Strategies to Treat Chronic Pain and Strengthen Impaired Descending Noradrenergic Inhibitory System


Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Descending Noradrenergic Inhibition from the LC
2.1. Physiological Role of the LC
2.2. Normal State
2.3. Early Stage of Neuropathic Pain
2.4. Chronic Neuropathic Pain
3. Gabapentinoids
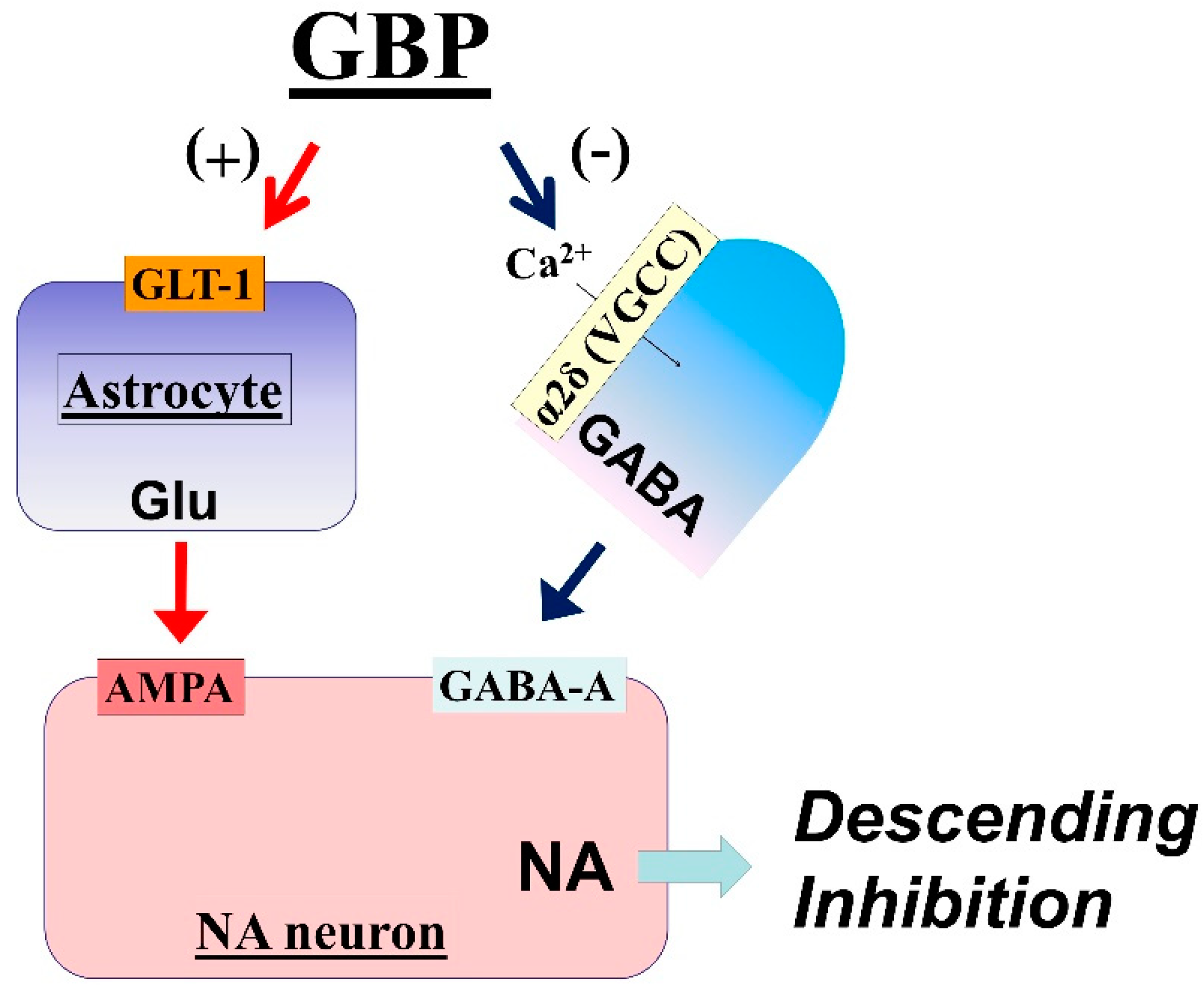
3.1. LC Is an Important Target of Gabapentin Analgesia
3.2. Mechanisms of LC Activation by Gabapentin
3.3. Impaired Gabapentin Analgesia in Chronic Neuropathic Pain
4. Antidepressants
4.1. Analgesic Mechanisms of Antidepressants for Neuropathic Pain
4.2. Actions of Antidepressants on the LC
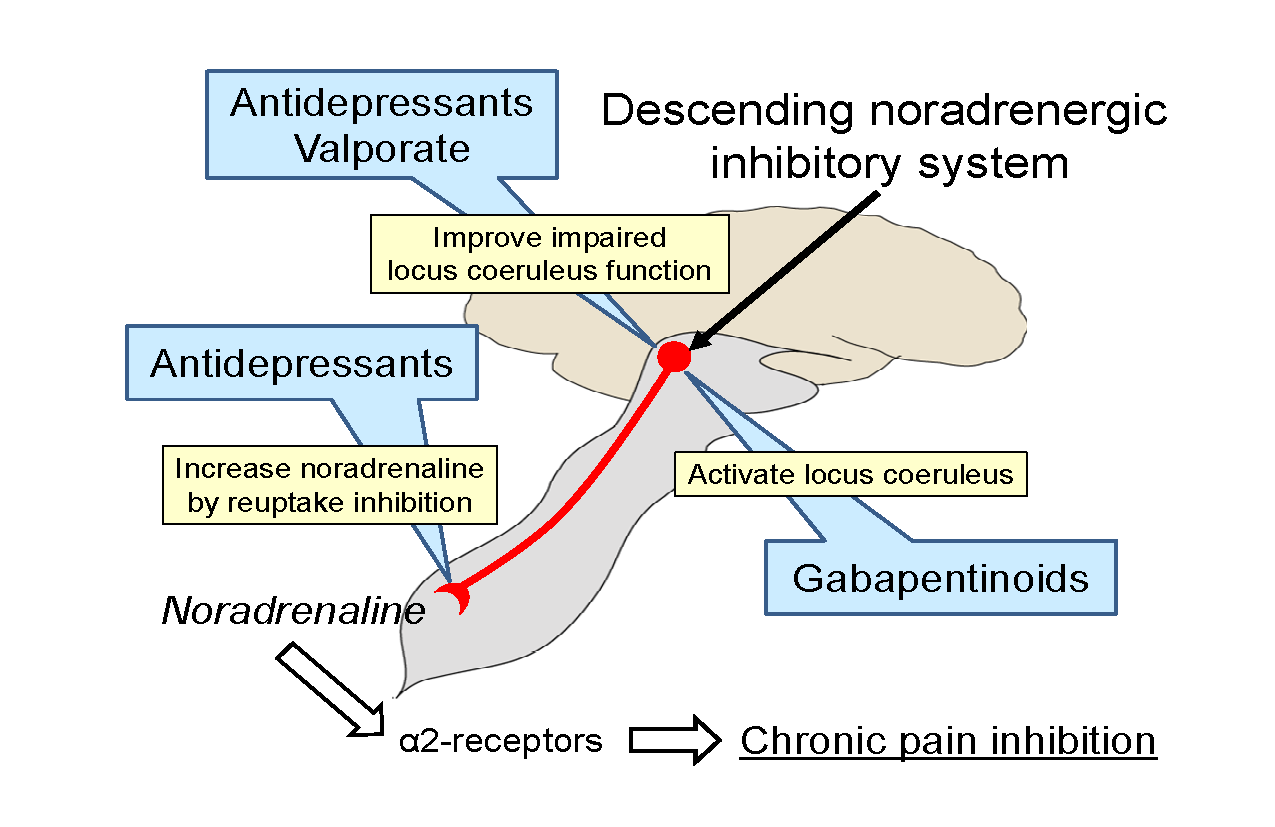
5. Strategies to Manage Neuropathic Pain on the Basis of Animal Studies
6. Conclusions
Funding
Conflicts of Interest
References
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Sindrup, S.H.; Jensen, T.S. The evidence for pharmacological treatment of neuropathic pain. Pain 2010, 150, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Attal, N.; Cruccu, G.; Baron, R.; Haanpää, M.; Hansson, P.; Jensen, T.S.; Nurmikko, T. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur. J. Neurol. 2010, 17, 1113-e88. [Google Scholar] [CrossRef]
- Dworkin, R.H.; O’Connor, B.; Backonja, M.; Farrar, J.T.; Finnerup, N.B.; Jensen, T.S.; Kalso, E.A.; Loeser, J.D.; Miaskowski, C.; Nurmikko, T.J.; et al. Pharmacologic management of neuropathic pain: Evidence-based recommendations. Pain 2007, 132, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Calandre, E.P.; Rico-Villademoros, F.; Slim, M. An update on pharmacotherapy for the treatment of fibromyalgia. Expert Opin. Pharmacother. 2015, 16, 1347–1368. [Google Scholar] [CrossRef] [PubMed]
- Proudfit, H.K.; Clark, F.M. The projections of locus coeruleus neurons to the spinal cord. Prog. Brain Res. 1991, 88, 123–141. [Google Scholar] [PubMed]
- Singewald, N.; Philippu, A. Release of neurotransmitters in the locus coeruleus. Prog. Neurobiol. 1998, 56, 237–267. [Google Scholar] [CrossRef]
- Loughlin, S.E.; Foote, S.L.; Grzanna, R. Efferent projections of nucleus locus coeruleus: Morphologic subpopulations have different efferent targets. Neuroscience 1986, 18, 307–319. [Google Scholar] [CrossRef]
- Hickey, L.; Li, Y.; Fyson, S.J.; Watson, T.C.; Perrins, R.; Hewinson, J.; Teschemacher, A.G.; Furue, H.; Lumb, B.M.; Pickering, A.E. Optoactivation of locus ceruleus neurons evokes bidirectional changes in thermal nociception in rats. J. Neurosci. 2014, 34, 4148–4160. [Google Scholar] [CrossRef]
- Millan, M.J. Descending control of pain. Prog. Neurobiol. 2002, 66, 355–474. [Google Scholar] [CrossRef]
- Llorca-Torralba, M.; Borges, G.; Neto, F.; Mico, J.A.; Berrocoso, E. Noradrenergic Locus Coeruleus pathways in pain modulation. Neuroscience 2016, 338, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.L.; Wu, Z.Z.; Zhou, H.Y.; Chen, S.R.; Zhang, H.M.; Li, D.P. Modulation of pain transmission by G-protein-coupled receptors. Pharmacol. Ther. 2008, 117, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Paqueron, X.; Conklin, D.; Eisenach, J.C. Plasticity in action of intrathecal clonidine to mechanical but not thermal nociception after peripheral nerve injury. Anesthesiology 2003, 99, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Saito, S.; Obata, H. Dexmedetomidine decreases hyperalgesia in neuropathic pain by increasing acetylcholine in the spinal cord. Neurosci. Lett. 2012, 529, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Clayton, B.A.; Johnson, J.E.; Eisenach, J.C. Brain derived nerve growth factor induces spinal noradrenergic fiber sprouting and enhances clonidine analgesia following nerve injury in rats. Pain 2008, 136, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Eisenach, J.C. Spinal alpha 2-adrenoceptor-mediated analgesia in neuropathic pain reflects brain-derived nerve growth factor and changes in spinal cholinergic neuronal function. Anesthesiology 2010, 113, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Eisenach, J.C.; DuPen, S.; Dubois, M.; Miguel, R.; Allin, D. Epidural clonidine analgesia for intractable cancer pain. The Epidural Clonidine Study Group. Pain 1995, 61, 391–399. [Google Scholar] [CrossRef]
- Sindrup, S.H.; Otto, M.; Finnerup, N.B.; Jensen, T.S. Antidepressants in the treatment of neuropathic pain. Basic Clin. Pharmacol. Toxicol. 2005, 96, 399–409. [Google Scholar] [CrossRef]
- Hayashida, K.; DeGoes, S.; Curry, R.; Eisenach, J.C. Gabapentin activates spinal noradrenergic activity in rats and humans and reduces hypersensitivity after surgery. Anesthesiology 2007, 106, 557–562. [Google Scholar] [CrossRef]
- Tanabe, M.; Takasu, K.; Takeuchi, Y.; Ono, H. Pain relief by gabapentin and pregabalin via supraspinal mechanisms after peripheral nerve injury. J. Neurosci. Res. 2008, 86, 3258–3264. [Google Scholar] [CrossRef]
- Kimura, M.; Suto, T.; Morado-Urbina, C.E.; Peters, C.M.; Eisenach, J.C.; Hayashida, K. Impaired pain-evoked analgesia after nerve injury in rats reflects altered glutamate regulation in the locus coeruleus. Anesthesiology 2015, 123, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Kimura, M.; Suto, T.; Eisenach, J.C.; Hayashida, K. Down-regulation of astroglial glutamate transporter-1 in the locus coeruleus impairs pain-evoked endogenous analgesia in rats. Neurosci. Lett. 2015, 608, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.N.; Rice, D.A.; McNair, P.J. Conditioned pain modulation in populations with chronic pain: A systematic review and meta-analysis. J. Pain 2012, 13, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Takasu, K.; Kasuya, N.; Shimizu, S.; Honda, M.; Ono, H. Role of descending noradrenergic system and spinal alpha2-adrenergic receptors in the effects of gabapentin on thermal and mechanical nociception after partial nerve injury in the mouse. Br. J. Pharmacol. 2005, 144, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Obata, H.; Nakajima, K.; Eisenach, J.C. Gabapentin acts within the locus coeruleus to alleviate neuropathic pain. Anesthesiology 2008, 109, 1077–1084. [Google Scholar] [CrossRef]
- Miyazaki, R.; Yamamoto, T. The efficacy of morphine, pregabalin, gabapentin, and duloxetine on mechanical allodynia is different from that on neuroma pain in the rat neuropathic pain model. Anesth. Analg. 2012, 115, 182–188. [Google Scholar] [CrossRef]
- Kitamura, R.; Andoh, T.; Mizoguchi, S.; Saito, Y.; Takahata, H.; Kuraishi, Y. Gabapentin inhibits bortezomib-induced mechanical allodynia through supraspinal action in mice. J. Pharmacol. Sci. 2014, 124, 502–510. [Google Scholar] [CrossRef]
- Gee, N.S.; Brown, J.P.; Dissanayake, V.U.; Offord, J.; Thurlow, R.; Woodruff, G.N. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J. Biol. Chem. 1996, 271, 5768–5776. [Google Scholar] [CrossRef]
- Li, C.Y.; Zhang, X.L.; Matthews, E.A.; Li, K.W.; Kurwa, A.; Boroujerdi, A.; Gross, J.; Gold, M.S.; Dickenson, A.H.; Feng, G.; et al. Calcium channel alpha2delta1 subunit mediates spinal hyperexcitability in pain modulation. Pain 2006, 125, 20–34. [Google Scholar] [CrossRef]
- Rauck, R.; Coffey, R.J.; Schultz, D.M.; Wallace, M.S.; Webster, L.R.; McCarville, S.E.; Grigsby, E.J.; Page, L.M. Intrathecal gabapentin to treat chronic intractable noncancer pain. Anesthesiology 2013, 119, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.P. Mechanisms of action of gabapentin. Rev. Neurol. 1997, 153 (Suppl. 1), S39–S45. [Google Scholar] [PubMed]
- Yoshizumi, M.; Parker, R.A.; Eisenach, J.C.; Hayashida, K. Gabapentin inhibits gamma-amino butyric acid release in the locus coeruleus but not in the spinal dorsal horn after peripheral nerve injury in rats. Anesthesiology 2012, 116, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Gotz, E.; Feuerstein, T.J.; Lais, A.; Meyer, D.K. Effects of gabapentin on release of gamma-aminobutyric acid from slices of rat neostriatum. Arzneimittelforschung 1993, 43, 636–638. [Google Scholar] [PubMed]
- Petroff, O.A.; Rothman, D.L.; Behar, K.L.; Lamoureux, D.; Mattson, R.H. The effect of gabapentin on brain gamma-aminobutyric acid in patients with epilepsy. Ann. Neurol. 1996, 39, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.P.; Gee, N.S.; Su, T.Z.; Kocsis, J.D.; Welty, D.F.; Brown, J.P.; Dooley, D.J.; Boden, P.; Singh, L. A summary of mechanistic hypotheses of gabapentin pharmacology. Epilepsy Res. 1998, 29, 233–249. [Google Scholar] [CrossRef]
- Brawek, B.; Loffler, M.; Weyerbrock, A.; Feuerstein, T.J. Effects of gabapentin and pregabalin on K+-evoked 3H-GABA and 3H-glutamate release from human neocortical synaptosomes. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 379, 361–369. [Google Scholar] [CrossRef]
- Takasu, K.; Ono, H.; Tanabe, M. Gabapentin produces PKA-dependent pre-synaptic inhibition of GABAergic synaptic transmission in LC neurons following partial nerve injury in mice. J. Neurochem. 2008, 105, 933–942. [Google Scholar] [CrossRef]
- Suto, T.; Severino, A.L.; Eisenach, J.C.; Hayashida, K. Gabapentin increases extracellular glutamatergic level in the locus coeruleus via astroglial glutamate transporter-dependent mechanisms. Neuropharmacology 2014, 81, 95–100. [Google Scholar] [CrossRef]
- Tanaka, S.; Tsuchida, A.; Kiuchi, Y.; Oguchi, K.; Numazawa, S.; Yoshida, T. GABAergic modulation of hippocampal glutamatergic neurons: An in vivo microdialysis study. Eur. J. Pharmacol. 2003, 465, 61–67. [Google Scholar] [CrossRef]
- Harte, M.; O’Connor, W.T. Evidence for a selective prefrontal cortical GABA(B) receptor-mediated inhibition of glutamate release in the ventral tegmental area: A dual probe microdialysis study in the awake rat. Neuroscience 2005, 130, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, M.; Eisenach, J.C.; Hayashida, K. Riluzole and gabapentinoids activate glutamate transporters to facilitate glutamate-induced glutamate release from cultured astrocytes. Eur. J. Pharmacol. 2012, 677, 87–92. [Google Scholar] [CrossRef]
- Kimura, M.; Eisenach, J.C.; Hayashida, K. Gabapentin loses efficacy over time after nerve injury in rats: Role of glutamate transporter-1 in the locus coeruleus. Pain 2016, 157, 2024–2032. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.D.; Calcutt, N.A.; Higuera, E.S.; Valder, C.R.; Song, Y.H.; Svensson, C.I.; Myers, R.R. Injury type-specific calcium channel alpha 2 delta-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J. Pharmacol. Exp. Ther. 2002, 303, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Fishbain, D.A.; Cutler, R.B.; Rosomoff, H.L.; Rosomoff, R.S. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta-analysis. Psychosom. Med. 1998, 60, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Onghena, P.; Van Houdenhove, B. Antidepressant-induced analgesia in chronic non-malignant pain: A meta-analysis of 39 placebo-controlled studies. Pain 1992, 49, 205–219. [Google Scholar] [CrossRef]
- Micó, J.A.; Ardid, D.; Berrocoso, E.; Eschalier, A. Antidepressants and pain. Trends Pharmacol. Sci. 2006, 27, 348–354. [Google Scholar] [CrossRef]
- Dharmshaktu, P.; Tayal, V.; Kalra, B.S. Efficacy of antidepressants as analgesics: A review. J. Clin. Pharmacol. 2012, 52, 6–17. [Google Scholar] [CrossRef]
- Berton, O.; Nestler, E.J. New approaches to antidepressant drug discovery: Beyond monoamines. Nat. Rev. Neurosci. 2006, 7, 137–151. [Google Scholar] [CrossRef]
- McQuay, H.J.; Tramèr, M.; Nye, B.A.; Carroll, D.; Wiffen, P.J.; Moore, R.A. A systematic review of antidepressants in neuropathic pain. Pain 1996, 68, 217–227. [Google Scholar] [CrossRef]
- Cook, R.J.; Sackett, D.L. The number needed to treat: A clinically useful measure of treatment effect. BMJ 1996, 310, 452–454. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Otto, M.; McQuay, H.J.; Jensen, T.S.; Sindrup, S.H. Algorithm for neuropathic pain treatment: An evidence based proposal. Pain 2005, 118, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Suto, T.; Saito, S.; Obata, H. Repeated administration of duloxetine suppresses neuropathic pain by accumulating effects of noradrenaline in the spinal cord. Anesth. Analg. 2018, 126, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Hiroki, T.; Suto, T.; Saito, S.; Obata, H. Repeated administration of amitriptyline in neuropathic pain: Modulation of the noradrenergic descending inhibitory system. Anesth. Analg. 2017, 125, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Obata, H.; Iriuchijima, N.; Saito, S. An increase in spinal cord noradrenaline is a major contributor to the antihyperalgesic effect of antidepressants after peripheral nerve injury in the rat. Pain 2012, 153, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Obata, H. Analgesic Mechanisms of Antidepressants for Neuropathic Pain. Int. J. Mol. Sci. 2017, 18, 2483. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; Stuhr-Hansen, N.; Zachariassen, L.G.; Koldsø, H.; Schiøtt, B.; Strømgaard, K.; Kristensen, A.S. Molecular basis for selective serotonin reuptake inhibition by the antidepressant agent fluoxetine (Prozac). Mol. Pharmacol. 2014, 85, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Owens, M.J.; Knight, D.L.; Nemeroff, C.B. Paroxetine binding to the rat norepinephrine transporter in vivo. Biol. Psychiatry 2000, 47, 842–845. [Google Scholar] [CrossRef]
- Chen, M.; Hoshino, H.; Saito, S.; Yang, Y.; Obata, H. Spinal dopaminergic involvement in the antihyperalgesic effect of antidepressants in a rat model of neuropathic pain. Neurosci. Lett. 2017, 649, 116–123. [Google Scholar] [CrossRef]
- Morón, J.A.; Brockington, A.; Wise, R.A.; Rocha, B.A.; Hope, B.T. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: Evidence from knock-out mouse lines. J. Neurosci. 2002, 22, 389–395. [Google Scholar] [CrossRef]
- Aston-Jones, G.; Bloom, F.E. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J. Neurosci. 1981, 1, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Tsuruoka, M.; Tamaki, J.; Maeda, M.; Hayashi, B.; Inoue, T. Biological implications of coeruleospinal inhibition of nociceptive processing in the spinal cord. Front. Integr. Neurosci. 2012, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Howorth, P.W.; Teschemacher, A.G.; Pickering, A.E. Retrograde adenoviral vector targeting of nociresponsive pontospinal noradrenergic neurons in the rat in vivo. J. Comp. Neurol. 2009, 512, 141–157. [Google Scholar] [CrossRef]
- Peters, C.M.; Hayashida, K.; Suto, T.; Houle, T.T.; Aschenbrenner, C.A.; Martin, T.J.; Eisenach, J.C. Individual differences in acute pain-induced endogenous analgesia predict time to resolution of postoperative pain in the rat. Anesthesiology 2015, 122, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, H.; Suto, T.; Saito, S.; Obata, H. Amitriptyline, but not pregabalin, reverses the attenuation of noxious stimulus-induced analgesia after nerve injury in rats. Anesth. Analg. 2016, 123, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Mateo, Y.; Fernández-Pastor, B.; Meana, J.J. Acute and chronic effects of desipramine and clorgyline on alpha(2)-adrenoceptors regulating noradrenergic transmission in the rat brain: A dual-probe microdialysis study. Br. J. Pharmacol. 2001, 133, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Grandoso, L.; Pineda, J.; Ugedo, L. Comparative study of the effects of desipramine and reboxetine on locus coeruleus neurons in rat brain slices. Neuropharmacology 2004, 46, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.M.; Weiss, J.M. Effects of chronic antidepressant drug administration and electroconvulsive shock on locus coeruleus electrophysiologic activity. Biol. Psychiatry 2001, 49, 117–129. [Google Scholar] [CrossRef]
- Alba-Delgado, C.; Mico, J.A.; Sánchez-Blázquez, P.; Berrocoso, E. Analgesic antidepressants promote the responsiveness of locus coeruleus neurons to noxious stimulation: Implications for neuropathic pain. Pain 2012, 153, 1438–1449. [Google Scholar] [CrossRef]
- Nibuya, M.; Morinobu, S.; Duman, R.S. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 1995, 15, 7539–7547. [Google Scholar] [CrossRef]
- Kajitani, N.; Hisaoka-Nakashima, K.; Morioka, N.; Okada-Tsuchioka, M.; Kaneko, M.; Kasai, M.; Shibasaki, C.; Nakata, Y.; Takebayashi, M. Antidepressant acts on astrocytes leading to an increase in the expression of neurotrophic/growth factors: Differential regulation of FGF-2 by noradrenaline. PLoS ONE 2012, 7, e51197. [Google Scholar] [CrossRef] [PubMed]
- Suto, T.; Kado, D.; Obata, H.; Saito, S. A tropomyosin receptor kinase B receptor activation in the locus coeruleus restores impairment of endogenous analgesia at a late stage following nerve injury in rats. J. Pain 2018. [Google Scholar] [CrossRef] [PubMed]
- Reimers, J.M.; Loweth, J.A.; Wolf, M.E. BDNF contributes to both rapid and homeostatic alterations in AMPA receptor surface expression innucleus accumbens medium spiny neurons. Eur. J. Neurosci. 2014, 39, 1159–1169. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayashida, K.-i.; Obata, H. Strategies to Treat Chronic Pain and Strengthen Impaired Descending Noradrenergic Inhibitory System. Int. J. Mol. Sci. 2019, 20, 822. https://doi.org/10.3390/ijms20040822
Hayashida K-i, Obata H. Strategies to Treat Chronic Pain and Strengthen Impaired Descending Noradrenergic Inhibitory System. International Journal of Molecular Sciences. 2019; 20(4):822. https://doi.org/10.3390/ijms20040822
Chicago/Turabian StyleHayashida, Ken-ichiro, and Hideaki Obata. 2019. "Strategies to Treat Chronic Pain and Strengthen Impaired Descending Noradrenergic Inhibitory System" International Journal of Molecular Sciences 20, no. 4: 822. https://doi.org/10.3390/ijms20040822
APA StyleHayashida, K.-i., & Obata, H. (2019). Strategies to Treat Chronic Pain and Strengthen Impaired Descending Noradrenergic Inhibitory System. International Journal of Molecular Sciences, 20(4), 822. https://doi.org/10.3390/ijms20040822