
Transglutaminase2: An Enduring Enzyme in Diabetes and Age-Related Metabolic Diseases


Abstract
1. Introduction
2. The Transglutaminase Enzyme Family
3. TG2 Expression and Aging
4. Molecular/Structural Features and Operation Mechanism
4.1. Mechanism of Activation and Inhibition
4.2. Interconversion of Active and Inactive Forms of TG2
4.3. Interaction with GTP
4.4. Inactivation of Human TG2 by ERp57 Protein
4.5. TG2 Kinase Activity
5. TG2 Roles under Normal and Diseased States
5.1. Protein Cross-Linking, Cell Membrane and ECM Stability
5.2. TG2 as an Inflammatory Biomarker
5.3. TG2 as Molecular Drug Target in Diabetes
5.4. TG2 in Pathogenesis of Fibroproliferative Disorders
5.5. Relationship of Other TGs to Fibrosis
5.6. TG2 in Angiogenesis and Tubule Formation
5.7. TG2 in Progression of Neurodegenerative Diseases
6. TG2 Inhibitors
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFU | Diabetic foot ulcer |
| MMPs | Matrix metalloproteinases |
| ECM | Extracellular matrix |
| TG2 | Transglutaminase |
| GTP | Guanosine triphosphate |
| GDP | Guanosine diphosphate |
| FXIII | Factor XIII |
| PLCδ1 | Phospholipase Cδ1 |
| Ghα | G proteinα |
| MALS | Multiangle light scattering |
| TRX | Thioredoxin |
| ERp57 | ER-resident protein 57 |
| PDI | Protein disulphide isomerase |
| QSOX1 | Quiescin sulfhydryl oxidase1 |
| TGF-β1 | Transforming growth factor |
| Fn | Fibronectin |
| ADSCs | Adipose-derived stem cells |
| TGF-3 | Transforming growth factor-3 |
| LPS | Lipopolysaccharide |
| IL-1β | Interleukin-1β |
| CSU | Spontaneous urticaria |
| AU | Acute urticaria |
| CD | Celiac disease |
| Ig | Immunoglobulin |
| ROS | Reactive oxygen species |
| VEGF | Vascular endothelial growth factor |
| MDC | Monodansylcadaverine |
| IKK | IkB kinase |
| OoC | Organ-on-a-chip |
| NFkB | Nuclear factor kappaB |
| FAK | Focal adhesion kinase |
| α-SMA | Alpha-smooth muscle actin |
| IPF | Idiopathic pulmonary fibrosis |
| VEGFR2 | Vascular endothelial growth factor receptor2 |
| EndMT | Endothelial-mesenchymal transition |
| ECs | Endothelial cells |
| OPCs | Oligodendrocytes precursor cells |
| Aβ | Amyloid-beta |
| GGEL | γ-glutamyl-ε-lysine |
| AD | Alzheimer’s diseases |
| HD | Huntington’s diseases |
| PD | Parkinson’s disease |
| SAR | Structure–activity relationship |
| DHI | 3-bromo-4,5-dihydroisoxazole |
| siRNA | interfering RNA |
| LTBP-1 | Latent TGF-β1 binding protein-1 |
| CAD | Coronary artery disease |
| UCEC | Uterine corpus endometrial carcinoma |
| BPD | Bronchopulmonary dysplasia |
| Rb | Retinoblastoma |
| IGFBP-3 | Insulin-like growth factor-binding protein-3 |
References
- Ong, K.L.; Stafford, L.K.; McLaughlin, S.A.; Boyko, E.J.; Vollset, S.E.; Smith, A.E.; Dalton, B.E.; Duprey, J.; Cruz, J.A.; Hagins, H. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023. [Google Scholar] [CrossRef] [PubMed]
- Kunkemoeller, B.; Kyriakides, T.R. Redox signaling in diabetic wound healing regulates extracellular matrix deposition. Antioxid. Redox Signal. 2017, 27, 823–838. [Google Scholar] [CrossRef] [PubMed]
- Westfall, A.E. Evaluating the Initial Management and Referral Process for Patients with Diabetic Foot Ulcers. Ph.D. Thesis, University of Kansas, Lawrence, KS, USA, 2019. [Google Scholar]
- Tavelli, B. Market and Business Potential Analysis of Wound Healing Products of Dermobor. Master’s Thesis, Fen Bilimleri Enstitüsü, Istanbul, Turkey, 2019. [Google Scholar]
- Janis, J.E.; Harrison, B. Wound healing: Part I. Basic science. Plast. Reconstr. Surg. 2016, 138, 9S–17S. [Google Scholar] [CrossRef]
- Chakroborty, D.; Goswami, S.; Basu, S.; Sarkar, C. Catecholamines in the regulation of angiogenesis in cutaneous wound healing. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 14093. [Google Scholar] [CrossRef]
- Liu, J.; Shu, B.; Zhou, Z.; Xu, Y.; Liu, Y.; Wang, P.; Xiong, K.; Xie, J. Involvement of miRNA203 in the proliferation of epidermal stem cells during the process of DM chronic wound healing through Wnt signal pathways. Stem Cell Res. Ther. 2020, 11, 348. [Google Scholar] [CrossRef] [PubMed]
- Sima, L.E.; Matei, D.; Condello, S. The Outside-In Journey of Tissue Transglutaminase in Cancer. Cells 2022, 11, 1779. [Google Scholar] [CrossRef]
- Doa’a, G.A.; Tranchant, C.C.; Al-Dwairi, A.; Alqudah, M.; Al-Shboul, O.; Hiram, R.; Allen, B.G.; Jaradat, S.; Alqbelat, J.; Abu-Zaiton, A.S. Implications of enigmatic transglutaminase 2 (TG2) in cardiac diseases and therapeutic developments. Biochem. Pharmacol. 2022, 201, 115104. [Google Scholar]
- Nurminskaya, M.V.; Belkin, A.M. Cellular functions of tissue transglutaminase. Int. Rev. Cell Mol. Biol. 2012, 294, 1–97. [Google Scholar]
- Shao, M.; Cao, L.; Shen, C.; Satpathy, M.; Chelladurai, B.; Bigsby, R.M.; Nakshatri, H.; Matei, D. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009, 69, 9192–9201. [Google Scholar] [CrossRef]
- Tatsukawa, H.; Hitomi, K. Role of transglutaminase 2 in cell death, survival, and fibrosis. Cells 2021, 10, 1842. [Google Scholar] [CrossRef]
- Wang, Z.; Telci, D.; Griffin, M. Importance of syndecan-4 and syndecan-2 in osteoblast cell adhesion and survival mediated by a tissue transglutaminase−fibronectin complex. Exp. Cell Res. 2011, 317, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Tempest, R.; Guarnerio, S.; Maani, R.; Cooper, J.; Peake, N. The biological and biomechanical role of transglutaminase-2 in the tumour microenvironment. Cancers 2021, 13, 2788. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, A.-M.; Liu, W. Transglutaminase 2 in cancer. Am. J. Cancer Res. 2015, 5, 2756. [Google Scholar] [PubMed]
- Caccamo, D.; Currò, M.; Ientile, R. Potential of transglutaminase 2 as a therapeutic target. Expert Opin. Ther. Targets 2010, 14, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, T.; Ota, M.; Nio, N.; Motoki, M. Comparison of substrate specificities of transglutaminases using synthetic peptides as acyl donors. Biosci. Biotechnol. Biochem. 2000, 64, 2608–2613. [Google Scholar] [CrossRef]
- Duarte, L.; Matte, C.R.; Bizarro, C.V.; Ayub, M.A.Z. Transglutaminases: Part I—Origins, sources, and biotechnological characteristics. World J. Microbiol. Biotechnol. 2020, 36, 15. [Google Scholar] [CrossRef]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef]
- Sane, D. Antibodies to tissue transglutaminase: An immune link between the gut, the coronaries and the myocardium? J. Intern. Med. 2008, 263, 1–3. [Google Scholar] [CrossRef]
- Aufenvenne, K.; Larcher, F.; Hausser, I.; Duarte, B.; Oji, V.; Nikolenko, H.; Del Rio, M.; Dathe, M.; Traupe, H. Topical enzyme-replacement therapy restores transglutaminase 1 activity and corrects architecture of transglutaminase-1-deficient skin grafts. Am. J. Hum. Genet. 2013, 93, 620–630. [Google Scholar] [CrossRef]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef]
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef]
- Grenard, P.; Bates, M.K.; Aeschlimann, D. Evolution of transglutaminase genes: Identification of a transglutaminase gene cluster on human chromosome 15q15: Structure of the gene encoding transglutaminase X and a novel gene family member, transglutaminase Z. J. Biol. Chem. 2001, 276, 33066–33078. [Google Scholar] [CrossRef]
- Jiang, W.G.; Ablin, R.J. Prostate transglutaminase: A unique transglutaminase and its role in prostate cancer. Biomark. Med. 2011, 5, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Candi, E.; Paradisi, A.; Terrinoni, A.; Pietroni, V.; Oddi, S.; Cadot, B.; Jogini, V.; Meiyappan, M.; Clardy, J.; Finazzi-Agro, A. Transglutaminase 5 is regulated by guanine–adenine nucleotides. Biochem. J. 2004, 381, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Tang, B.S.; Lan, W.; Song, N.N.; Huang, Y.; Zhang, L.; Guan, W.J.; Shi, Y.T.; Shen, L.; Jiang, H. Distribution of transglutaminase 6 in the central nervous system of adult mice. Anat. Rec. 2013, 296, 1576–1587. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Hu, K.; Frantz, S.; Jaffer, F.A.; Tung, C.-H.; Hiller, K.-H.; Voll, S.; Nordbeck, P.; Sosnovik, D.; Gattenlöhner, S. Factor XIII deficiency causes cardiac rupture, impairs wound healing, and aggravates cardiac remodeling in mice with myocardial infarction. Circulation 2006, 113, 1196–1202. [Google Scholar] [CrossRef]
- Muszbek, L.; Yee, V.C.; Hevessy, Z. Blood coagulation factor XIII: Structure and function. Thromb. Res. 1999, 94, 271–305. [Google Scholar] [CrossRef]
- Wang, Z.; Collighan, R.J.; Gross, S.R.; Danen, E.H.; Orend, G.; Telci, D.; Griffin, M. RGD-independent cell adhesion via a tissue transglutaminase-fibronectin matrix promotes fibronectin fibril deposition and requires syndecan-4/2 and α5β1 integrin co-signaling. J. Biol. Chem. 2010, 285, 40212–40229. [Google Scholar] [CrossRef] [PubMed]
- Bass, M.D.; Morgan, M.R.; Roach, K.A.; Settleman, J.; Goryachev, A.B.; Humphries, M.J. p190RhoGAP is the convergence point of adhesion signals from α5β1 integrin and syndecan-4. J. Cell Biol. 2008, 181, 1013–1026. [Google Scholar] [CrossRef]
- Verderio, E.A.; Telci, D.; Okoye, A.; Melino, G.; Griffin, M. A novel RGD-independent cell adhesion pathway mediated by fibronectin-bound tissue transglutaminase rescues cells from anoikis. J. Biol. Chem. 2003, 278, 42604–42614. [Google Scholar] [CrossRef]
- Stoffels, J.M.; Zhao, C.; Baron, W. Fibronectin in tissue regeneration: Timely disassembly of the scaffold is necessary to complete the build. Cell. Mol. Life Sci. 2013, 70, 4243–4253. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Collighan, R.J.; Pytel, K.; Rathbone, D.L.; Li, X.; Griffin, M. Characterization of heparin-binding site of tissue transglutaminase: Its importance in cell surface targeting, matrix deposition, and cell signaling. J. Biol. Chem. 2012, 287, 13063–13083. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y. Transglutaminase 2 in inflammation. Front. Biosci. Landmark 2006, 11, 3026–3035. [Google Scholar] [CrossRef]
- Su, T.; Qin, X.-Y.; Furutani, Y. Transglutaminase 2 as a marker for inflammation and therapeutic target in sepsis. Int. J. Mol. Sci. 2021, 22, 1897. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, U.; Ferlosio, A.; Arcuri, G.; Spagnoli, L.G.; Orlandi, A. Transglutaminase 2 as a biomarker of osteoarthritis: An update. Amino Acids 2013, 44, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Bains, W. Transglutaminse 2 and EGGL, the protein cross-link formed by transglutaminse 2, as therapeutic targets for disabilities of old age. Rejuvenation Res. 2013, 16, 495–517. [Google Scholar] [CrossRef]
- Santhanam, L.; Tuday, E.C.; Webb, A.K.; Dowzicky, P.; Kim, J.H.; Oh, Y.J.; Sikka, G.; Kuo, M.; Halushka, M.K.; Macgregor, A.M. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ. Res. 2010, 107, 117–125. [Google Scholar] [CrossRef]
- Nemes, Z.; Fésüs, L.; Égerházi, A.; Keszthelyi, A.; Degrell, I.M. Nε (γ-glutamyl) lysine in cerebrospinal fluid marks Alzheimer type and vascular dementia. Neurobiol. Aging 2001, 22, 403–406. [Google Scholar] [CrossRef]
- Dedeoglu, A.; Kubilus, J.K.; Jeitner, T.M.; Matson, S.A.; Bogdanov, M.; Kowall, N.W.; Matson, W.R.; Cooper, A.J.; Ratan, R.R.; Beal, M.F. Therapeutic effects of cystamine in a murine model of Huntington’s disease. J. Neurosci. 2002, 22, 8942–8950. [Google Scholar] [CrossRef]
- Lin, C.H.S.; Chen, J.; Zhang, Z.; Johnson, G.V.; Cooper, A.J.; Feola, J.; Bank, A.; Shein, J.; Ruotsalainen, H.J.; Pihlajaniemi, T.A. Endostatin and transglutaminase 2 are involved in fibrosis of the aging kidney. Kidney Int. 2016, 89, 1281–1292. [Google Scholar] [CrossRef]
- Hong, G.U.; Ro, J.Y.; Bae, Y.; Kwon, I.-H.; Park, G.-H.; Choi, Y.H.; Choi, J.-H. Association of TG2 from mast cells and chronic spontaneous urticaria pathogenesis. Ann. Allergy Asthma Immunol. 2016, 117, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Lee, K.B.; Son, Y.H.; Shin, J.; Lee, J.-H.; Kim, H.-J.; Hong, A.-Y.; Bae, H.W.; Kwon, M.-A.; Lee, W.J. Transglutaminase 2 mediates UV-induced skin inflammation by enhancing inflammatory cytokine production. Cell Death Dis. 2017, 8, e3148. [Google Scholar] [CrossRef]
- Rosenthal, A.K.; Gohr, C.M.; Uzuki, M.; Masuda, I. Osteopontin promotes pathologic mineralization in articular cartilage. Matrix Biol. 2007, 26, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Macaione, V.; Aguennouz, M.; Mazzeo, A.; De Pasquale, M.; Russo, M.; Toscano, A.; De Luca, G.; Di Giorgio, R.M.; Vita, G.; Rodolico, C. Expression of transglutaminase 2 does not differentiate focal myositis from generalized inflammatory myopathies. Acta Neurol. Scand. 2008, 117, 393–398. [Google Scholar] [CrossRef]
- Su, C.C.; Su, T.R.; Lai, J.C.; Tsay, G.J.; Lin, H.K. Elevated transglutaminase-2 expression in the epidermis of psoriatic skin and its role in the skin lesion development. J. Dermatol. 2017, 44, 699–702. [Google Scholar] [CrossRef]
- Wang, Z.; Griffin, M. TG2, a novel extracellular protein with multiple functions. Amino Acids 2012, 42, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.; Khosla, C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol. Ther. 2007, 115, 232–245. [Google Scholar] [CrossRef]
- Benn, M.C.; Weber, W.; Klotzsch, E.; Vogel, V.; Pot, S.A. Tissue transglutaminase in fibrosis—More than an extracellular matrix cross-linker. Curr. Opin. Biomed. Eng. 2019, 10, 156–164. [Google Scholar] [CrossRef]
- Zhang, J.; Guttmann, R.P.; Johnson, G.V. Tissue transglutaminase is an in situ substrate of calpain: Regulation of activity. J. Neurochem. 1998, 71, 240–247. [Google Scholar] [CrossRef]
- Fok, J.Y.; Ekmekcioglu, S.; Mehta, K. Implications of tissue transglutaminase expression in malignant melanoma. Mol. Cancer Ther. 2006, 5, 1493–1503. [Google Scholar] [CrossRef]
- Bianchi, N.; Beninati, S.; Bergamini, C.M. Spotlight on the transglutaminase 2 gene: A focus on genomic and transcriptional aspects. Biochem. J. 2018, 475, 1643–1667. [Google Scholar] [CrossRef]
- Pinilla, E.; Comerma-Steffensen, S.; Prat-Duran, J.; Rivera, L.; Matchkov, V.V.; Buus, N.H.; Simonsen, U. Transglutaminase 2 inhibitor LDN 27219 age-dependently lowers blood pressure and improves endothelium-dependent vasodilation in resistance arteries. Hypertension 2021, 77, 216–227. [Google Scholar] [CrossRef]
- Stamnaes, J.; Pinkas, D.M.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 2010, 285, 25402–25409. [Google Scholar] [CrossRef]
- Oh, K.; Park, H.-B.; Byoun, O.-J.; Shin, D.-M.; Jeong, E.M.; Kim, Y.W.; Kim, Y.S.; Melino, G.; Kim, I.-G.; Lee, D.-S. Epithelial transglutaminase 2 is needed for T cell interleukin-17 production and subsequent pulmonary inflammation and fibrosis in bleomycin-treated mice. J. Exp. Med. 2011, 208, 1707–1719. [Google Scholar] [CrossRef]
- Piper, J.L.; Gray, G.M.; Khosla, C. High selectivity of human tissue transglutaminase for immunoactive gliadin peptides: Implications for celiac sprue. Biochemistry 2002, 41, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.-S.; Lin, C.-J.; Greenberg, C.S. Role of tissue transglutaminase-2 (TG2)-mediated aminylation in biological processes. Amino Acids 2017, 49, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Facchiano, A.; Facchiano, F. Transglutaminases and their substrates in biology and human diseases: 50 years of growing. Amino Acids 2009, 36, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Aeschlimann, D.; Paulsson, M. Cross-linking of laminin-nidogen complexes by tissue transglutaminase. A novel mechanism for basement membrane stabilization. J. Biol. Chem. 1991, 266, 15308–15317. [Google Scholar] [CrossRef]
- Case, A.; Stein, R.L. Kinetic analysis of the action of tissue transglutaminase on peptide and protein substrates. Biochemistry 2003, 42, 9466–9481. [Google Scholar] [CrossRef] [PubMed]
- Odii, B.O.; Coussons, P. Biological functionalities of transglutaminase 2 and the possibility of its compensation by other members of the transglutaminase family. Sci. World J. 2014, 2014, 714561. [Google Scholar] [CrossRef]
- Han, B.-G.; Cho, J.-W.; Cho, Y.D.; Jeong, K.-C.; Kim, S.-Y.; Lee, B.I. Crystal structure of human transglutaminase 2 in complex with adenosine triphosphate. Int. J. Biol. Macromol. 2010, 47, 190–195. [Google Scholar] [CrossRef]
- Jang, T.-H.; Lee, D.-S.; Choi, K.; Jeong, E.M.; Kim, I.-G.; Kim, Y.W.; Chun, J.N.; Jeon, J.-H.; Park, H.H. Crystal structure of transglutaminase 2 with GTP complex and amino acid sequence evidence of evolution of GTP binding site. PLoS ONE 2014, 9, e107005. [Google Scholar] [CrossRef] [PubMed]
- Begg, G.E.; Holman, S.R.; Stokes, P.H.; Matthews, J.M.; Graham, R.M.; Iismaa, S.E. Mutation of a critical arginine in the GTP-binding site of transglutaminase 2 disinhibits intracellular cross-linking activity. J. Biol. Chem. 2006, 281, 12603–12609. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Haylor, J.L.; Hau, Z.; Jones, R.A.; Vickers, M.E.; Wagner, B.; Griffin, M.; Saint, R.E.; Coutts, I.G.; El Nahas, A.M. Transglutaminase inhibition ameliorates experimental diabetic nephropathy. Kidney Int. 2009, 76, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Dihazi, H.; Dihazi, G.H.; Bibi, A.; Eltoweissy, M.; Mueller, C.A.; Asif, A.R.; Rubel, D.; Vasko, R.; Mueller, G.A. Secretion of ERP57 is important for extracellular matrix accumulation and progression of renal fibrosis, and is an early sign of disease onset. J. Cell Sci. 2013, 126, 3649–3663. [Google Scholar] [PubMed]
- Michael, C.Y.; Melkonian, A.V.; Ousey, J.A.; Khosla, C. Endoplasmic reticulum–resident protein 57 (ERp57) oxidatively inactivates human transglutaminase 2. J. Biol. Chem. 2018, 293, 2640–2649. [Google Scholar]
- Mishra, S.; Murphy, L.J. Tissue transglutaminase has intrinsic kinase activity: Identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J. Biol. Chem. 2004, 279, 23863–23868. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Murphy, L.J. Phosphorylation of transglutaminase 2 by PKA at Ser216 creates 14-3-3 binding sites. Biochem. Biophys. Res. Commun. 2006, 347, 1166–1170. [Google Scholar] [CrossRef]
- Mishra, S.; Melino, G.; Murphy, L.J. Transglutaminase 2 kinase activity facilitates protein kinase A-induced phosphorylation of retinoblastoma protein. J. Biol. Chem. 2007, 282, 18108–18115. [Google Scholar] [CrossRef]
- Mishra, S.; Murphy, L.J. The p53 oncoprotein is a substrate for tissue transglutaminase kinase activity. Biochem. Biophys. Res. Commun. 2006, 339, 726–730. [Google Scholar] [CrossRef]
- Mishra, S.; Saleh, A.; Espino, P.S.; Davie, J.R.; Murphy, L.J. Phosphorylation of histones by tissue transglutaminase. J. Biol. Chem. 2006, 281, 5532–5538. [Google Scholar] [CrossRef]
- Lee, K.-H.; Lee, N.; Lim, S.; Jung, H.; Ko, Y.-G.; Park, H.-Y.; Jang, Y.; Lee, H.; Hwang, K.-C. Calreticulin inhibits the MEK1, 2-ERK1, 2 pathway in α1-adrenergic receptor/Gh-stimulated hypertrophy of neonatal rat cardiomyocytes. J. Steroid Biochem. Mol. Biol. 2003, 84, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Katt, W.P.; Antonyak, M.A.; Cerione, R.A. Opening up about tissue transglutaminase: When conformation matters more than enzymatic activity. Med One 2018, 3, e180011. [Google Scholar] [PubMed]
- Hinz, B.; McCulloch, C.A.; Coelho, N.M. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp. Cell Res. 2019, 379, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Chau, G.; Walraven, M.; Boo, S.; Koehler, A.; Chow, M.L.; Olsen, A.L.; Im, M.; Lodyga, M.; Wells, R.G. The fibronectin ED-A domain enhances recruitment of latent TGF-β-binding protein-1 to the fibroblast matrix. J. Cell Sci. 2018, 131, jcs201293. [Google Scholar] [CrossRef] [PubMed]
- Troilo, H.; Steer, R.; Collins, R.F.; Kielty, C.M.; Baldock, C. Independent multimerization of Latent TGFβ Binding Protein-1 stabilized by cross-linking and enhanced by heparan sulfate. Sci. Rep. 2016, 6, 34347. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, M.; Cao, L.; Pincheira, R.; Emerson, R.; Bigsby, R.; Nakshatri, H.; Matei, D. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res. 2007, 67, 7194–7202. [Google Scholar] [CrossRef] [PubMed]
- Grose, R.; Hutter, C.; Bloch, W.; Thorey, I.; Watt, F.M.; Fässler, R.; Brakebusch, C.; Werner, S. A crucial role of β1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development 2002, 129, 2303–2315. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, N.; Hu, L.; Xi, Y.; Mi, W.; Ma, Y. Integrin β1 in adipose-derived stem cells accelerates wound healing via activating PI3K/AKT pathway. Tissue Eng. Regen. Med. 2020, 17, 183–192. [Google Scholar] [CrossRef]
- Cardoso, I.; Østerlund, E.C.; Stamnaes, J.; Iversen, R.; Andersen, J.T.; Jørgensen, T.J.; Sollid, L.M. Dissecting the interaction between transglutaminase 2 and fibronectin. Amino Acids 2017, 49, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Jambrovics, K.; Uray, I.P.; Keresztessy, Z.; Keillor, J.W.; Fésüs, L.; Balajthy, Z. Transglutaminase 2 programs differentiating acute promyelocytic leukemia cells in all-trans retinoic acid treatment to inflammatory stage through NF-κB activation. Haematologica 2019, 104, 505. [Google Scholar] [CrossRef]
- Csomós, K.; Német, I.; Fésüs, L.; Balajthy, Z. Tissue transglutaminase contributes to the all-trans-retinoic acid–induced differentiation syndrome phenotype in the NB4 model of acute promyelocytic leukemia. Blood J. Am. Soc. Hematol. 2010, 116, 3933–3943. [Google Scholar] [CrossRef] [PubMed]
- Tonutti, E.; Bizzaro, N. Antitissue Transglutaminase and Antiendomysial Antibodies. In Autoantibodies; Elsevier: Amsterdam, The Netherlands, 2014; pp. 463–470. [Google Scholar]
- Lee, Y.-J.; Jung, S.-H.; Kim, S.-H.; Kim, M.-S.; Lee, S.; Hwang, J.; Kim, S.-Y.; Kim, Y.-M.; Ha, K.-S. Essential Role of Transglutaminase 2 in Vascular Endothelial Growth Factor–Induced Vascular Leakage in the Retina of Diabetic Mice. Diabetes 2016, 65, 2414–2428. [Google Scholar] [CrossRef]
- Bhatt, M.P.; Lim, Y.-C.; Hwang, J.; Na, S.; Kim, Y.-M.; Ha, K.-S. C-peptide prevents hyperglycemia-induced endothelial apoptosis through inhibition of reactive oxygen species–mediated transglutaminase 2 activation. Diabetes 2013, 62, 243–253. [Google Scholar] [CrossRef]
- Park, D.; Choi, S.S.; Ha, K.-S. Transglutaminase 2: A multi-functional protein in multiple subcellular compartments. Amino Acids 2010, 39, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Skill, N.J.; Johnson, T.S.; Coutts, I.G.; Saint, R.E.; Fisher, M.; Huang, L.; El Nahas, A.M.; Collighan, R.J.; Griffin, M. Inhibition of transglutaminase activity reduces extracellular matrix accumulation induced by high glucose levels in proximal tubular epithelial cells. J. Biol. Chem. 2004, 279, 47754–47762. [Google Scholar] [CrossRef]
- Al-Basher, G.; Al-Otibi, F. Biological activity of olive leaf extract and regulation of tissue transglutaminase expression in diabetic wound healing. Int. J. Pharmacol. 2018, 14, 963–972. [Google Scholar] [CrossRef]
- Kumar, S.; Mehta, K. Tissue transglutaminase constitutively activates HIF-1α promoter and nuclear factor-κB via a non-canonical pathway. PLoS ONE 2012, 7, e49321. [Google Scholar] [CrossRef]
- Ejiugwo, M.; Rochev, Y.; Gethin, G.; O’Connor, G. Toward Developing Immunocompetent Diabetic Foot Ulcer-on-a-Chip Models for Drug Testing. Tissue Eng. Part C Methods 2021, 27, 77–88. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, W.; Kang, J.; Li, M.; Huang, J.; Ke, Q.; Kim, H.S.; Xu, B.; Kwak, S.-S. Overexpression of alfalfa Orange gene in tobacco enhances carotenoid accumulation and tolerance to multiple abiotic stresses. Plant Physiol. Biochem. 2018, 130, 613–622. [Google Scholar] [CrossRef]
- Philp, C.J.; Siebeke, I.; Clements, D.; Miller, S.; Habgood, A.; John, A.E.; Navaratnam, V.; Hubbard, R.B.; Jenkins, G.; Johnson, S.R. Extracellular matrix cross-linking enhances fibroblast growth and protects against matrix proteolysis in lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 594–603. [Google Scholar] [CrossRef]
- Olsen, K.C.; Sapinoro, R.; Kulkarni, A.A.; Kottmann, M.; Ringo, K.; Strong, E.; Johnson, G.V.; Phipps, R.P.; Sime, P.J. Tissue transglutaminase promotes pulmonary fibrosis through multiple mechanisms including AKT activation. In A58. Animal Models of Pulmonary Fibrosis; American Thoracic Society: New York, NY, USA, 2010; p. A1963. [Google Scholar]
- Bhedi, C.D.; Nasirova, S.; Toksoz, D.; Warburton, R.R.; Morine, K.J.; Kapur, N.K.; Galper, J.B.; Preston, I.R.; Hill, N.S.; Fanburg, B.L. Glycolysis regulated transglutaminase 2 activation in cardiopulmonary fibrogenic remodeling. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 930. [Google Scholar] [CrossRef]
- Chen, H.-H.; Zhao, P.; Zhao, W.-X.; Tian, J.; Guo, W.; Xu, M.; Zhang, C.; Lu, R. Stachydrine ameliorates pressure overload-induced diastolic heart failure by suppressing myocardial fibrosis. Am. J. Transl. Res. 2017, 9, 4250. [Google Scholar] [PubMed]
- Kim, D.-S.; Park, K.-S.; Kim, S.-Y. Silencing of TGase 2 sensitizes breast cancer cells to apoptosis by regulation of survival factors. Front. Biosci. Landmark 2009, 14, 2514–2521. [Google Scholar] [CrossRef][Green Version]
- Boroughs, L.K.; Antonyak, M.A.; Cerione, R.A. A novel mechanism by which tissue transglutaminase activates signaling events that promote cell survival. J. Biol. Chem. 2014, 289, 10115–10125. [Google Scholar] [CrossRef] [PubMed]
- Haroon, Z.A.; Hettasch, J.M.; Lai, T.S.; Dewhirst, M.W.; Greenberg, C.S. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999, 13, 1787–1795. [Google Scholar] [CrossRef]
- Jones, R.; Kotsakis, P.; Johnson, T.; Chau, D.; Ali, S.; Melino, G.; Griffin, M. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006, 13, 1442–1453. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217, e2019010. [Google Scholar] [CrossRef]
- Olsen, K.C.; Sapinoro, R.E.; Kottmann, R.; Kulkarni, A.A.; Iismaa, S.E.; Johnson, G.V.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Transglutaminase 2 and its role in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Su, Y.; Palanski, B.A.; Fujikura, K.; Garcia, M.J.; Frangogiannis, N.G. Pharmacologic inhibition of the enzymatic effects of tissue transglutaminase reduces cardiac fibrosis and attenuates cardiomyocyte hypertrophy following pressure overload. J. Mol. Cell. Cardiol. 2018, 117, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Stuckey, D.J.; Murdoch, C.E.; Camelliti, P.; Lip, G.Y.; Griffin, M. Cardiac fibrosis can be attenuated by blocking the activity of transglutaminase 2 using a selective small-molecule inhibitor. Cell Death Dis. 2018, 9, 613. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, H.; Fan, Q.; Wang, L.; Wang, Y.; Li, G. Multifunctional nanocatalyst-based ultrasensitive detection of human tissue transglutaminase 2. Biosens. Bioelectron. 2016, 83, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Nemes, Z.; Steinert, P.M. Bricks and mortar of the epidermal barrier. Exp. Mol. Med. 1999, 31, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Iismaa, S.E. The prostate-specific protein, transglutaminase 4 (TG4), is an autoantigen associated with male subfertility. Ann. Transl. Med. 2016, 4 (Suppl. 1), S35. [Google Scholar] [CrossRef]
- Furutani, Y.; Kato, A.; Fibriani, A.; Hirata, T.; Kawai, R.; Jeon, J.-H.; Fujii, Y.; Kim, I.-G.; Kojima, S.; Hirose, S. Identification, evolution, and regulation of expression of Guinea pig trappin with an unusually long transglutaminase substrate domain. J. Biol. Chem. 2005, 280, 20204–20215. [Google Scholar] [CrossRef] [PubMed]
- Al-U’datt, D.a.G.; Tranchant, C.C.; Al-Husein, B.; Hiram, R.; Al-Dwairi, A.; AlQudah, M.; Al-Shboul, O.; Jaradat, S.; Alqbelat, J.; Almajwal, A. Involvement and possible role of transglutaminases 1 and 2 in mediating fibrotic signalling, collagen cross-linking and cell proliferation in neonatal rat ventricular fibroblasts. PLoS ONE 2023, 18, e0281320. [Google Scholar] [CrossRef]
- Chen, S.; Ma, J.; Chi, J.; Zhang, B.; Zheng, X.; Chen, J.; Liu, J. Roles and potential clinical implications of tissue transglutaminase in cardiovascular diseases. Pharmacol. Res. 2022, 177, 106085. [Google Scholar] [CrossRef]
- Tatsukawa, H.; Tani, Y.; Otsu, R.; Nakagawa, H.; Hitomi, K. Global identification and analysis of isozyme-specific possible substrates crosslinked by transglutaminases using substrate peptides in mouse liver fibrosis. Sci. Rep. 2017, 7, 45049. [Google Scholar] [CrossRef]
- Curci, D.; Dillon, S.T.; Gu, X.; Winter, H.; Libermann, T.A. Proteome-Wide Analysis Using SOMAscan Identifies and Validates Epidermal Growth Factor as a Disease Marker of Collagenous Gastritis. Gastro Hep Adv. 2022, 1, 689–702. [Google Scholar] [CrossRef]
- Doa’a, G.A.; Tranchant, C.C.; Alu’datt, M.; Abusara, S.; Al-Dwairi, A.; AlQudah, M.; Al-Shboul, O.; Hiram, R.; Altuntas, Y.; Jaradat, S. Inhibition of transglutaminase 2 (TG2) ameliorates ventricular fibrosis in isoproterenol-induced heart failure in rats. Life Sci. 2023, 321, 121564. [Google Scholar]
- An, G.; Meka, C.R.; Bright, S.P.; Veltri, R.W. Human prostate-specific transglutaminase gene: Promoter cloning, tissue-specific expression, and down-regulation in metastatic prostate cancer. Urology 1999, 54, 1105–1111. [Google Scholar] [CrossRef]
- Dean, M.D. Genetic disruption of the copulatory plug in mice leads to severely reduced fertility. PLoS Genet. 2013, 9, e1003185. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.G.; Ablin, R.J.; Kynaston, H.G.; Mason, M.D. The prostate transglutaminase (TGase-4, TGaseP) regulates the interaction of prostate cancer and vascular endothelial cells, a potential role for the ROCK pathway. Microvasc. Res. 2009, 77, 150–157. [Google Scholar] [CrossRef]
- Ablin, R.J.; Owen, S.; Jiang, W.G. Prostate transglutaminase (TGase-4) induces epithelial-to-mesenchymal transition in prostate cancer cells. Anticancer Res. 2017, 37, 481–487. [Google Scholar] [CrossRef]
- Shinde, A.V.; Dobaczewski, M.; De Haan, J.J.; Saxena, A.; Lee, K.-K.; Xia, Y.; Chen, W.; Su, Y.; Hanif, W.; Kaur Madahar, I. Tissue transglutaminase induction in the pressure-overloaded myocardium regulates matrix remodelling. Cardiovasc. Res. 2017, 113, 892–905. [Google Scholar] [CrossRef]
- Steppan, J.; Sikka, G.; Jandu, S.; Barodka, V.; Halushka, M.K.; Flavahan, N.A.; Belkin, A.M.; Nyhan, D.; Butlin, M.; Avolio, A. Exercise, vascular stiffness, and tissue transglutaminase. J. Am. Heart Assoc. 2014, 3, e000599. [Google Scholar] [CrossRef] [PubMed]
- Tatsukawa, H.; Otsu, R.; Tani, Y.; Wakita, R.; Hitomi, K. Isozyme-specific comprehensive characterization of transglutaminase-crosslinked substrates in kidney fibrosis. Sci. Rep. 2018, 8, 7306. [Google Scholar] [CrossRef] [PubMed]
- Burhan, I.; Furini, G.; Lortat-Jacob, H.; Atobatele, A.G.; Scarpellini, A.; Schroeder, N.; Atkinson, J.; Maamra, M.; Nutter, F.H.; Watson, P. Interplay between transglutaminases and heparan sulphate in progressive renal scarring. Sci. Rep. 2016, 6, 31343. [Google Scholar] [CrossRef]
- Witsch, T.J.; Niess, G.; Sakkas, E.; Likhoshvay, T.; Becker, S.; Herold, S.; Mayer, K.; Vadász, I.; Roberts, J.D.; Seeger, W. Transglutaminase 2: A new player in bronchopulmonary dysplasia? Eur. Respir. J. 2014, 44, 109–121. [Google Scholar] [CrossRef]
- Zhang, S.; Yao, H.-F.; Li, H.; Su, T.; Jiang, S.-H.; Wang, H.; Zhang, Z.-G.; Dong, F.-Y.; Yang, Q.; Yang, X.-M. Transglutaminases are oncogenic biomarkers in human cancers and therapeutic targeting of TGM2 blocks chemoresistance and macrophage infiltration in pancreatic cancer. Cell. Oncol. 2023, 46, 1473–1492. [Google Scholar] [CrossRef]
- Hielscher, A.; Ellis, K.; Qiu, C.; Porterfield, J.; Gerecht, S. Fibronectin deposition participates in extracellular matrix assembly and vascular morphogenesis. PLoS ONE 2016, 11, e0147600. [Google Scholar] [CrossRef]
- Hubmacher, D.; Apte, S.S. The biology of the extracellular matrix: Novel insights. Curr. Opin. Rheumatol. 2013, 25, 65. [Google Scholar] [CrossRef]
- Forlino, A.; Marini, J.C. Osteogenesis imperfecta. Lancet 2016, 387, 1657–1671. [Google Scholar] [CrossRef]
- Faye, C.; Inforzato, A.; Bignon, M.; Hartmann, D.J.; Muller, L.; Ballut, L.; Olsen, B.R.; Day, A.J.; Ricard-Blum, S. Transglutaminase-2: A new endostatin partner in the extracellular matrix of endothelial cells. Biochem. J. 2010, 427, 467–475. [Google Scholar] [CrossRef]
- Bell, S.E.; Mavila, A.; Salazar, R.; Bayless, K.J.; Kanagala, S.; Maxwell, S.A.; Davis, G.E. Differential gene expression during capillary morphogenesis in 3D collagen matrices: Regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 2001, 114, 2755–2773. [Google Scholar] [CrossRef] [PubMed]
- Dardik, R.; Loscalzo, J.; Inbal, A. Factor XIII (FXIII) and angiogenesis. J. Thromb. Haemost. 2006, 4, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Perez, M.; Lee, E.-S.; Kojima, S.; Griffin, M. The functional relationship between transglutaminase 2 and transforming growth factor β1 in the regulation of angiogenesis and endothelial–mesenchymal transition. Cell Death Dis. 2017, 8, e3032. [Google Scholar] [CrossRef]
- Niger, C.; Beazley, K.E.; Nurminskaya, M. Induction of chondrogenic differentiation in mesenchymal stem cells by TGF-beta cross-linked to collagen-PLLA [poly (L-lactic acid)] scaffold by transglutaminase 2. Biotechnol. Lett. 2013, 35, 2193–2199. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Z.; Perez, M.; Caja, S.; Melino, G.; Johnson, T.; Lindfors, K.; Griffin, M. A novel extracellular role for tissue transglutaminase in matrix-bound VEGF-mediated angiogenesis. Cell Death Dis. 2013, 4, e808. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.; Mahoney, S.-A.; Haynes, L. Transglutaminase C in cerebellar granule neurons: Regulation and localization of substrate cross-linking. Neuroscience 1995, 65, 1063–1076. [Google Scholar] [CrossRef]
- Lesort, M.; Chun, W.; Johnson, G.; Ferrante, R. Tissue transglutaminase is increased in Huntington’s disease brain. J. Neurochem. 1999, 73, 2018–2027. [Google Scholar]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef]
- Espitia Pinzon, N.; Van Mierlo, H.; De Jonge, J.C.; Brevé, J.J.; Bol, J.G.; Drukarch, B.; Van Dam, A.-M.; Baron, W. Tissue transglutaminase promotes early differentiation of oligodendrocyte progenitor cells. Front. Cell. Neurosci. 2019, 13, 281. [Google Scholar] [CrossRef] [PubMed]
- Currò, M.; Ferlazzo, N.; Condello, S.; Caccamo, D.; Ientile, R. Transglutaminase 2 silencing reduced the beta-amyloid-effects on the activation of human THP-1 cells. Amino Acids 2010, 39, 1427–1433. [Google Scholar] [CrossRef]
- Gundemir, S.; Monteagudo, A.; Akbar, A.; Keillor, J.W.; Johnson, G.V. The complex role of transglutaminase 2 in glioblastoma proliferation. Neuro-Oncol. 2017, 19, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Halverson, R.A.; Lewis, J.; Frausto, S.; Hutton, M.; Muma, N.A. Tau protein is cross-linked by transglutaminase in P301L tau transgenic mice. J. Neurosci. 2005, 25, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Nemes, Z.; Devreese, B.; Steinert, P.M.; Beeumen, J.V.; Fésüs, L. Cross-linking of ubiquitin, HSP27, parkin and α-synuclein by γ-glutamyl-ε-lysine bonds in Alzheimer’s neurofibrillary tangles. FASEB J. 2004, 18, 1135–1137. [Google Scholar] [CrossRef] [PubMed]
- Jeitner, T.M.; Matson, W.R.; Folk, J.E.; Blass, J.P.; Cooper, A.J. Increased levels of γ-glutamylamines in Huntington disease CSF. J. Neurochem. 2008, 106, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Hartley, D.M.; Zhao, C.; Speier, A.C.; Woodard, G.A.; Li, S.; Li, Z.; Walz, T. Transglutaminase induces protofibril-like amyloid β-protein assemblies that are protease-resistant and inhibit long-term potentiation. J. Biol. Chem. 2008, 283, 16790–16800. [Google Scholar] [CrossRef]
- Tucholski, J.; Roth, K.A.; Johnson, G.V. Tissue transglutaminase overexpression in the brain potentiates calcium-induced hippocampal damage. J. Neurochem. 2006, 97, 582–594. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Pinto, J.T.; Krasnikov, B.F.; Horswill, M.; Cooper, A.J. Transglutaminases and neurodegeneration. J. Neurochem. 2009, 109, 160–166. [Google Scholar] [CrossRef]
- McConoughey, S.J.; Basso, M.; Niatsetskaya, Z.V.; Sleiman, S.F.; Smirnova, N.A.; Langley, B.C.; Mahishi, L.; Cooper, A.J.; Antonyak, M.A.; Cerione, R.A. Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol. Med. 2010, 2, 349–370. [Google Scholar] [CrossRef]
- Munsie, L.; Caron, N.; Atwal, R.S.; Marsden, I.; Wild, E.J.; Bamburg, J.R.; Tabrizi, S.J.; Truant, R. Mutant huntingtin causes defective actin remodeling during stress: Defining a new role for transglutaminase 2 in neurodegenerative disease. Hum. Mol. Genet. 2011, 20, 1937–1951. [Google Scholar] [CrossRef]
- Yunes-Medina, L.; Paciorkowski, A.; Nuzbrokh, Y.; Johnson, G.V. Depletion of transglutaminase 2 in neurons alters expression of extracellular matrix and signal transduction genes and compromises cell viability. Mol. Cell. Neurosci. 2018, 86, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Badarau, E.; Wang, Z.; Rathbone, D.L.; Costanzi, A.; Thibault, T.; Murdoch, C.E.; El Alaoui, S.; Bartkeviciute, M.; Griffin, M. Development of potent and selective tissue transglutaminase inhibitors: Their effect on TG2 function and application in pathological conditions. Chem. Biol. 2015, 22, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Alhassan, E.; Yadav, A.; Kelly, C.P.; Mukherjee, R. Novel nondietary therapies for celiac disease. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-R.; Song, J.-H.; Ahn, J.-H.; Lee, G.-S.; Ahn, H.; Yoon, S.-I.; Kang, S.G.; Kim, P.-H.; Jeon, S.-M.; Choi, E.-J. Antiviral and anti-inflammatory activity of budesonide against human rhinovirus infection mediated via autophagy activation. Antivir. Res. 2018, 151, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Mäki, M.; Lundin, K.E.; Isola, J.; Friesing-Sosnik, T.; Taavela, J.; Popp, A.; Koskenpato, J.; Langhorst, J.; Hovde, Ø. A randomized trial of a transglutaminase 2 inhibitor for celiac disease. N. Engl. J. Med. 2021, 385, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Palanski, B.A.; Khosla, C. Cystamine and disulfiram inhibit human transglutaminase 2 via an oxidative mechanism. Biochemistry 2018, 57, 3359–3363. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.-H.; Lee, H.-J.; Jang, G.-Y.; Kim, C.-W.; Shin, D.-M.; Cho, S.-Y.; Yeo, E.-J.; Park, S.-C.; Kim, I.-G. Different inhibition characteristics of intracellular transglutaminase activity by cystamine and cysteamine. Exp. Mol. Med. 2004, 36, 576–581. [Google Scholar] [CrossRef][Green Version]
- Elli, L.; Ciulla, M.M.; Busca, G.; Roncoroni, L.; Maioli, C.; Ferrero, S.; Bardella, M.T.; Bonura, A.; Paliotti, R.; Terrani, C. Beneficial effects of treatment with transglutaminase inhibitor cystamine on the severity of inflammation in a rat model of inflammatory bowel disease. Lab. Investig. 2011, 91, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.T.; Khomenko, T.; Szabo, S.; McLaren, G.D.; Denton, T.T.; Krasnikov, B.F.; Jeitner, T.M.; Cooper, A.J. Measurement of sulfur-containing compounds involved in the metabolism and transport of cysteamine and cystamine. Regional differences in cerebral metabolism. J. Chromatogr. B 2009, 877, 3434–3441. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.-C.; Chen, Y.-C.; Lai, W.-X.; Chiang, S.-Y.; Huang, C.-Y.; Tzang, B.-S. Beneficial effects of treatment with cystamine on brain in NZB/W F1 mice. Eur. J. Pharmacol. 2008, 591, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Tzang, B.S.; Hsu, T.C.; Kuo, C.Y.; Chen, T.Y.; Chiang, S.Y.; Li, S.L.; Kao, S.H. Cystamine attenuates lupus-associated apoptosis of ventricular tissue by suppressing both intrinsic and extrinsic pathways. J. Cell. Mol. Med. 2012, 16, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Aeschlimann, D.; Paulsson, M. Transglutaminases: Protein cross-linking enzymes in tissues and body fluids. Thromb. Haemost. 1994, 71, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y. New insights into development of transglutaminase 2 inhibitors as pharmaceutical lead compounds. Med. Sci. 2018, 6, 87. [Google Scholar] [CrossRef]
- Choi, K.; Siegel, M.; Piper, J.L.; Yuan, L.; Cho, E.; Strnad, P.; Omary, B.; Rich, K.M.; Khosla, C. Chemistry and biology of dihydroisoxazole derivatives: Selective inhibitors of human transglutaminase 2. Chem. Biol. 2005, 12, 469–475. [Google Scholar] [CrossRef]
- Penumatsa, K.C.; Toksoz, D.; Warburton, R.R.; Kharnaf, M.; Preston, I.R.; Kapur, N.K.; Khosla, C.; Hill, N.S.; Fanburg, B.L. Transglutaminase 2 in pulmonary and cardiac tissue remodeling in experimental pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L752–L762. [Google Scholar] [CrossRef]
- Akbar, A.; McNeil, N.M.; Albert, M.R.; Ta, V.; Adhikary, G.; Bourgeois, K.; Eckert, R.L.; Keillor, J.W. Structure–activity relationships of potent, targeted covalent inhibitors that abolish both the transamidation and GTP binding activities of human tissue transglutaminase. J. Med. Chem. 2017, 60, 7910–7927. [Google Scholar] [CrossRef]
- Song, M.; Hwang, H.; Im, C.Y.; Kim, S.-Y. Recent Progress in the Development of Transglutaminase 2 (TGase2) Inhibitors: Miniperspective. J. Med. Chem. 2017, 60, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Maamra, M.; Benayad, O.M.; Matthews, D.; Kettleborough, C.; Atkinson, J.; Cain, K.; Bon, H.; Brand, H.; Parkinson, M.; Watson, P.F. Transglutaminase 2: Development of therapeutic antibodies reveals four inhibitory epitopes and confirms extracellular function in fibrotic remodelling. Br. J. Pharmacol. 2022, 179, 2697–2712. [Google Scholar] [CrossRef] [PubMed]
- Dyer, L.M.; Schooler, K.P.; Ai, L.; Klop, C.; Qiu, J.; Robertson, K.D.; Brown, K.D. The transglutaminase 2 gene is aberrantly hypermethylated in glioma. J. Neuro-Oncol. 2011, 101, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Kim, W.-J.; Demircan, B.; Dyer, L.M.; Bray, K.J.; Skehan, R.R.; Massoll, N.A.; Brown, K.D. The transglutaminase 2 gene (TGM2), a potential molecular marker for chemotherapeutic drug sensitivity, is epigenetically silenced in breast cancer. Carcinogenesis 2008, 29, 510–518. [Google Scholar] [CrossRef]
- Attarwala, H.Z.; Suri, K.; Amiji, M.M. Co-silencing of tissue transglutaminase-2 and interleukin-15 genes in a celiac disease mimetic mouse model using a nanoparticle-in-microsphere oral system. Mol. Pharm. 2021, 18, 3099–3107. [Google Scholar] [CrossRef]
- Attarwala, H.; Clausen, V.; Chaturvedi, P.; Amiji, M.M. Cosilencing intestinal transglutaminase-2 and interleukin-15 using gelatin-based nanoparticles in an in vitro model of celiac disease. Mol. Pharm. 2017, 14, 3036–3044. [Google Scholar] [CrossRef]





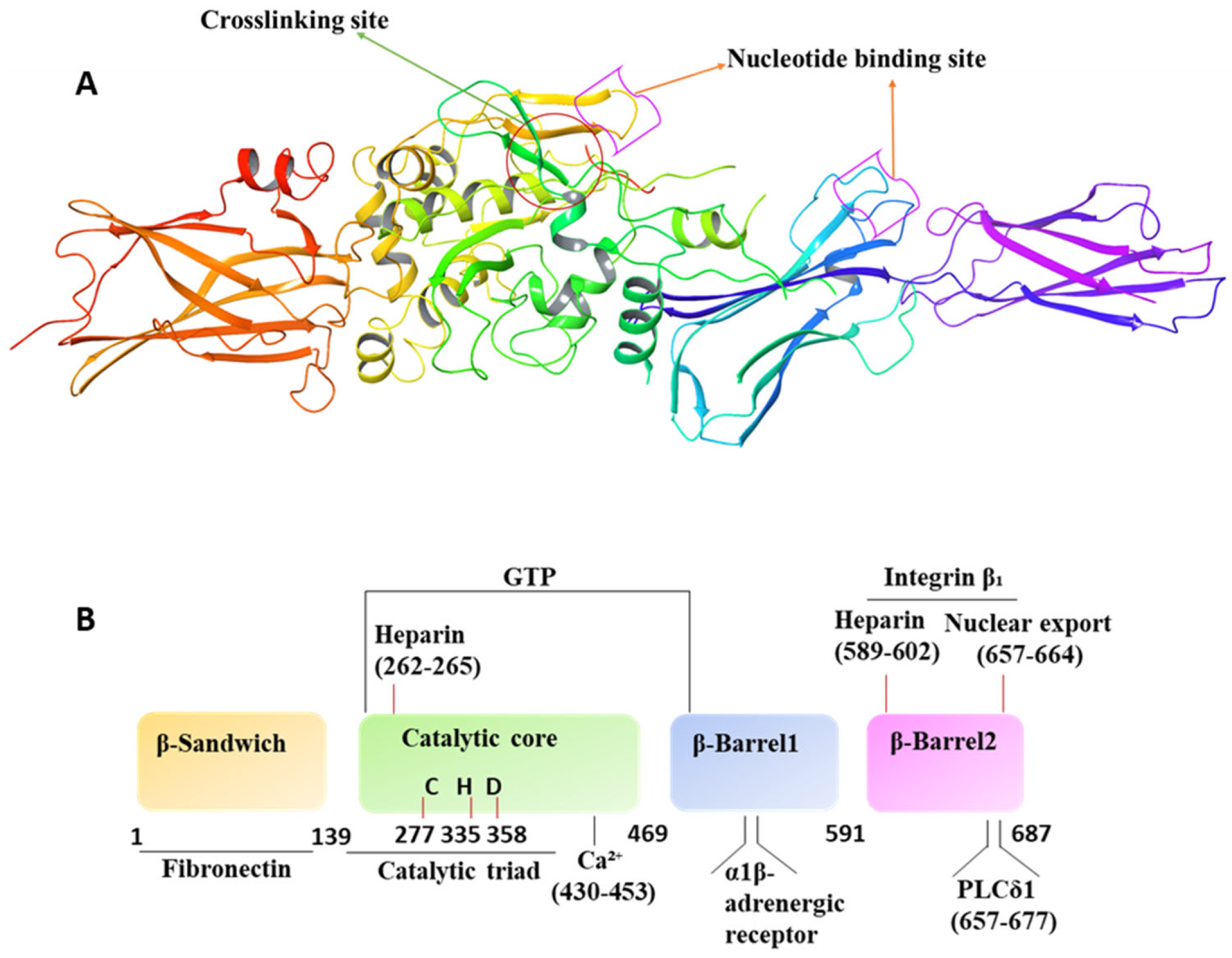{kind=link}
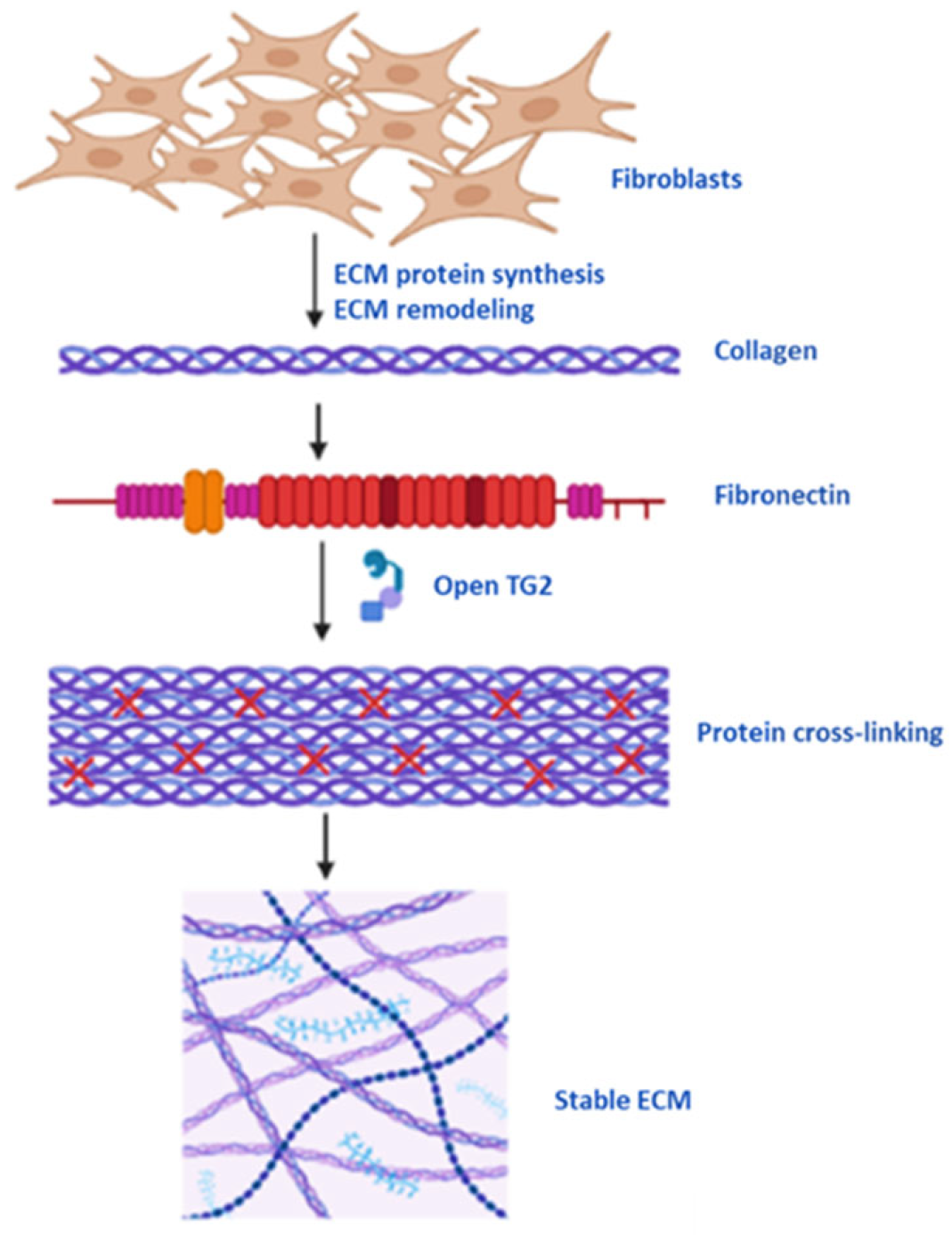{kind=link}
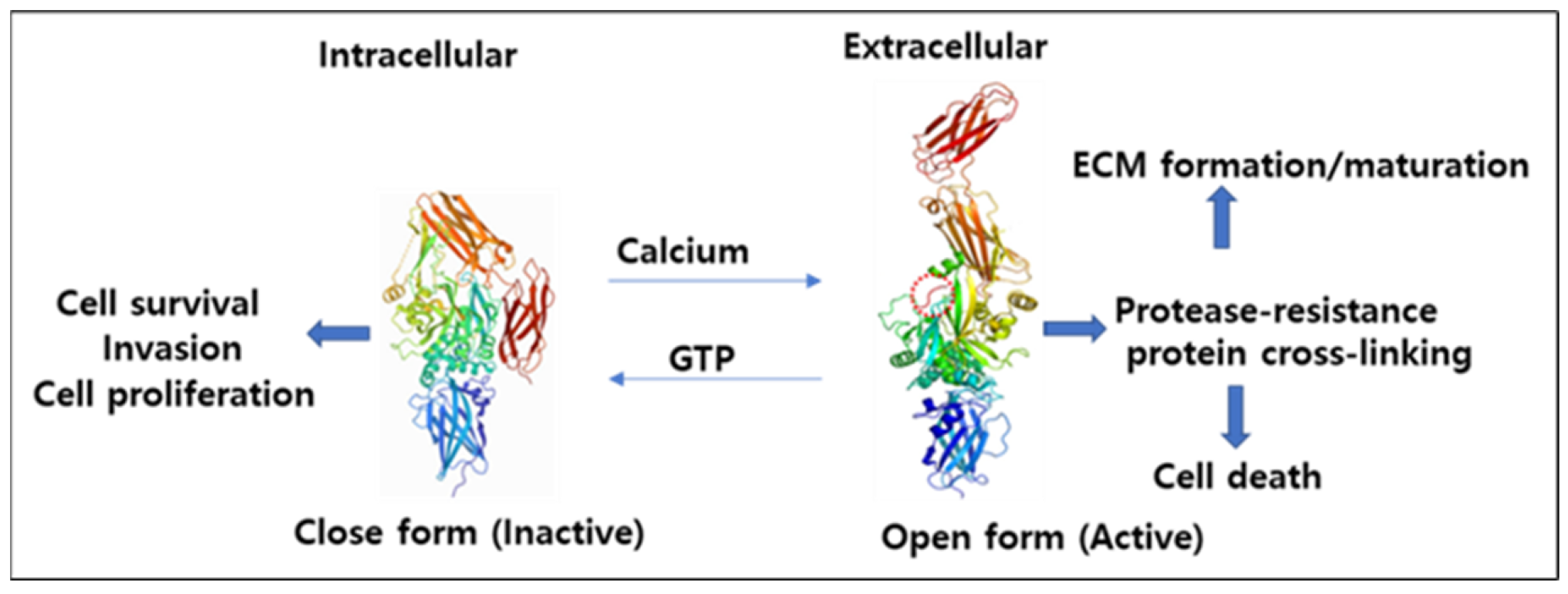{kind=link}
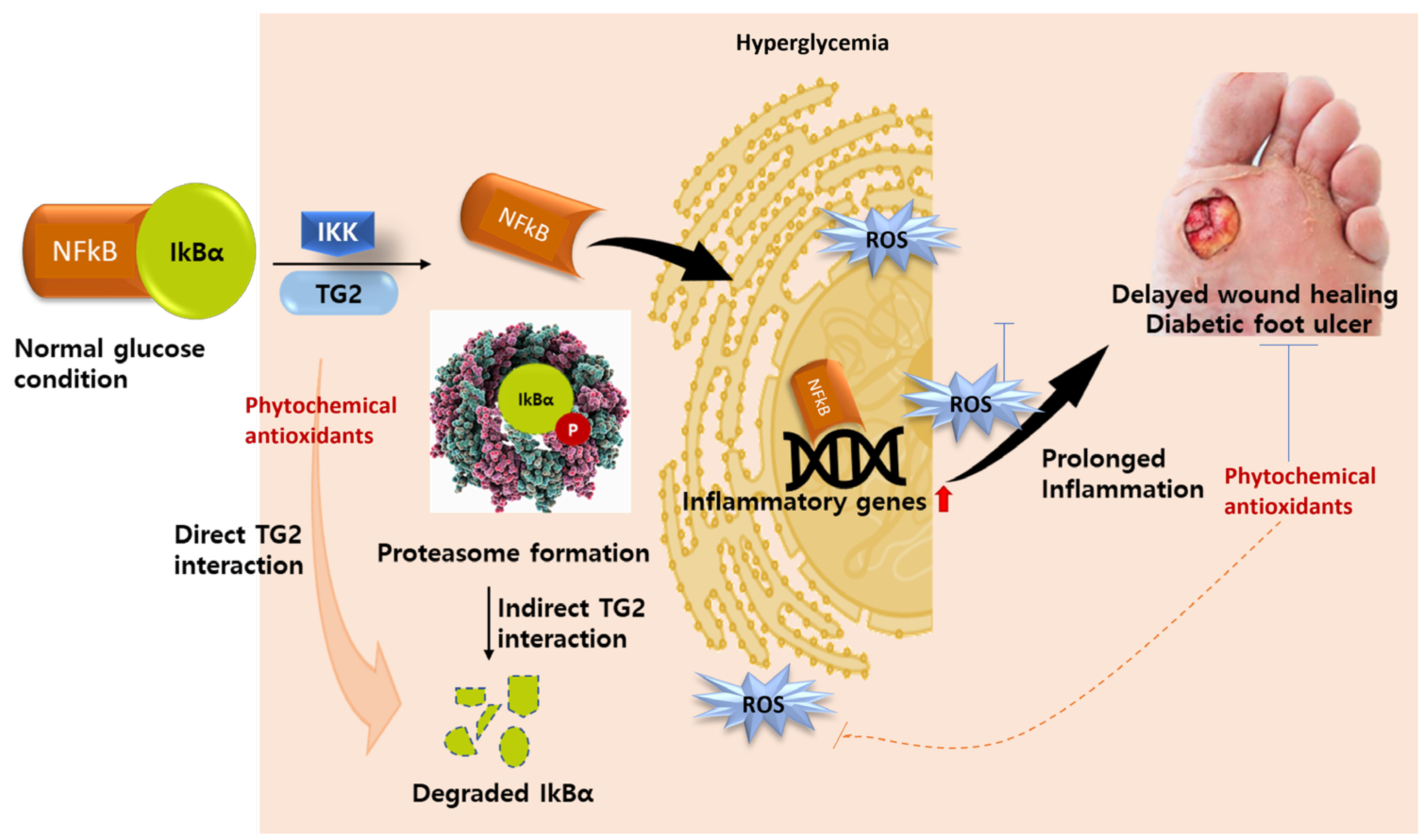{kind=link}
| Enzyme Type | Cellular Location | Function | Tissue Distribution | Pathological Conditions | Reference |
|---|---|---|---|---|---|
| TG1 | Vesicles, plasma membrane | Water-resistance barrier, prevention of pathogens | Keratinocytes | Collodion babies at birth, lifelong pronounced scaling, dramatically increased trans-epidermal water loss | [21] |
| TG2 | ECM, nucleus, mitochondria, plasma membrane | Cell differentiation, inflammation, cell death, tissue regeneration, ECM assembly | Fibroblasts, vascular endothelium, smooth muscle cells, ECM of a variety of tissues, and the arterial walls | Delayed wound healing, endothelial-basement-membrane biogenesis and enhanced fibrosis and keratinization | [30,31] |
| TG3 | ECM, cell membrane, cytoplasm | Cell differentiation | Hair follicle, epidermis, brain, mucosa, small intestine, and skin prostatic and glandular fluids | Fibrosis, tumor | [19,32] |
| TG4 | ECM, Cell membrane | Cell differentiation, tissue regeneration, ECM assembly | Skin, prostate tissues, fallopian tubes, vagina, adrenal gland, intestine, lungs and urinary bladder | Fibrosis | [33] |
| TG5 | Cell membrane | Cell differentiation, migration | Keratinocytes | Keratinization, cornification of cells | [34] |
| TG6 | Cell membrane, ECM, cytoplasm | Cell differentiation and proliferation | Astrocytes, lungs, testes, keratinocytes, central nervous system (CNS) | Multiple sclerosis (MS) | [27,35] |
| TG7 | Cell membrane, ECM | Cell differentiation and proliferation | Skin epidermis, brain, testicles, lungs | Fibrosis | [18,19] |
| (FXIII) | ECM, cytoplasm, plasma | ECM turnover | Plasma | Delayed wound healing, blood coagulation and prolonged inflammation, | [28,29] |
| Erythrocyte membrane protein band 4.2 | Cytoskeleton of the RBCs | Stabilizes shape and restricts deformability of erythrocytes via interaction with spectrin, protein 3, glycophorin C, ankyrin and actin | Blood | Hereditary spherocytosis | [36,37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, N.; Kim, S.-Y. Transglutaminase2: An Enduring Enzyme in Diabetes and Age-Related Metabolic Diseases. Kinases Phosphatases 2024, 2, 67-92. https://doi.org/10.3390/kinasesphosphatases2010005
Yadav N, Kim S-Y. Transglutaminase2: An Enduring Enzyme in Diabetes and Age-Related Metabolic Diseases. Kinases and Phosphatases. 2024; 2(1):67-92. https://doi.org/10.3390/kinasesphosphatases2010005
Chicago/Turabian StyleYadav, Neera, and Sun-Yeou Kim. 2024. "Transglutaminase2: An Enduring Enzyme in Diabetes and Age-Related Metabolic Diseases" Kinases and Phosphatases 2, no. 1: 67-92. https://doi.org/10.3390/kinasesphosphatases2010005
APA StyleYadav, N., & Kim, S.-Y. (2024). Transglutaminase2: An Enduring Enzyme in Diabetes and Age-Related Metabolic Diseases. Kinases and Phosphatases, 2(1), 67-92. https://doi.org/10.3390/kinasesphosphatases2010005