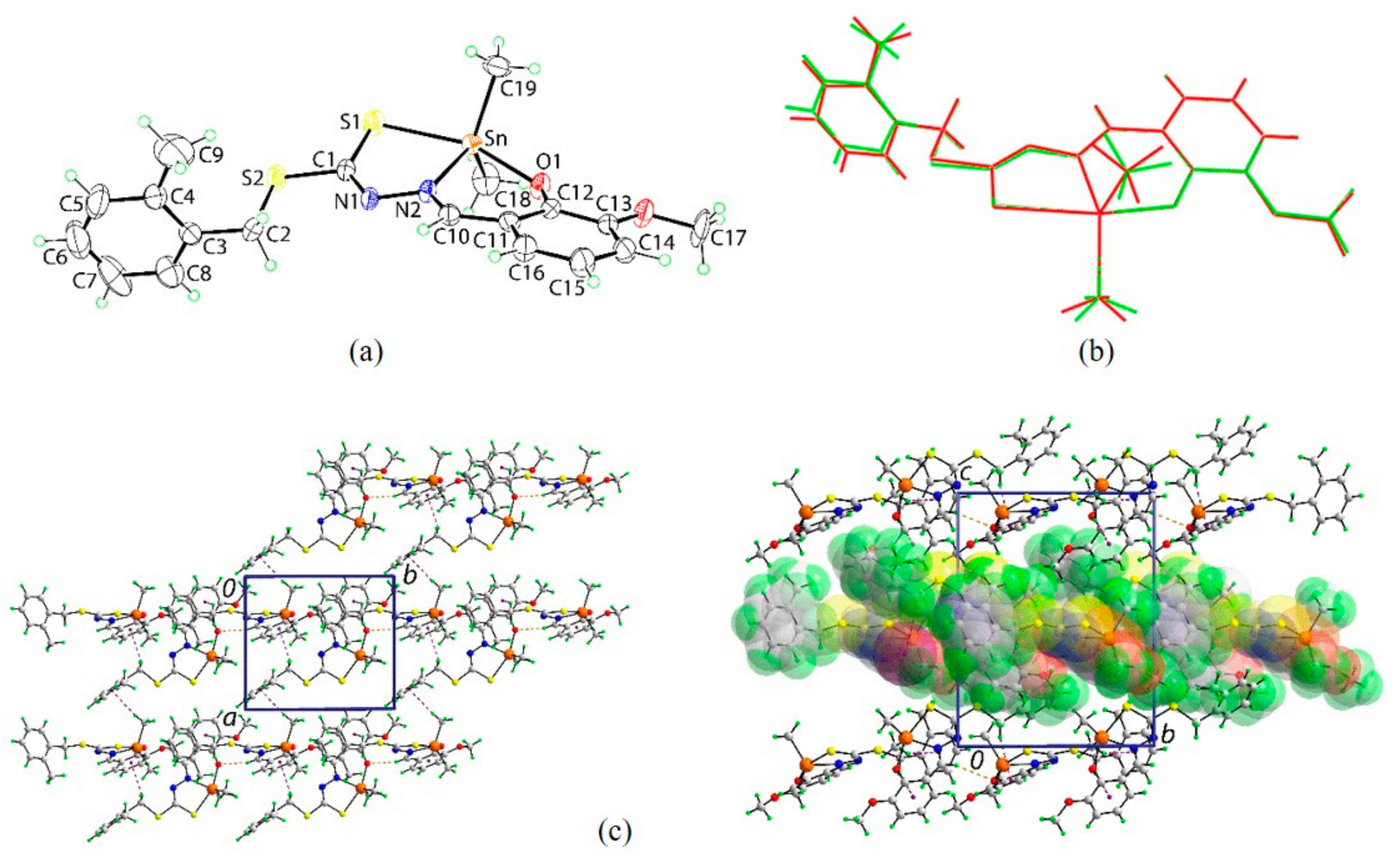
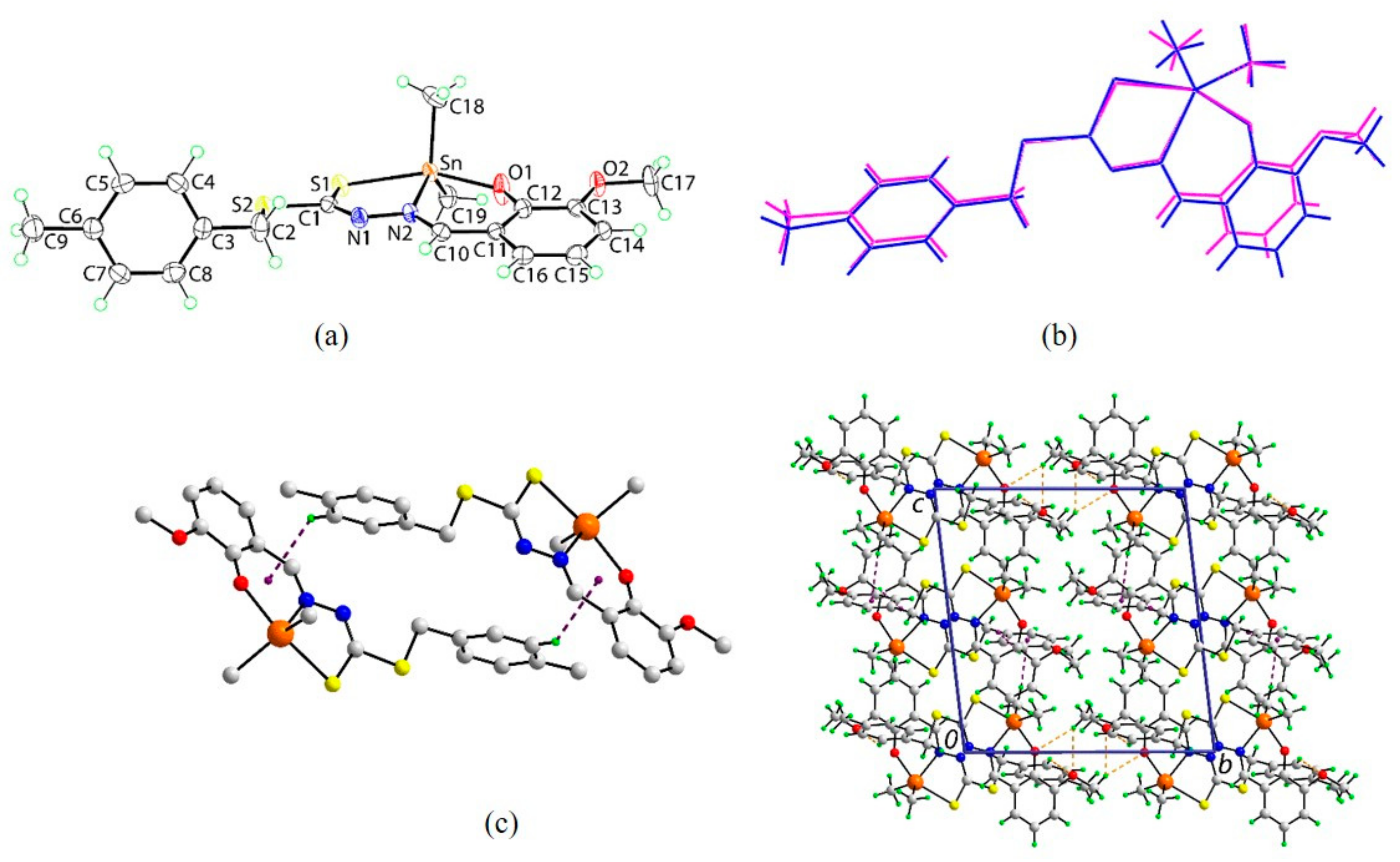
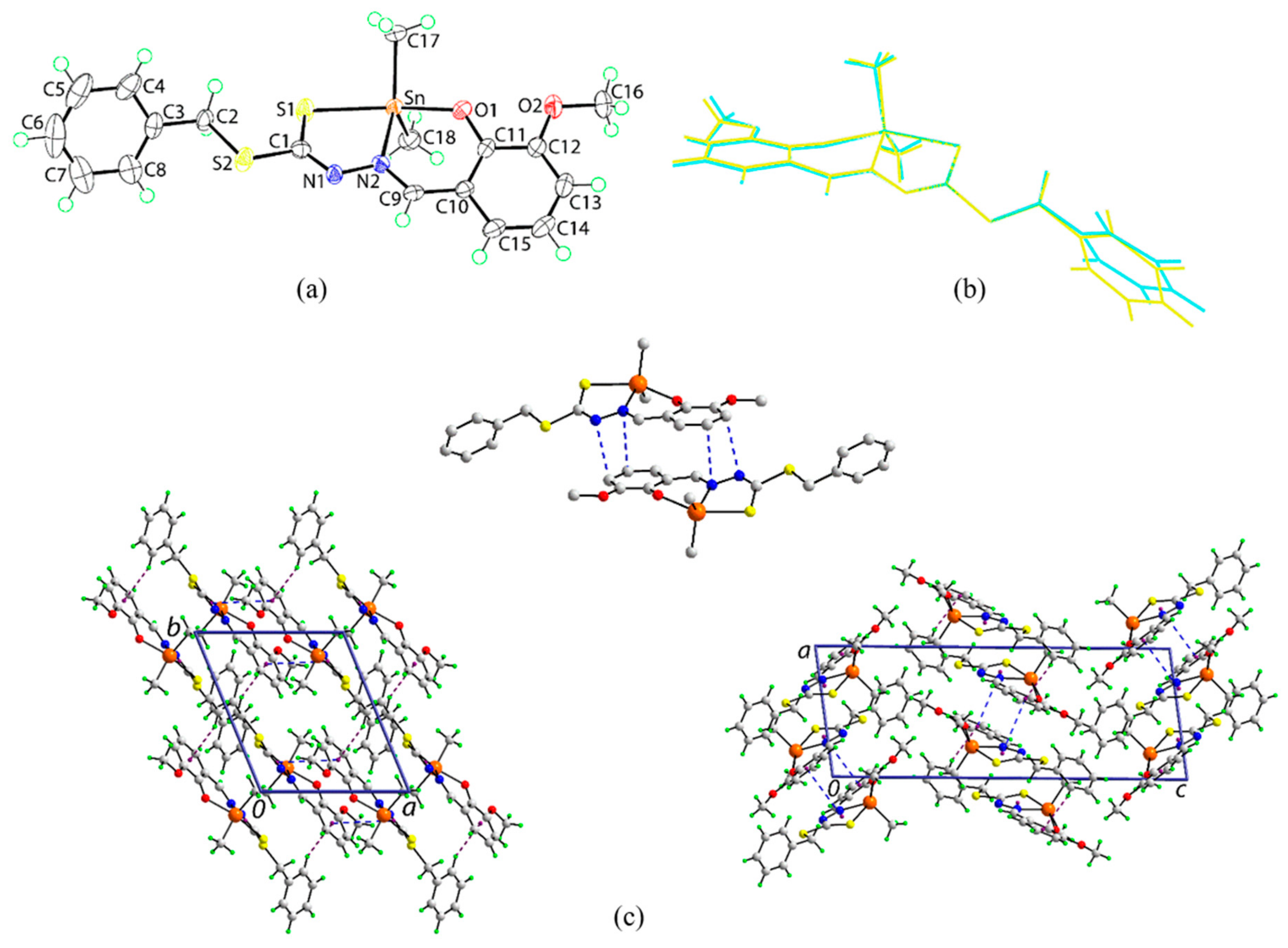
2.5. X-ray Crystallography
2.6. UV-Vis Absorption Spectroscopy
2.7. Biological Assays
2.7.1. Cytotoxicity
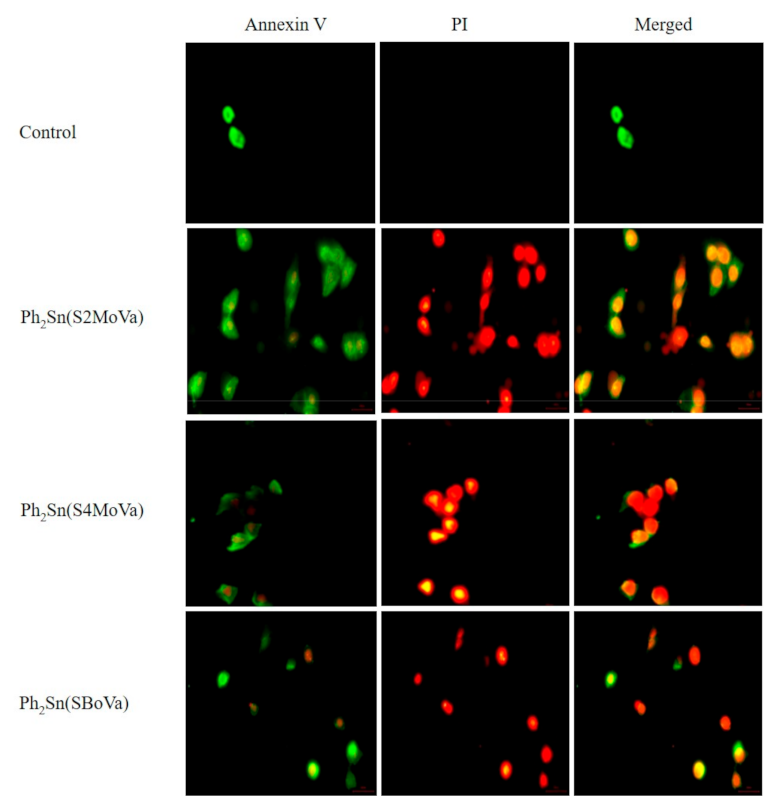
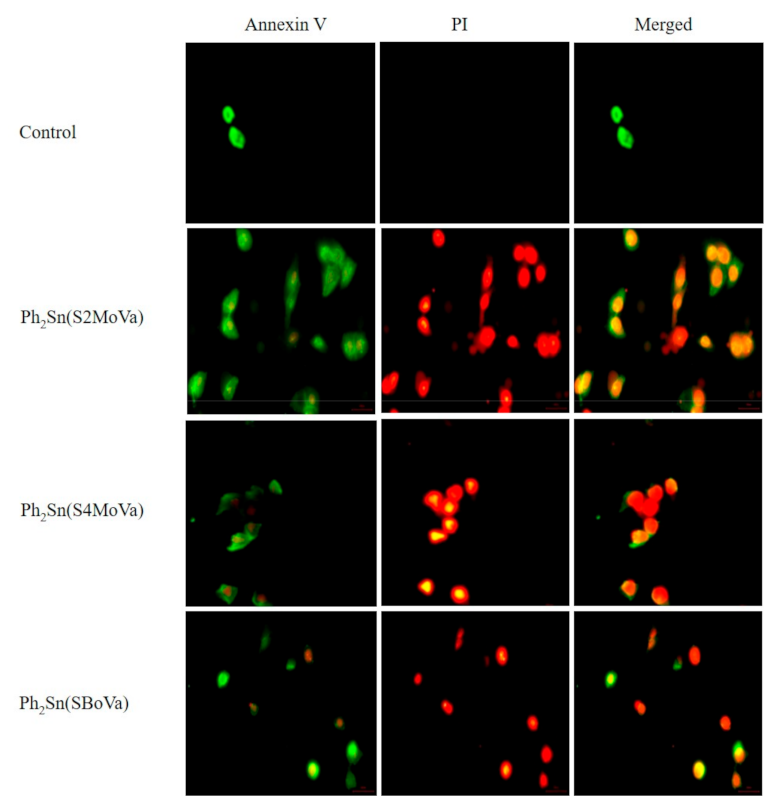
2.7.2. Annexin V Assays of Ph2Sn(S2MoVa)Ph2, Ph2Sn(S4MoVa), and Ph2Sn(SBoVa)-Treated RT-112 Cells
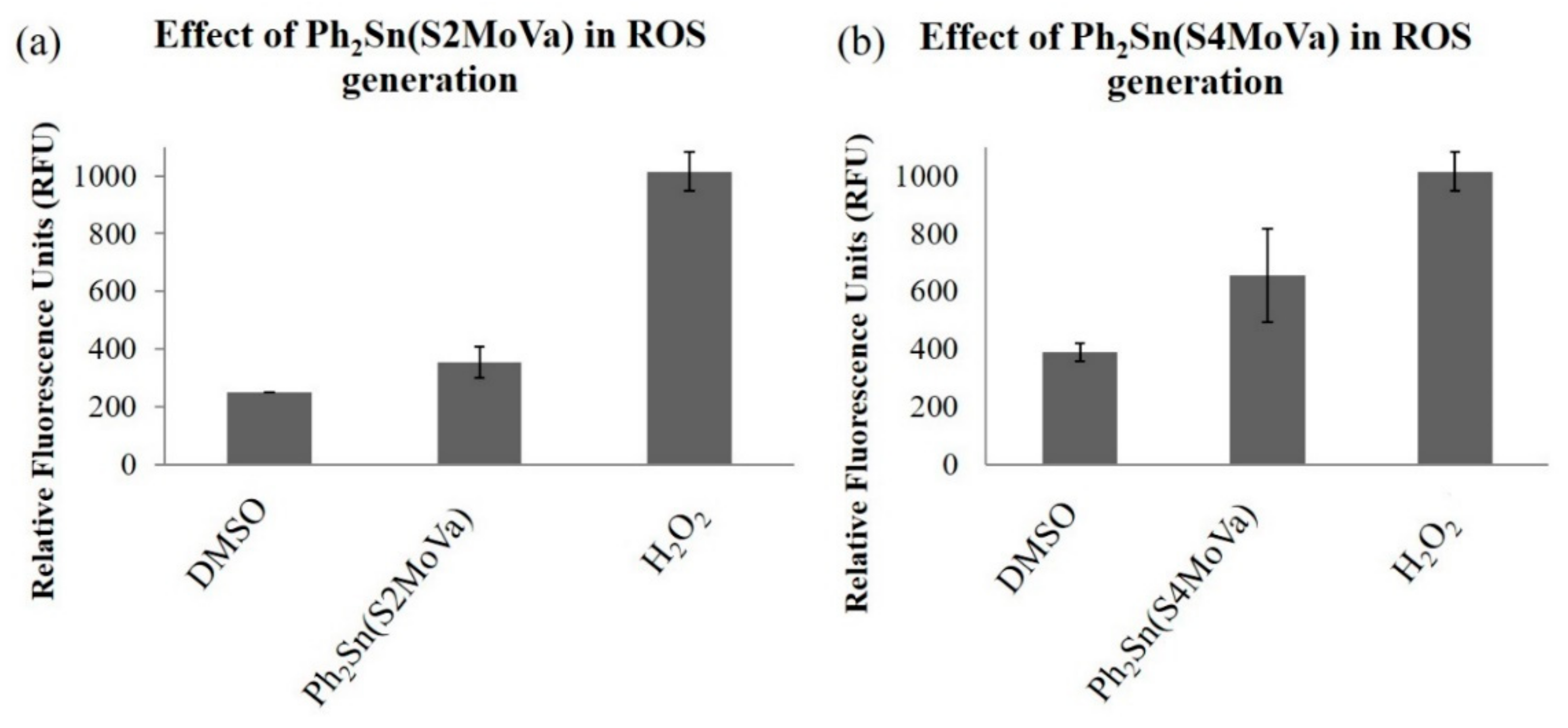
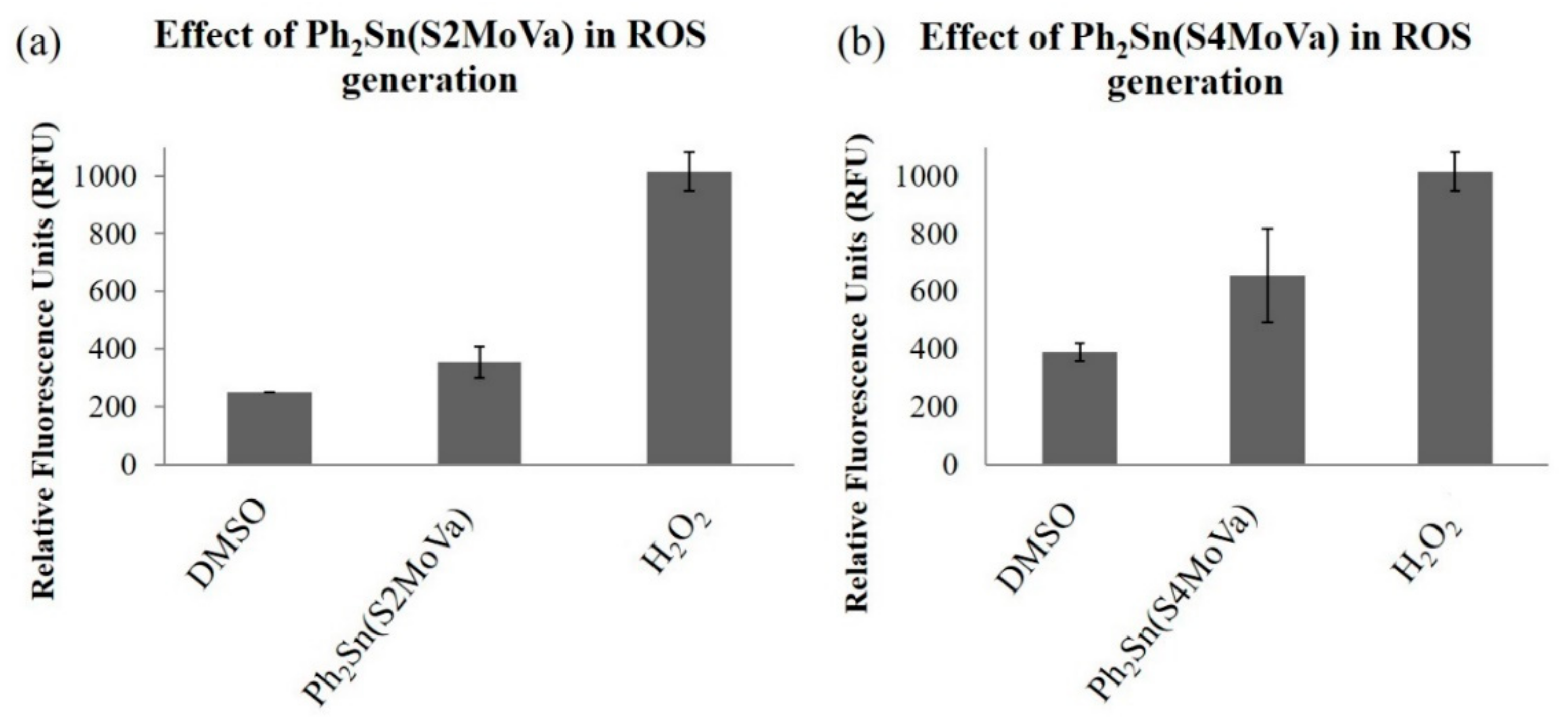
2.7.3. Measurement of Reactive Oxygen Species (ROS) in RT-112 Cells Treated with Ph2Sn(S2MoVa) and Ph2Sn(S4MoVa)
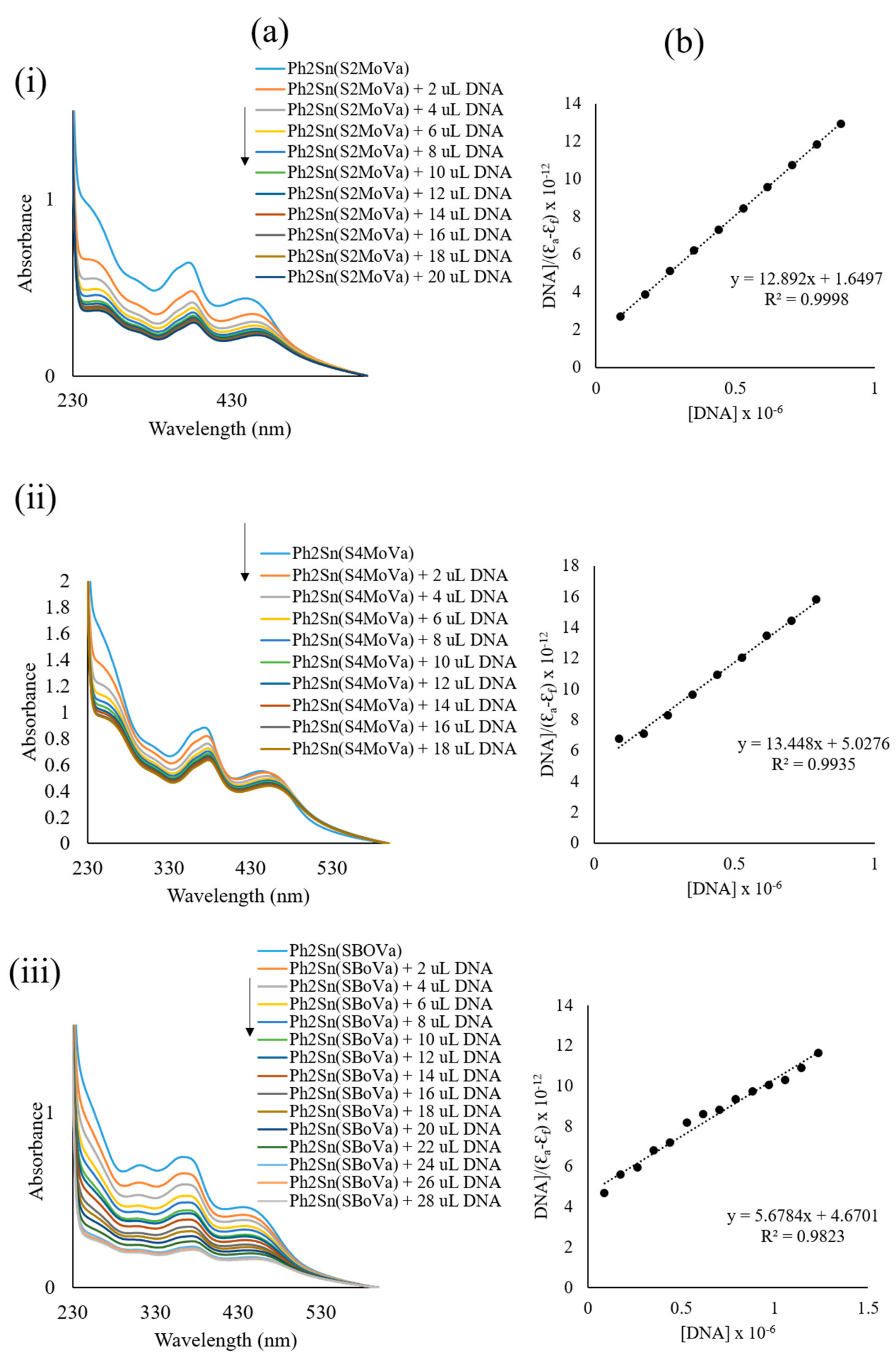
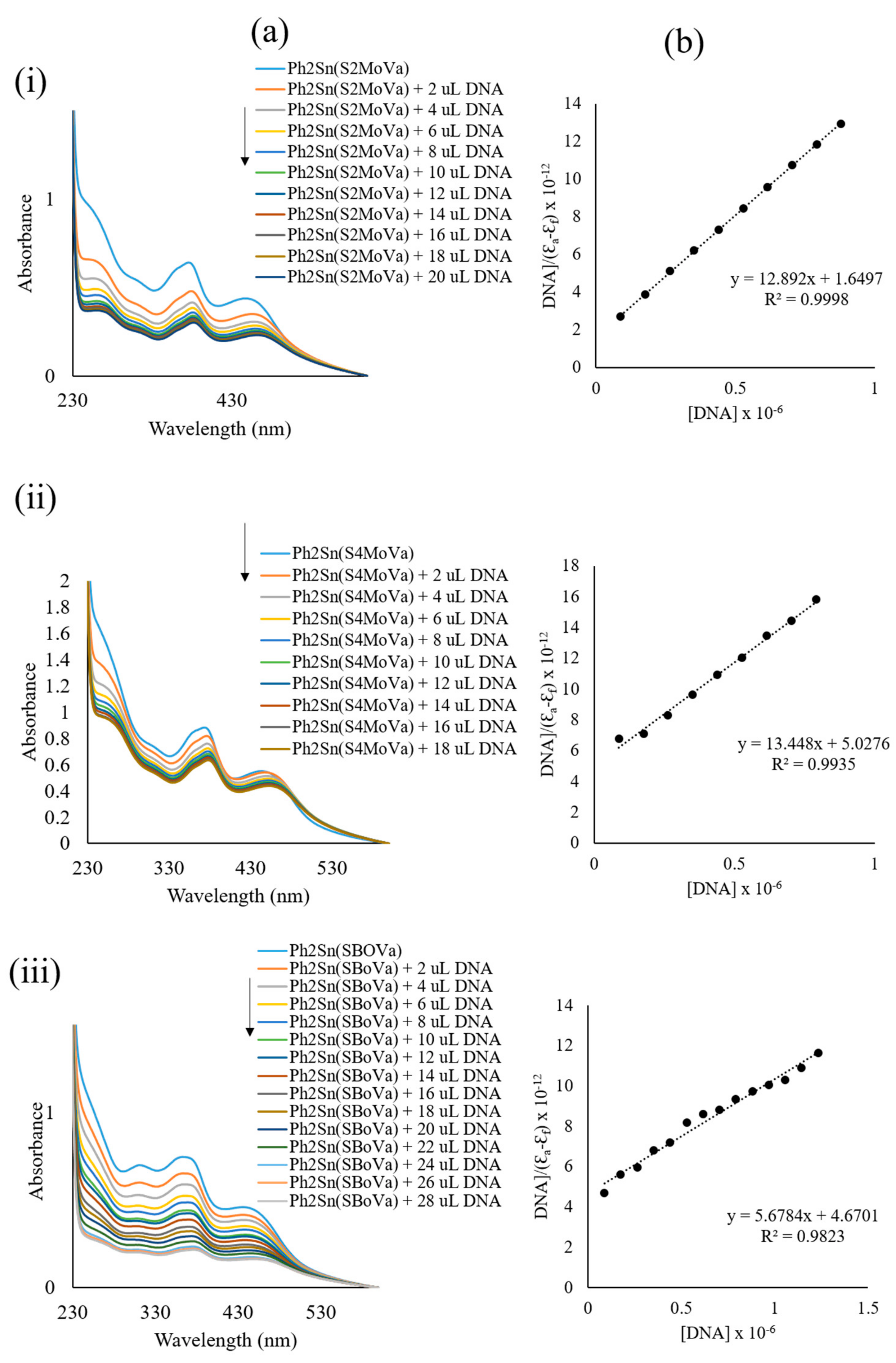
2.7.4. DNA Interaction Studies
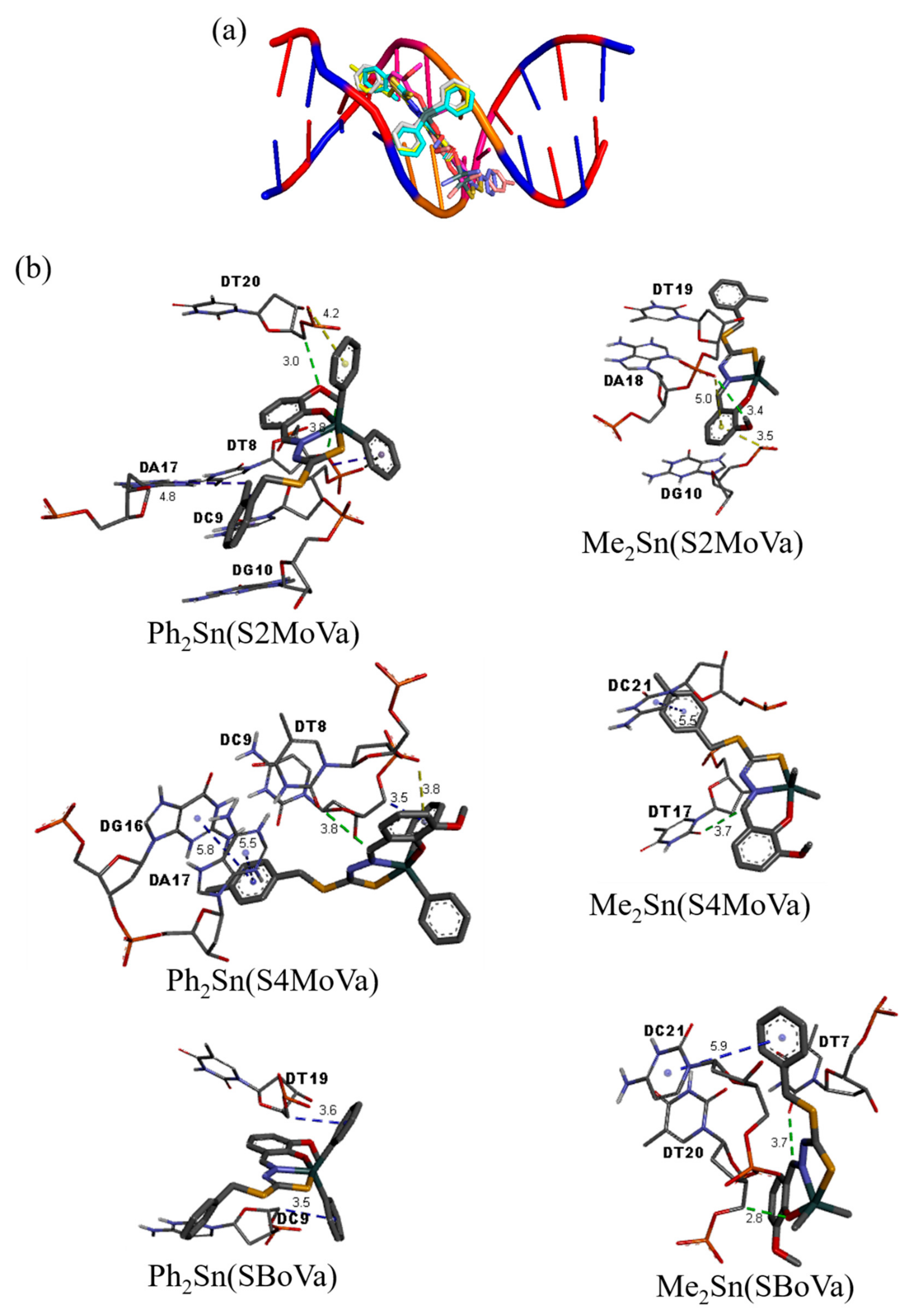
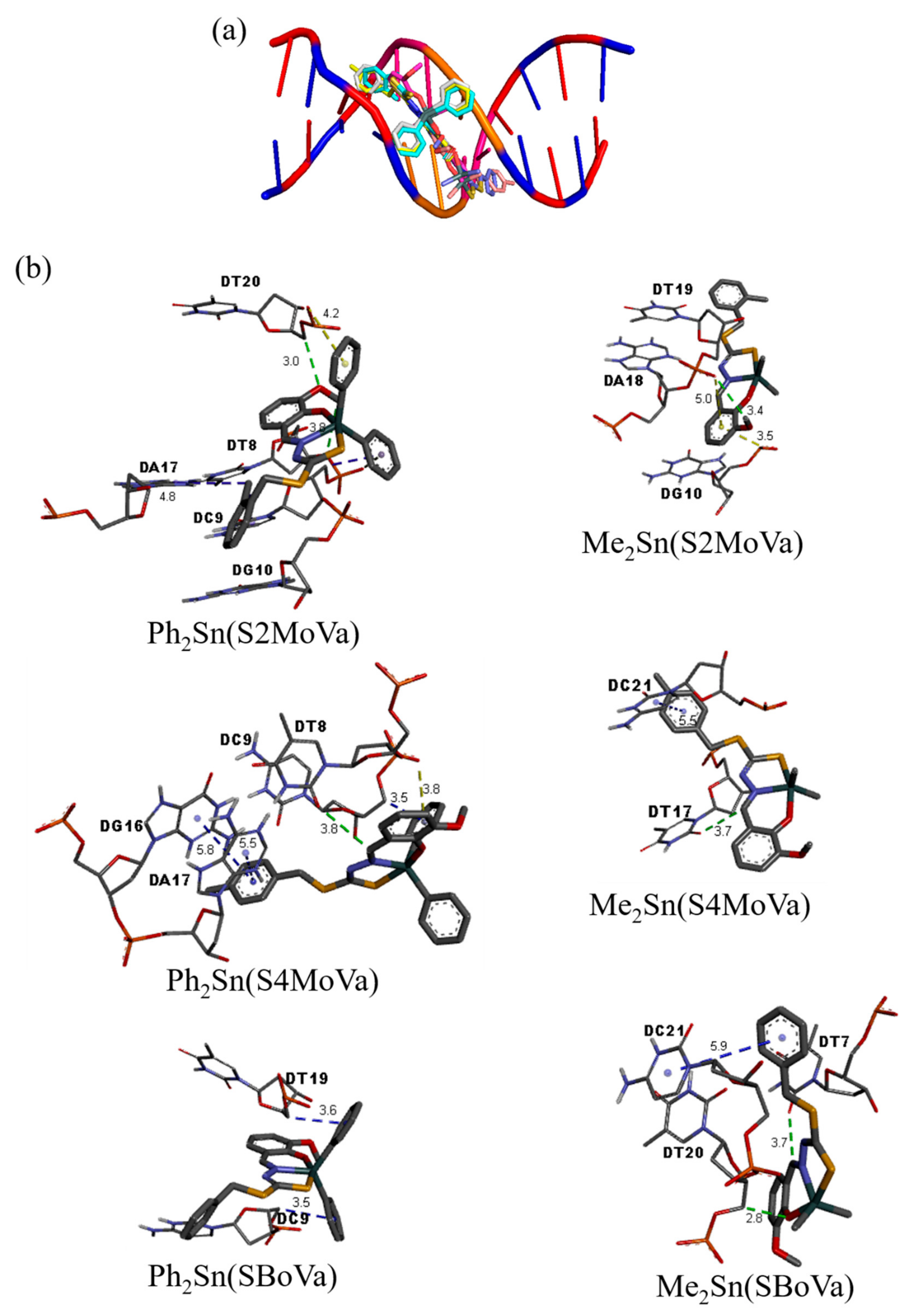
2.8. Molecular Docking Studies
3. Experimental
3.1. Physical Measurements
3.2. Materials
3.3. Synthesis
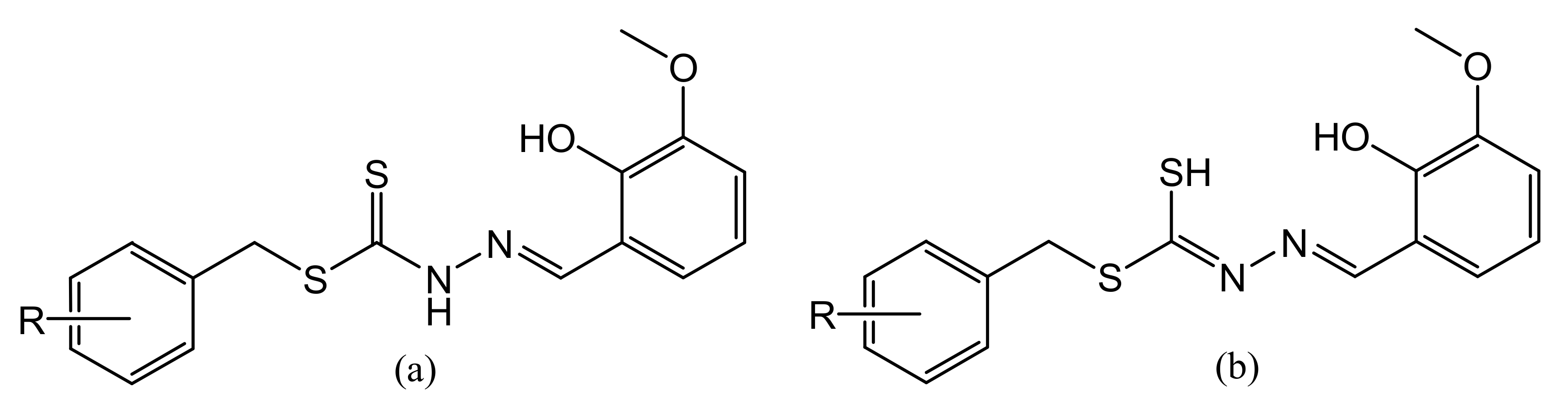
3.3.1. S-N-R-Benzyldithiocarbazates (N = 2,3, R = Methyl)
3.3.2. S-2-Methybenzyl-β-N-(2-hydroxy-3-methoxybenzylmethylene) Dithiocarbazate (S2MoVaH)
3.3.3. S-4-Methybenzyl-β-N-(2-hydroxy-3-methoxybenzylmethylene)dithiocarbazate (S4MoVaH)
3.3.4. S-Benzyl-β-N-(2-hydroxy-3-methoxybenzylmethylene) Dithiocarbazate (SBoVaH)
3.3.5. Diphenyltin(IV) Compounds
3.3.6. Dimethyltin(IV) Compounds
3.4. Single Crystal X-ray Structure Determination
3.5. Computational Methods
3.5.1. Density Functional Theory (DFT) Calculations
3.5.2. Molecular Docking Studies
3.6. Biological Assays
3.6.1. MTT Assays
Cell Culture and Stock Solutions
In Vitro Growth Inhibition Assay
3.6.2. Quantification of Apoptosis by Annexin V
3.6.3. Measurement of Reactive Oxygen Species (ROS)
3.6.4. DNA Binding Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boseley, S. Available online: https://www.theguardian.com/society/2014/feb/03/worldwide-cancer-cases-soar-next-20-years (accessed on 20 February 2018).
- Global Cancer Observatory. Available online: http://gco.iarc.fr/ (accessed on 20 February 2018).
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Galanski, M. Recent Developments in the Field of Anticancer Platinum Complexes. Recent Pat. Anticancer Drug Discov. 2006, 1, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Sirajuddin, M.; Ali, S.; Mckee, V.; Zaib, S.; Iqbal, J. Organotin(IV) carboxylate derivatives as a new addition to anticancer and antileishmanial agents: Design, physicochemical characterization and interaction with Salmon sperm DNA. RSC Adv. 2014, 4, 57505–57521. [Google Scholar] [CrossRef]
- Subbaraj, P.; Ramu, A.; Raman, N.; Dharmaraja, J. Synthesis, characterization, DNA interaction and pharmacological studies of substituted benzophenone derived Schiff base metal(II) complexes. J. Saudi Chem. Soc. 2015, 19, 207–216. [Google Scholar] [CrossRef]
- Jain, M.; Gaur, S.; Singh, V.P.; Singh, R. V Organosilicon (IV) and organotin (IV) complexes as biocides and nematicides: Synthetic, spectroscopic and biological studies of N∩N donor sulfonamide imine and its chelates. Main Gr. Met. Compd. 2004, 18, 73–82. [Google Scholar] [CrossRef]
- Liu, K.; Yan, H.; Chang, G.; Li, Z.; Niu, M.; Hong, M. Organotin(IV) complexes derived from hydrazone Schiff base: Synthesis, crystal structure, in vitro cytotoxicity and DNA/BSA interactions. Inorg. Chim. Acta 2017, 464, 137–146. [Google Scholar] [CrossRef]
- Salam, M.A.; Hussein, M.A.; Ramli, I.; Islam, S. Synthesis, structural characterization and evaluation of biological activity of organotin(IV) complexes with 2-hydroxy-5-methoxybenzaldehyde-N(4)-methylthiosemicarbazone. J. Organomet. Chem. 2016, 813, 71–77. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, N.K. Spectral and thermal studies with anti-fungal aspects of some organotin(IV) complexes with nitrogen and sulphur donor ligands derived from 2-phenylethylamine. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 71, 669–675. [Google Scholar] [CrossRef]
- Tariq, M.; Ali, S.; Muhammad, N.; Shah, N.A.; Sirajuddin, M.; Tahir, M.N.; Khalid, N.; Khan, M.R. Biological screening, DNA interaction studies, and catalytic activity of organotin(IV) 2-(4-ethylbenzylidene) butanoic acid derivatives: Synthesis, spectroscopic characterization, and X-ray structure. J. Coord. Chem. 2014, 67, 323–340. [Google Scholar] [CrossRef]
- Kumar, A.; Chaudhary, P.; Singh, R.; Kaushik, N.K. Organotin(IV) complexes of thiohydrazones of phenethylamine: Synthesis, characterization, biological and thermal study. Main Gr. Chem. 2016, 15, 163–178. [Google Scholar]
- Bacchi, A.; Bonardi, A.; Carcelli, M.; Mazza, P.; Pelagatti, P.; Pelizzi, C.; Pelizzi, G.; Solinas, C.; Zani, F. Organotin complexes with pyrrole-2,5-dicarboxaldehyde bis (acylhydrazones). Synthesis, structure, antimicrobial activity and genotoxicity. J. Inorg. Biochem. 1998, 69, 101–112. [Google Scholar] [CrossRef]
- Win, Y.F.; Choong, C.S.; Dang, J.C.; Iqbal, M.A.; Quah, C.K.; Majid, A.M.S.A.; Teoh, S.G. Polymeric seven-coordinated organotin(IV) complexes derived from 5-amino-2-chlorobenzoic acid and in vitro anti-cancer studies. J. Coord. Chem. 2014, 67, 3401–3413. [Google Scholar] [CrossRef]
- Malhotra, R.; Ravesh, A.; Singh, V. Synthesis, characterization, antimicrobial activities and QSAR studies of organotin(IV) complexes. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Ali, S.; Mckee, V.; Sohail, M.; Pasha, H. Potentially bioactive organotin (IV) compounds: Synthesis, characterization, in vitro bioactivities and interaction with SS-DNA. Eur. J. Med. Chem. 2014, 84, 343–363. [Google Scholar] [CrossRef] [PubMed]
- Fent, K. Ecotoxicology of organotin compounds. Crit. Rev. Toxicol. 1996, 26, 1–117. [Google Scholar] [CrossRef] [PubMed]
- Evan, C.J. Industrial Uses of Tin Chemicals. In Chemistry of Tin, 2nd ed.; Smith, P.J., Ed.; Blackie Academic & Professional: Glasgow, UK, 1998; pp. 442–474. [Google Scholar]
- Akbar Ali, M.; Huq Mirza, A.; Kok Wei, L.; Bernhardt, P.V.; Atchade, O.; Song, X.; Eng, G.; May, L. Synthesis and characterization of pentagonal bipyramidal organotin(IV) complexes of 2,6-diacetylpyridine Schiff bases of S-alkyl- and aryldithiocarbazates. J. Coord. Chem. 2010, 63, 1194–1206. [Google Scholar] [CrossRef]
- Nath, M. Tin Chemistry: Fundamentals, Frontiers, and Applications; Davies, A.G., Gielen, M., Pannell, K.H., Tiekink, E.R.T., Eds.; John Wiley & Sons: West Sussex, UK, 2008; pp. 413–492. [Google Scholar]
- Sedaghat, T.; Golalzadeh, A.; Motamedi, H. Diorganotin Complexes with N(4)-Phenylthiosemicarbazones: Synthesis, Spectroscopic Characterization, and Antibacterial Activity. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 1694–1702. [Google Scholar] [CrossRef]
- Gielen, M. Organotin compounds and their therapeutic potential: A report from the Organometallic Chemistry Department of the Free University of Brussels. Appl. Organomet. Chem. 2002, 16, 481–494. [Google Scholar] [CrossRef]
- Gielen, M.; Biesemans, M.; Willem, R. Organotin compounds: From kinetics to stereochemistry and antitumour activities. Appl. Organomet. Chem. 2005, 19, 440–450. [Google Scholar] [CrossRef]
- Nath, M.; Saini, P.K. Chemistry and applications of organotin(IV) complexes of Schiff bases. Dalton Trans. 2011, 40, 7077–7121. [Google Scholar] [CrossRef]
- Rehman, W.; Yasmeen, R.; Rahim, F.; Waseem, M.; Guo, C.Y.; Hassan, Z.; Rashid, U.; Ayub, K. Synthesis biological screening and molecular docking studies of some tin(IV) Schiff base adducts. J. Photochem. Photobiol. B Biol. 2016, 164, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Sirajuddin, M.; Ali, S.; Tahir, M.N. Pharmacological investigation of mono-, di- and tri-organotin(IV) derivatives of carbodithioates: Design, spectroscopic characterization, interaction with SS-DNA and POM analyses. Inorg. Chim. Acta 2016, 439, 145–158. [Google Scholar] [CrossRef]
- Basu Baul, T.S.; Masharing, C.; Ruisi, G.; Jirásko, R.; Holčapek, M.; de Vos, D.; Wolstenholme, D.; Linden, A. Self-assembly of extended Schiff base amino acetate skeletons, 2-{[(2Z)-(3-hydroxy-1-methyl-2-butenylidene)]amino}phenylpropionate and 2-{[(E)-1-(2-hydroxyaryl)alkylidene]amino}phenylpropionate skeletons incorporating organotin(IV) moieties: Synthesis, spectroscopic characterization, crystal structures, and in vitro cytotoxic activity. J. Organomet. Chem. 2007, 692, 4849–4862. [Google Scholar]
- Katsoulakou, E.; Tiliakos, M.; Papaefstathiou, G.; Terzis, A.; Raptopoulou, C.; Geromichalos, G.; Papazisis, K.; Papi, R.; Pantazaki, A.; Kyriakidis, D.; et al. Diorganotin(IV) complexes of dipeptides containing the α-aminoisobutyryl residue (Aib): Preparation, structural characterization, antibacterial and antiproliferative activities of [(n-Bu)2Sn(H-1L)] (LH = H-Aib-L-Leu-OH, H-Aib-L-Ala-OH). J. Inorg. Biochem. 2008, 102, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.-N.; Qi, Z.-D.; Ouyang, Y.-W.; Liao, F.-L.; Zhang, Y.; Liu, Y. Studies on interaction between Vitamin B12 and human serum albumin. J. Pharm. Biomed. Anal. 2008, 47, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Sielecki, T.M.; Boylan, J.F.; Benfield, P.A.; Trainor, G.L. Cyclin-dependent kinase inhibitors: Useful targets in cell cycle regulation. J. Med. Chem. 2000, 43, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rudorf, W. Reactions of Carbon Disulfide with C-nucleophiles. Sulfur Rep. 1991, 11, 51–141. [Google Scholar] [CrossRef]
- Wiecek, J.; Dokorou, V.; Ciunik, Z.; Kovala-Demertzi, D. Organotin complexes of pyruvic acid thiosemicarbazone: Synthesis, crystal structures and antiproliferative activity of neutral and cationic diorganotin complexes. Polyhedron 2009, 28, 3298–3304. [Google Scholar] [CrossRef]
- Arjmand, F.; Parveen, S.; Tabassum, S.; Pettinari, C. Organo-tin antitumor compounds: Their present status in drug development and future perspectives. Inorg. Chim. Acta 2014, 423, 26–37. [Google Scholar] [CrossRef]
- Song, X.; Zapata, A.; Eng, G. Organotins and quantitative-structure activity/property relationships. J. Organomet. Chem. 2006, 691, 1756–1760. [Google Scholar] [CrossRef]
- Nath, M.; Pokharia, S.; Yadav, R. Organotin(IV) complexes of amino acids and peptides. Coord. Chem. Rev. 2001, 215, 99–149. [Google Scholar] [CrossRef]
- Chandrasekhar, V.; Nagendran, S.; Baskar, V. Organotin assemblies containing Sn-O bonds. Coord. Chem. Rev. 2002, 235, 1–52. [Google Scholar] [CrossRef]
- Rehman, W.; Baloch, M.K.; Badshah, A. Synthesis, spectral characterization and bio-analysis of some organotin(IV) complexes. Eur. J. Med. Chem. 2008, 43, 2380–2385. [Google Scholar] [CrossRef] [PubMed]
- Oztaş, N.A.; Yenişehirli, G.; Ancin, N.; Oztaş, S.G.; Ozcan, Y.; Ide, S. Synthesis, characterization, biological activities of dimethyltin(IV) complexes of Schiff bases with ONO-type donors. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2009, 72, 929–935. [Google Scholar] [CrossRef]
- López-Torres, E.; Zani, F.; Mendiola, M.A. Antimicrobial activity of organotin(IV) complexes with the ligand benzil bis(benzoylhydrazone) and 4,4′-bipyridyl as coligand. J. Inorg. Biochem. 2011, 105, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Bergamascbi, G.; Bonardi, A.; Leporati, E.; Mazza, P.; Pehzgatti, P.; Pelizzi, C.; Pelizzi, G.; Argiielles, M.C.R.; Zani, F. Organotin Complexes with Pyrrole-2-carboxaldehyde Monoacylhydrazones. Synthesis, Spectroscopic Properties, Antimicrobial Activity, and Genotoxicity. J. Inorg. Biochem. 1997, 68, 295–305. [Google Scholar] [CrossRef]
- Baul, T.S.B. Antimicrobial activity of organotin(IV) compounds: A review. Appl. Organomet. Chem. 2008, 22, 195–204. [Google Scholar] [CrossRef]
- Tarafder, M.T.H.; Chew, K.; Crouse, K.A.; Ali, A.M.; Yamin, B.M.; Fun, H.K. Synthesis and characterization of Cu(II), Ni(II) and Zn(II) metal complexes of bidentate NS isomeric Schiff bases derived from S-methyldithiocarbazate (SMDTC): Bioactivity of the bidentate NS isomeric Schiff bases, some of their Cu(II), Ni(II) and Zn(II). Polyhedron 2002, 21, 2683–2690. [Google Scholar] [CrossRef]
- Jeffrey, P.M.; Damian, M.; Radom, L. An Evaluation of Harmonic Vibrational Frequency Scale Factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar]
- Elsayed, S.A.; Noufal, A.M.; El-Hendawy, A.M. Synthesis, structural characterization and antioxidant activity of some vanadium(IV), Mo(VI)/(IV) and Ru(II) complexes of pyridoxal Schiff base derivatives. J. Mol. Struct. 2017, 1144, 120–128. [Google Scholar] [CrossRef]
- Malhotra, R.; Singh, J.P.; Dudeja, M.; Dhindsa, K.S. Ligational behavior of N-substituted acid hydrazides towards transition metals and potentiation of their microbiocidal activity. J. Inorg. Biochem. 1992, 46, 119–127. [Google Scholar] [CrossRef]
- Ali, M.A.; Majumder, S.M.M.-H.; Butcher, R.J.; Jasinski, J.P.; Jasinski, J.M. The preparation and characterization of bis-chelated nickel(II) complexes of the 6-methylpyridine-2-carboxaldehyde Schiff bases of S-alkyldithiocarbazates and the X-ray crystal structure of the bis {S-methyl-β-N-(6-methylpyrid-2-yl)-methylenedithiocarba. Polyhedron 1997, 16, 2749–2754. [Google Scholar] [CrossRef]
- Ali, M.A.; Mirza, A.H.; Butcher, R.J. Synthesis and characterization of copper(II) complexes of the methylpyruvate Schiff base of S-methyldithiocarbazate (Hmpsme) and the X-crystal structures of Hmpsme and [Cu (mpsme)Cl]. Polyhedron 2001, 20, 1037–1043. [Google Scholar] [CrossRef]
- Ali, M.A.; Mirza, A.H.; Tan, A.L.; Wei, L.K.; Bernhardt, P.V. The preparation and characterization of seven-coordinate tin(IV) complexes of the 2,6-diacetylpyridine Schiff bases of S-alkyl/aryl-dithiocarbazates and the X-ray crystal structure of the [Sn(dapsme)I2] complex (dapsme=doubly deprotonated form of the 2,6-diacetylpyridine Schiff base of S-methyldithiocarbazate). Polyhedron 2004, 23, 2405–2412. [Google Scholar]
- Mıhçıokur, Ö.; Özpozan, T. Molecular structure, vibrational spectroscopic analysis (IR & Raman), HOMO-LUMO and NBO analysis of anti-cancer drug sunitinib using DFT method. J. Mol. Struct. 2017, 1149, 27–41. [Google Scholar]
- Yusof, E.N.M.; Jotani, M.M.; Tiekink, E.R.T.; Ravoof, T.B.S.A. 2-[(1E)-({[(Benzylsulfanyl)methanethioyl]amino}-imino)methyl]-6-methoxyphenol: Crystal structure and Hirshfeld surface analysis. Acta Crystallogr. Sect. E Crystallogr. Commun. 2016, 72, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Mirza, A.H.; Butcher, R.J.; Crouse, K.A. The preparation, characterization and biological activity of palladium(II) and platinum(II) complexes of tridentate NNS ligands derived from S-methyl- and S-benzyldithiocarbazates and the X-ray crystal structure of the [Pd(mpasme)Cl] complex. Transit. Met. Chem. 2006, 31, 79–87. [Google Scholar] [CrossRef]
- Naqeebullah; Farina, Y.; Chan, K.M.; Mun, L.K.; Rajab, N.F.; Ooi, T.C. Diorganotin(IV) derivatives of N-methyl p-fluorobenzo-hydroxamic acid: Preparation, spectral characterization, X-ray diffraction studies and antitumor activity. Molecules 2013, 18, 8696–8711. [Google Scholar] [CrossRef]
- Holeček, J.; Nádvorník, M.; Handlíř, K.; Lyčka, A. 13C and 119Sn NMR spectra of Di-n-butyltin(IV) compounds. J. Organomet. Chem. 1986, 315, 299–308. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Zhang, R.-F.; Zhang, S.-L.; Cheng, S.; Li, Q.-L.; Ma, C.-L. Syntheses, structures and anti-tumor activity of four new organotin(IV) carboxylates based on 2-thienylselenoacetic acid. Dalton Trans. 2016, 45, 8412–8421. [Google Scholar] [CrossRef]
- Barreiro, S.; Durán-Carril, M.L.; Viqueira, J.; Sousa-Pedrares, A.; García-Vázquez, J.A.; Romero, J. Structural studies and bioactivity of diorganotin(IV) complexes of pyridin-2-thionato derivatives. J. Organomet. Chem. 2015, 791, 155–162. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, J.; Tian, G.; Zhang, R. Syntheses, crystal structures and coordination modes of tri- and di-organotin derivatives with 2-mercapto-4-methylpyrimidine. J. Organomet. Chem. 2005, 690, 519–533. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds containing Nitrogen-Sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[l,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) Perchlorate. J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar] [CrossRef]
- Milčič, M.K.; Medakovič, V.B.; Sredojevič, D.N.; Juranič, N.O.; Zarič, S.D. Electron delocalization mediates the metal-dependent capacity for CH/π interactions of acetylacetonato chelates. Inorg. Chem. 2006, 45, 4755–4763. [Google Scholar] [CrossRef] [PubMed]
- Tiekink, E.R.T. Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists. Coord. Chem. Rev. 2017, 345, 209–228. [Google Scholar] [CrossRef]
- Chen, D.; Lai, C.S.; Tiekink, E.R.T. Crystal structures of 2,2′-bipyridine adducts of two cadmium O-alkyl dithiocarbonates: Rationalisation of disparate coordination geometries based on different crystal packing environments. Z. Kristallogr. 2002, 217, 747–752. [Google Scholar] [CrossRef]
- Malenov, D.P.; Janjić, G.V.; Medaković, V.B.; Hall, M.B.; Zarić, S.D. Noncovalent bonding: Stacking interactions of chelate rings of transition metal complexes. Coord. Chem. Rev. 2017, 345, 318–341. [Google Scholar] [CrossRef]
- Akbar Ali, M.; Mirza, A.H.; Hamid, M.H.S.A.; Bernhardt, P.V. Diphenyltin(IV) complexes of the 2-quinolinecarboxaldehyde Schiff bases of S-methyl- and S-benzyldithiocarbazate (Hqaldsme and Hqaldsbz): X-ray crystal structures of Hqaldsme and two conformers of its diphenyltin(IV) complex. Polyhedron 2005, 24, 383–390. [Google Scholar] [CrossRef]
- Anto, R.J.; Sukumaran, K.; Kuttan, G.; Rao, M.N.A.; Subbaraju, V.; Kuttan, R. Anticancer and antioxidant activity of synthetic chalcones and related compounds. Cancer Lett. 1995, 97, 33–37. [Google Scholar] [CrossRef]
- González-garcía, C.; Mata, A.; Zani, F.; Mendiola, M.A.; López-torres, E. Synthesis and antimicrobial activity of tetradentate ligands bearing hydrazone and/or thiosemicarbazone motifs and their diorganotin (IV) complexes. J. Inorg. Biochem. 2016, 163, 118–130. [Google Scholar] [CrossRef]
- Andree, H.A.M.; Reutelingsperger, C.P.M.; Hauptmann, R.; Hemker, H.C.; Hermens, W.T.; Willems, G.M. Binding of vascular anticoagulant alpha (VAC alpha) to planar phospholipid bilayers. J. Biol. Chem. 1990, 265, 4923–4928. [Google Scholar] [PubMed]
- Gea, R.; Ma, W.-H.; Li, Y.-L.; Li, Q.-S. Apoptosis induced neurotoxicity of Di-n-butyl-di-(4-chlorobenzohydroxamato) Tin(IV) via mitochondria-mediated pathway in PC12 cells. Toxicol. In Vitro 2013, 27, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.H.; Hulett, M.D.; Parish, C.R. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ. 2010, 17, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Prosser, K.E.; Chang, S.W.; Saraci, F.; Le, P.H.; Walsby, C.J. Anticancer copper pyridine benzimidazole complexes: ROS generation, biomolecule interactions, and cytotoxicity. J. Inorg. Biochem. 2017, 167, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Fani, S.; Kamalidehghan, B.; Lo, K.M.; Hashim, N.M.; Chow, K.M.; Ahmadipour, F. Synthesis, structural characterization, and anticancer activity of a monobenzyltin compound against MCF-7 breast cancer cells. Drug Des. Dev. Ther. 2015, 9, 6191–6201. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Bukhari, I.H.; Ali, S.; Shahzadi, S.; Shahid, M.; Munawar, K.S. Synthesis and spectroscopic and thermogravimetric characterization of heterobimetallic complexes with Sn(IV) and Pd(II); DNA binding, alkaline phosphatase inhibition and biological activity studies. J. Coord. Chem. 2015, 68, 662–677. [Google Scholar] [CrossRef]
- Hussain, S.; Ali, S.; Shahzadi, S.; Tahir, M.N.; Shahid, M. Synthesis, characterization, biological activities, crystal structure and DNA binding of organotin(IV) 5-chlorosalicylates. J. Coord. Chem. 2015, 68, 2369–2387. [Google Scholar] [CrossRef]
- Yadav, S.; Yousuf, I.; Usman, M.; Ahmad, M.; Arjmand, F.; Tabassum, S. Synthesis and spectroscopic characterization of diorganotin(IV) complexes of N′-(4-hydroxypent-3-en-2-ylidene)isonicotinohydrazide: Chemotherapeutic potential validation by in vitro interaction studies with DNA/HSA, DFT, molecular docking and cytotoxic activity. RSC Adv. 2015, 5, 50673–50690. [Google Scholar]
- Ali, M.A.; Tarafder, M.T. Metal complexes of sulphur and nitrogen-containing ligands: Complexes of S-benzyldithiocarbazate and a Schiff base formed by its condensation with pyridine-2-carboxaldehyde. J. Inorg. Nucl. Chem. 1977, 39, 1785–1791. [Google Scholar]
- Ravoof, T.B.S.A.; Crouse, K.A.; Tahir, M.I.M.; Rosli, R.; Watkin, D.J.; How, F.N.F. Synthesis, Characterisation and Biological Activities of 2-Methylbenzyl 2-(dipyridin-2-yl methylene)hydrazinecarbodithioate. J. Chem. Crystallogr. 2011, 41, 491–495. [Google Scholar] [CrossRef]
- Omar, S.A.; Ravoof, T.B.S.A.; Tahir, M.I.M.; Crouse, K.A. Synthesis and characterization of mixed-ligand copper(II) saccharinate complexes containing tridentate NNS Schiff bases. X-ray crystallographic analysis of the free ligands and one complex. Transit. Met. Chem. 2013, 39, 119–126. [Google Scholar] [CrossRef]
- Yusof, E.N.M.; Ravoof, T.B.S.A.; Tiekink, E.R.T.; Veerakumarasivam, A.; Crouse, K.A.; Tahir, M.I.M.; Ahmad, H. Synthesis, characterization and biological evaluation of transition metal complexes derived from N, S bidentate ligands. Int. J. Mol. Sci. 2015, 16, 11034–11054. [Google Scholar] [CrossRef] [PubMed]
- Yusof, E.N.M.; Ravoof, T.B.S.A.; Jamsari, J.; Tiekink, E.R.T.; Veerakumarasivam, A.; Crouse, K.A.; Tahir, M.I.M.; Ahmad, H. Synthesis, characterization and biological studies of S-4-methylbenzyl-β-N-(2-furylmethylene)dithiocarbazate (S4MFuH) its Zn2+, Cu2+, Cd2+ and Ni2+ complexes. Inorg. Chim. Acta 2015, 438, 85–93. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlis PRO; Agilent Technologies Inc.: Santa Clara, CA, USA, 2015. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND, Crystal Impact GbR: Bonn, Germany, 2006.
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Roy, D.; Todd, K.; John, M. GaussView, Ver 5.0.9; Semichem, Inc.: Shawnee Mission, KS, USA, 2009.
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Tsai, H.-Y. Synthesis, X-ray Structure, Spectroscopic Properties and DFT Studies of a Novel Schiff Base. Int. J. Mol. Sci. 2014, 15, 18706–18724. [Google Scholar] [CrossRef] [PubMed]
- Scalmani, G.; Frisch, M.J.; Mennucci, B.; Tomasi, J.; Cammi, R.; Barone, V. Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 2006, 124, 94107. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to Isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software News and Updates AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- ADL: Parameters for Docking with Metal Ions in Receptor. Available online: http://autodock.1369657.n2.nabble.com/ADL-Parameters-fordocking-with-metal-ions-in-receptor-td2505649.html (accessed on 7 September 2016).
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sakoff, J.A.; Ackland, S.P. Thymidylate synthase inhibition induces S-phase arrest, biphasic mitochondrial alterations and caspase-dependent apoptosis in leukaemia cells. Cancer Chemother. Pharmacol. 2000, 46, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.M.; Ruiz Van Haperen, V.W.T.; Veerman, G.; Kuiper, C.M.; Peters, G.J. Synergistic interaction between cisplatin and gemcitabine in vitro. Clin. Cancer Res. 1996, 2, 521–530. [Google Scholar] [PubMed]
- Sudan, S.; Rupasinghe, H.V. Antiproliferative activity of long chain acylated esters of quercetin-3-O-glucoside in hepatocellular carcinoma HepG2 cells. Exp. Biol. Med. 2015, 240, 1452–1464. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Chang, G.; Li, R.; Niu, M. Anti-proliferative activity and DNA/BSA interactions of five mono- or di-organotin(IV) compounds derived from 2-hydroxy-N′-[(2-hydroxy-3-methoxyphenyl)methylidene]-benzohydrazone. New J. Chem. 2016, 40, 7889–7900. [Google Scholar] [CrossRef]
- Ganeshpandian, M.; Loganathan, R.; Suresh, E.; Riyasdeen, A.; Akbarsha, M.A.; Palaniandavar, M. New ruthenium(II) arene complexes of anthracenyl-appended diazacycloalkanes: Effect of ligand intercalation and hydrophobicity on DNA and protein binding and cleavage and cytotoxicity. Dalton Trans. 2014, 43, 1203–1219. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, R.; Ramakrishnan, S.; Suresh, E.; Riyasdeen, A.; Akbarsha, M.A.; Palaniandavar, M. Mixed ligand copper(II) complexes of N,N-bis(benzimidazol-2-ylmethyl)amine (BBA) with diimine co-ligands: Efficient chemical nuclease and protease activities and cytotoxicity. Inorg. Chem. 2012, 51, 5512–5532. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Me2Sn(S2MoVa) | Me2Sn(S4MoVa) | Me2Sn(SBoVa) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Bond lengths | molecule a | molecule b | B3LYP | molecule a | molecule b | B3LYP | molecule a | molecule b | B3LYP |
| LanL2DZ/6-311G(d,p) | LanL2DZ/6-311G(d,p) | LanL2DZ/6-311G(d,p) | |||||||
| Sn–S1 | 2.545(2) | 2.542(2) | 2.576 | 2.5391(8) | 2.5309(7) | 2.566 | 2.5789(11) | 2.5607(10) | 2.590 |
| Sn–O1 | 2.092(6) | 2.102(6) | 2.085 | 2.090(2) | 2.214(2) | 2.078 | 2.103(3) | 2.093(3) | 2.078 |
| Sn–N2 | 2.188(6) | 2.192(6) | 2.285 | 2.223(2) | 2.088(2) | 2.232 | 2.194(3) | 2.205(3) | 2.226 |
| C1–S1 | 1.726(9) | 1.717(8) | 1.751 | 1.736(3) | 1.734(3) | 1.753 | 1.725(4) | 1.726(4) | 1.748 |
| C1–S2 | 1.744(8) | 1.747(8) | 1.772 | 1.752(3) | 1.757(3) | 1.771 | 1.771(4) | 1.763(4) | 1.783 |
| N1–N2 | 1.394(9) | 1.406(9) | 1.389 | 1.402(3) | 1.409(3) | 1.388 | 1.397(5) | 1.395(4) | 1.383 |
| C1–N1 | 1.304(11) | 1.273(10) | 1.293 | 1.289(4) | 1.287(4) | 1.293 | 1.298(5) | 1.291(5) | 1.296 |
| C(x)–N2 | 1.308(9) | 1.306(9) | 1.313 | 1.309(4) | 1.307(4) | 1.312 | 1.311(5) | 1.304(5) | 1.307 |
| Bond angles | |||||||||
| S1–Sn–O1 | 157.89(17) | 158.25(17) | 159.5 | 158.51(7) | 155.88(7) | 159.7 | 155.82(9) | 154.24(10) | 159.8 |
| S1–Sn–N2 | 77.35(18) | 76.94(18) | 77.6 | 77.33(7) | 77.78(6) | 77.8 | 76.70(9) | 77.13(8) | 77.3 |
| O1–Sn–N2 | 82.2(2) | 82.3(2) | 82.0 | 82.13(9) | 82.15(8) | 82.2 | 82.76(11) | 82.09(12) | 82.4 |
| C18–Sn–C(y) | 128.9(5) | 128.2(5) | 123.9 | 122.58(14) | 123.05(14) | 123.9 | 127.70(18) | 124.8(2) | 123.7 |
| S1–C1–S2 | 112.1(5) | 111.9(5) | 113.4 | 111.46(17) | 111.81(17) | 113.4 | 120.2(2) | 120.1(2) | 120.8 |
| S1–C1–N1 | 129.1(6) | 128.9(6) | 128.2 | 129.1(2) | 129.6(2) | 128.2 | 128.7(3) | 128.3(3) | 127.7 |
| S2–C1–N1 | 118.9(7) | 119.3(6) | 118.4 | 119.5(2) | 118.6(2) | 118.4 | 111.1(3) | 111.6(3) | 111.5 |
| Compounds | Growth Inhibition Concentration, GI50 (µM) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EJ-28 | RT-112 | HT29 | U87 | MCF-7 | A2780 | H460 | A431 | Du145 | BE2-C | SJ-G2 | MIA | MCF10A | |
| S2MoVaH | 3.4 ± 0.47 | 4.6 ± 0.45 | 3.9 ± 0.71 | 4.3 ± 0.30 | 2.7 ± 0.21 | 3.2 ± 0.15 | 4.3 ± 0.12 | 3.8 ± 0.03 | 4.1 ± 0.09 | 3.1 ± 0.00 | 3.0 ± 0.09 | 4.9 ± 0.37 | 3.1 ± 0.07 |
| Ph2Sn(S2MoVa) | 0.35 ± 0.05 | 0.31 ± 0.05 | 1.8 ± 0.13 | 4.4 ± 1.7 | 1.4 ± 0.12 | 0.23 ± 0.01 | 2.8 ± 0.00 | 2.1 ± 0.09 | 1.7 ± 0.07 | 0.47 ± 0.05 | 0.84 ± 0.09 | 0.34 ± 0.02 | 1.2 ± 0.30 |
| Me2Sn(S2MoVa) | 2.5 ± 0.78 | 2.4 ± 0.77 | 2.7 ± 0.10 | 3.6 ± 0.23 | 3.1 ± 0.12 | 3.1 ± 0.06 | 3.3 ± 0.13 | 3.0 ± 0.03 | 3.9 ± 0.03 | 2.9 ± 0.03 | 2.5 ± 0.30 | 3.9 ± 0.07 | 3.0 ± 0.09 |
| S4MoVaH | 3.6 ± 0.44 | 3.2 ± 0.51 | 8.0 ± 3.5 | 3.9 ± 0.00 | 3.5 ± 0.00 | 3.1 ± 0.40 | 5.5 ± 0.73 | 4.2 ± 0.74 | 5.4 ± 0.23 | 3.6 ± 0.27 | 3.3 ± 0.87 | 11 ± 2.3 | 3.5 ± 0.23 |
| Ph2Sn(S4MoVa) | 3.1 ± 0.37 | 1.7 ± 0.49 | 0.87 ± 0.22 | 1.3 ± 0.40 | 0.82 ± 0.14 | 0.35 ± 0.07 | 1.9 ± 0.23 | 1.4 ± 0.38 | 1.5 ± 0.31 | 0.53 ± 0.01 | 1.0 ± 0.16 | 0.39 ± 0.03 | 1.1 ± 0.37 |
| Me2Sn(S4MoVa) | >5.0 | >5.0 | 6.9 ± 3.5 | 3.3 ± 0.18 | 3.3 ± 0.32 | 3.1 ± 0.63 | 4.7 ± 0.46 | 4.2 ± 0.34 | 4.2 ± 0.10 | 3.3 ± 0.23 | 3.1 ± 0.55 | 8.9 ± 1.30 | 3.3 ± 0.15 |
| SBoVaH | 3.1 ± 0.20 | 3.7 ± 0.57 | 2.2 ± 0.033 | 3.1 ± 0.26 | 2.5 ± 0.12 | 3.0 ± 0.06 | 3.3 ± 0.21 | 3.0 ± 0.15 | 3.4 ± 0.30 | 2.8 ± 0.20 | 2.4 ± 0.26 | 4.5 ± 0.25 | 3.0 ± 0.07 |
| Ph2Sn(SBoVa) | 0.32 ± 0.05 | 0.58 ± 0.29 | 1.2 ± 0.22 | 1.1 ± 0.39 | 0.90 ± 0.16 | 0.23 ± 0.03 | 2.2 ± 0.00 | 1.6 ± 0.07 | 1.4 ± 0.33 | 0.50 ± 0.05 | 0.74 ± 0.15 | 0.43 ± 0.04 | 1.3 ± 0.20 |
| Me2Sn(SBoVa) | 3.4 ± 0.30 | 2.4 ± 0.33 | 2.8 ± 0.15 | 4.5 ± 0.19 | 2.5 ± 0.21 | 3.2 ± 0.03 | 3.8 ± 0.10 | 3.2 ± 0.03 | 3.2 ± 0.13 | 2.7 ± 0.13 | 2.1 ± 0.12 | 5.0 ± 0.09 | 2.8 ± 0.41 |
| Cisplatin | 3.97 ± 0.48 | 3.56 ± 0.88 | 11.0 ± 2.0 | 4.0 ± 1.0 | 6.5 ± 0.8 | 1.0 ± 0.1 | 0.9 ± 0.2 | 2.4 ± 0.3 | 1.2 ± 0.1 | 1.9 ± 0.2 | 0.4 ± 0.1 | 8.0 ± 1.0 | nd |
| Compounds | Binding Constant (Kb) | Hypochromism, (%) | Bathochromism Shift (nm) | Gibbs Free Energy (kJ mol−1) |
|---|---|---|---|---|
| Ph2Sn(S2MoVa) | 6.21 × 105 | 41.06 | 6 | −33.1 |
| Me2Sn(S2MoVa) | 3.46 × 105 | 37.97 | 10 | −31.6 |
| Ph2Sn(S4MoVa) | 2.67 × 105 | 28.25 | 4 | −31.0 |
| Me2Sn(S4MoVa) | 4.15 × 105 | 43.72 | 7 | −32.1 |
| Ph2Sn(SBoVa) | 1.21 × 106 | 70.72 | 12 | −34.7 |
| Me2Sn(SBoVa) | 3.91 × 106 | 53.26 | 1 | −37.6 |
| Complex | Final Intermolecular Energy, kcal mol−1 | Final Total Internal Energy (2), kcal mol−1 | Torsional Free Energy (3), kcal mol−1 | Unbound System’s Energy [=(2)] (4), kcal mol−1 | Estimated Free Energy of Binding [(1) + (2) + (3) − (4)], kcal mol−1 | Estimated Free Energy of Binding, kJ mol−1 | ||
|---|---|---|---|---|---|---|---|---|
| vdW + Hbond + desolv. Energy | Electrostatic Energy | Total (1) | ||||||
| Ph2Sn(S2MoVa) | −11.46 | 0.00 | −11.47 | −2.19 | 1.79 | −2.19 | −9.68 | −40.50 |
| Me2Sn(S2MoVa) | −10.27 | −0.03 | −10.30 | 0.57 | 1.19 | −0.57 | −9.10 | −38.07 |
| Ph2Sn(S4MoVa) | −11.87 | 0.01 | −11.86 | −1.82 | 1.79 | −1.82 | −10.07 | −42.13 |
| Me2Sn(S4MoVa) | −10.84 | 0.00 | −10.83 | −0.53 | 1.19 | −0.53 | −9.64 | −40.33 |
| Ph2Sn(SBoVa) | −11.07 | 0.03 | −11.04 | −1.86 | 1.79 | −1.86 | −9.25 | −38.70 |
| Me2Sn(SBoVa) | −10.58 | −0.01 | −10.59 | −0.54 | 1.19 | −0.54 | −9.40 | −39.33 |
| Compound | Me2Sn(S2MoVa) | Me2Sn(S4MoVa) | Me2Sn(SBoVa) |
|---|---|---|---|
| Formula | C19H22N2O2S2Sn | C19H22N2O2S2Sn | C18H20N2O2S2Sn |
| Formula weight | 493.19 | 493.19 | 479.19 |
| Crystal colour | light-yellow | light-yellow | light-yellow |
| Crystal size/mm3 | 0.03 × 0.05 × 0.10 | 0.05 × 0.13 × 0.20 | 0.03 × 0.05 × 0.10 |
| Crystal system | Monoclinic | Triclinic | Triclinic |
| Space group | Pn | P | P |
| a/Å | 11.3716(9) | 10.5908(4) | 8.9408(4) |
| b/Å | 12.0262(8) | 13.9243(5) | 10.4182(4) |
| c/Å | 16.3644(15) | 14.3974(5) | 22.6034(12) |
| α/° | 90 | 95.596(3) | 97.713(4) |
| β/° | 109.285(9) | 93.693(3) | 94.600(4) |
| γ/° | 90 | 102.116(2) | 111.634(4) |
| V/Å3 | 2112.4(3) | 2058.11(13) | 1920.11(16) |
| Z | 4 | 4 | 4 |
| Dc/g cm−3 | 1.551 | 1.592 | 1.658 |
| F(000) | 992 | 992 | 960 |
| μ(MoKα)/mm−1 | 1.422 | 1.46 | 1.562 |
| Measured data | 6348 | 18090 | 15229 |
| θ range/° | 3.6–29.4 | 3.5–29.4 | 3.4–29.4 |
| Unique data | 6348 | 9402 | 8720 |
| Observed data (I ≥ 2.0σ(I)) | 5090 | 7605 | 6671 |
| No. parameters | 478 | 477 | 457 |
| R, obs. data; all data | 0.037; 0.064 | 0.034; 0.070 | 0.041; 0.083 |
| a; b in weighting scheme | 0.022; 0 | 0.026; 0.240 | 0.032; 0.781 |
| Rw, obs. data; all data | 0.055; 0.074 | 0.049; 0.078 | 0.062; 0.092 |
| GoF | 0.99 | 1.01 | 1.01 |
| Range of residual electron | |||
| density peaks/eÅ−3 | −0.56–0.66 | −0.46–0.75 | −1.03–0.63 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Md Yusof, E.N.; M. Latif, M.A.; M. Tahir, M.I.; Sakoff, J.A.; Simone, M.I.; Page, A.J.; Veerakumarasivam, A.; Tiekink, E.R.T.; B. S. A. Ravoof, T. o-Vanillin Derived Schiff Bases and Their Organotin(IV) Compounds: Synthesis, Structural Characterisation, In-Silico Studies and Cytotoxicity. Int. J. Mol. Sci. 2019, 20, 854. https://doi.org/10.3390/ijms20040854
Md Yusof EN, M. Latif MA, M. Tahir MI, Sakoff JA, Simone MI, Page AJ, Veerakumarasivam A, Tiekink ERT, B. S. A. Ravoof T. o-Vanillin Derived Schiff Bases and Their Organotin(IV) Compounds: Synthesis, Structural Characterisation, In-Silico Studies and Cytotoxicity. International Journal of Molecular Sciences. 2019; 20(4):854. https://doi.org/10.3390/ijms20040854
Chicago/Turabian StyleMd Yusof, Enis Nadia, Muhammad A. M. Latif, Mohamed I. M. Tahir, Jennette A. Sakoff, Michela I. Simone, Alister J. Page, Abhi Veerakumarasivam, Edward R. T. Tiekink, and Thahira B. S. A. Ravoof. 2019. "o-Vanillin Derived Schiff Bases and Their Organotin(IV) Compounds: Synthesis, Structural Characterisation, In-Silico Studies and Cytotoxicity" International Journal of Molecular Sciences 20, no. 4: 854. https://doi.org/10.3390/ijms20040854
APA StyleMd Yusof, E. N., M. Latif, M. A., M. Tahir, M. I., Sakoff, J. A., Simone, M. I., Page, A. J., Veerakumarasivam, A., Tiekink, E. R. T., & B. S. A. Ravoof, T. (2019). o-Vanillin Derived Schiff Bases and Their Organotin(IV) Compounds: Synthesis, Structural Characterisation, In-Silico Studies and Cytotoxicity. International Journal of Molecular Sciences, 20(4), 854. https://doi.org/10.3390/ijms20040854