

CK2 Inhibitors Targeting Inside and Outside the Catalytic Box


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. An Overview of CK2
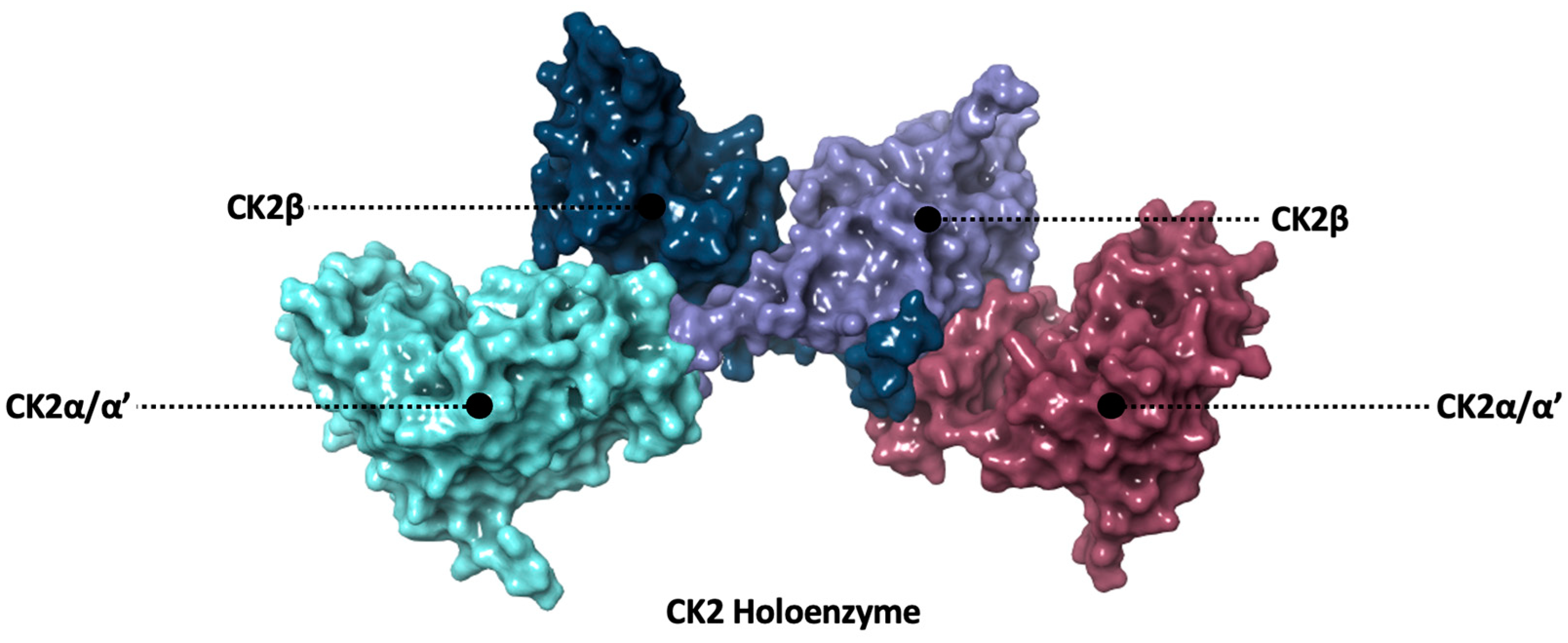
1.2. Structure of CK2
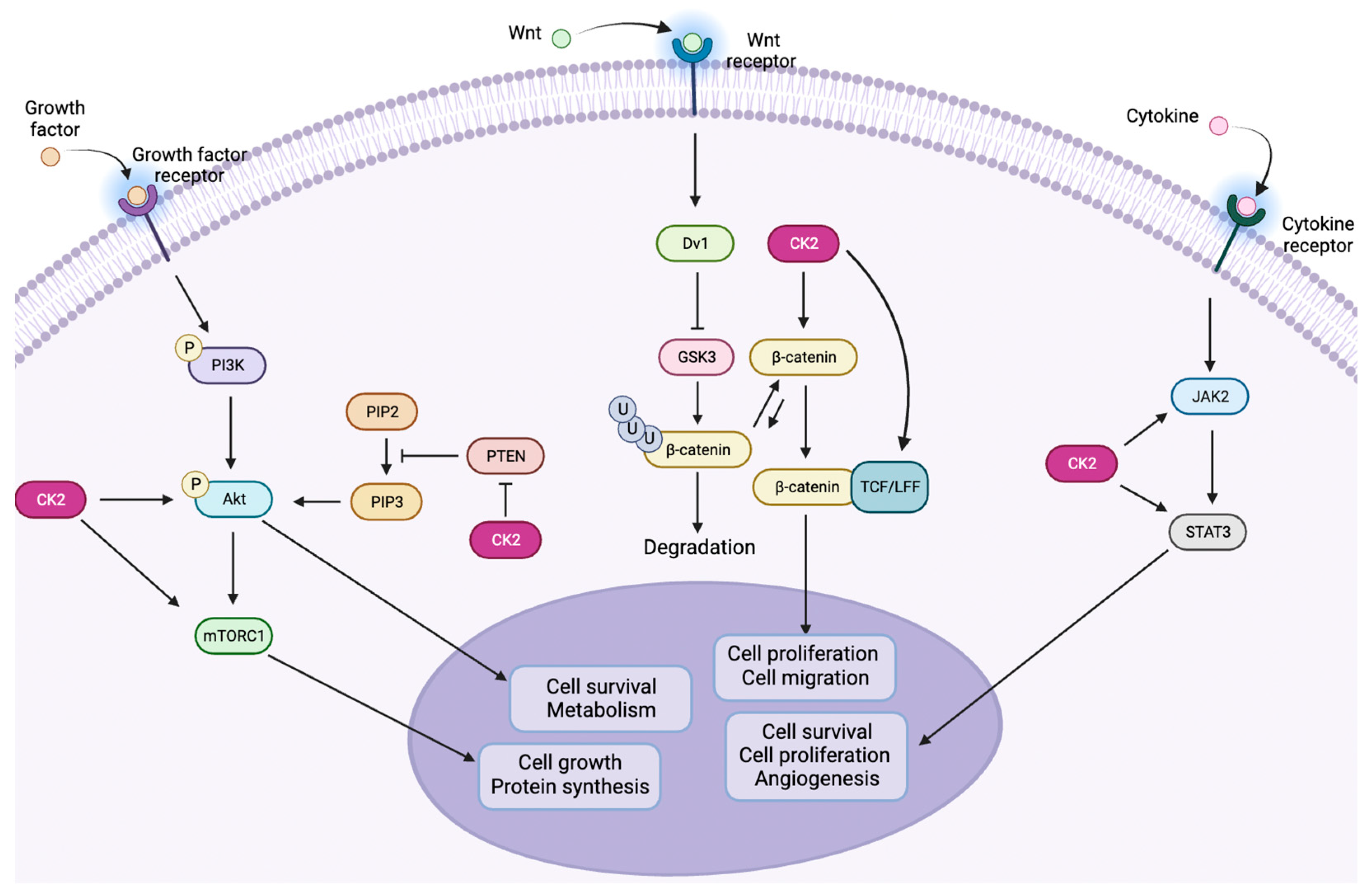
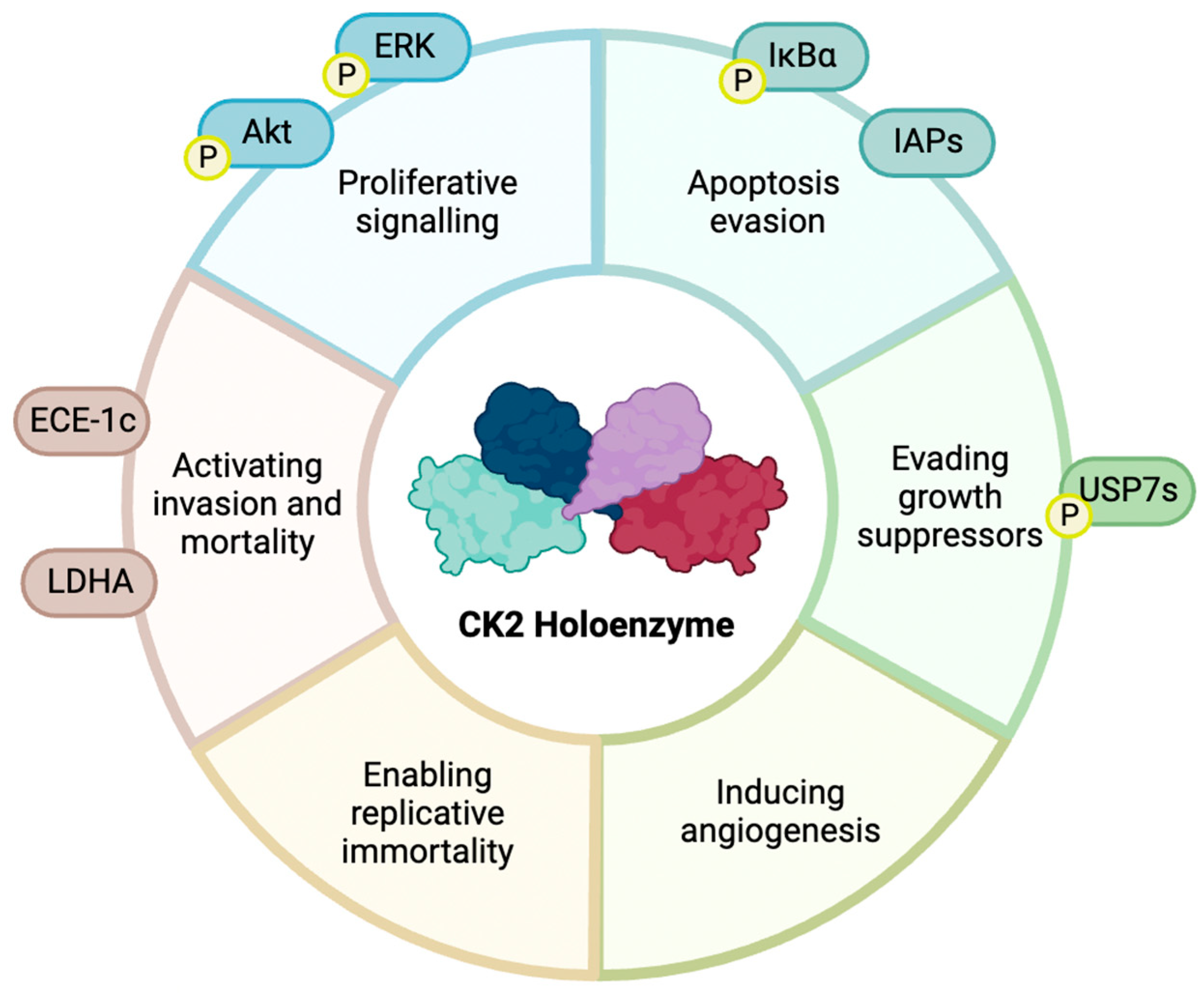
1.3. CK2 in Cancer
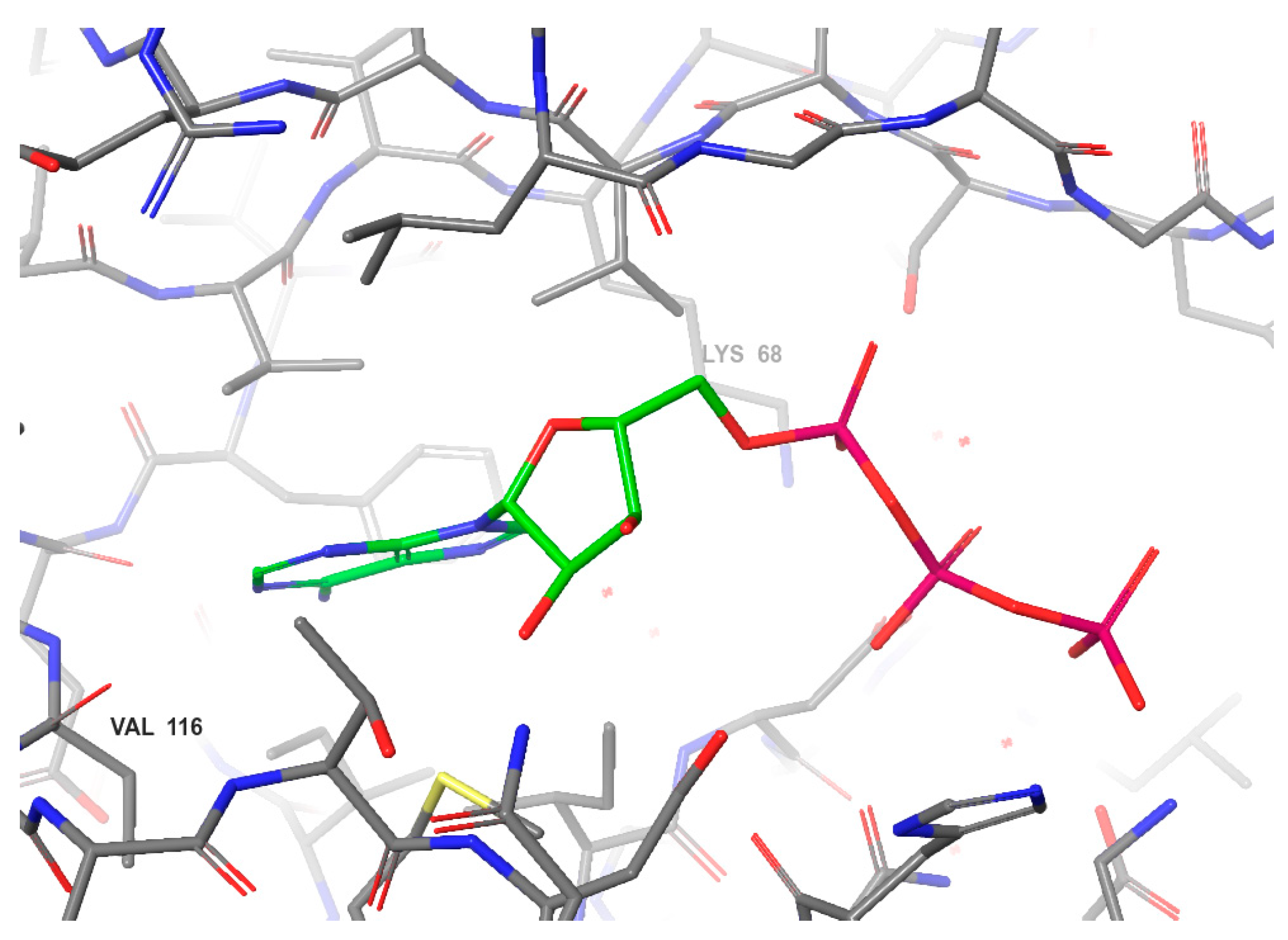
2. Targeting CK2 within the ATP Site
2.1. ATP-Competitive Inhibition
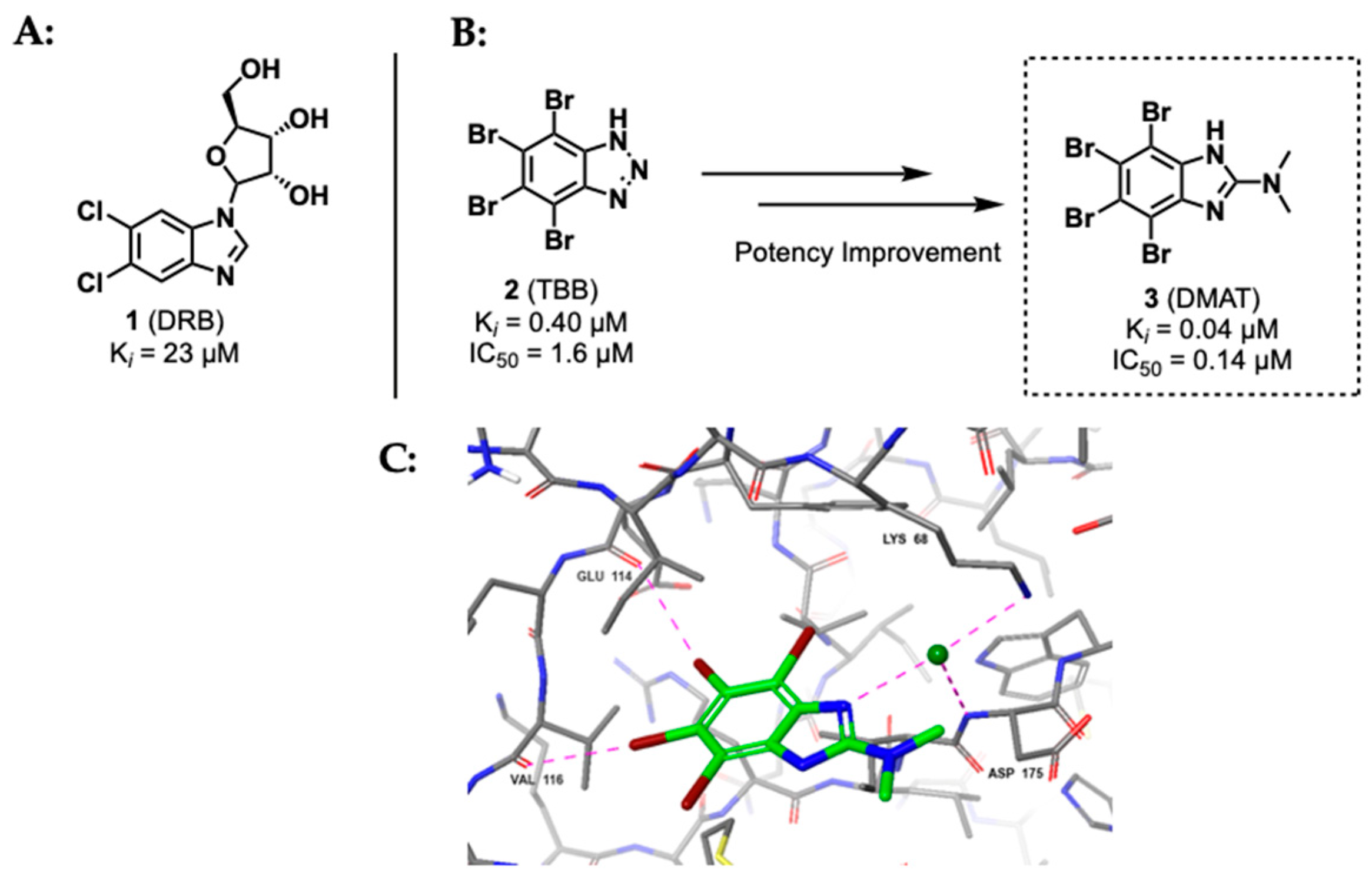
2.1.1. Polyhalogenated Benzimidazole and Benzotriazole Derivatives
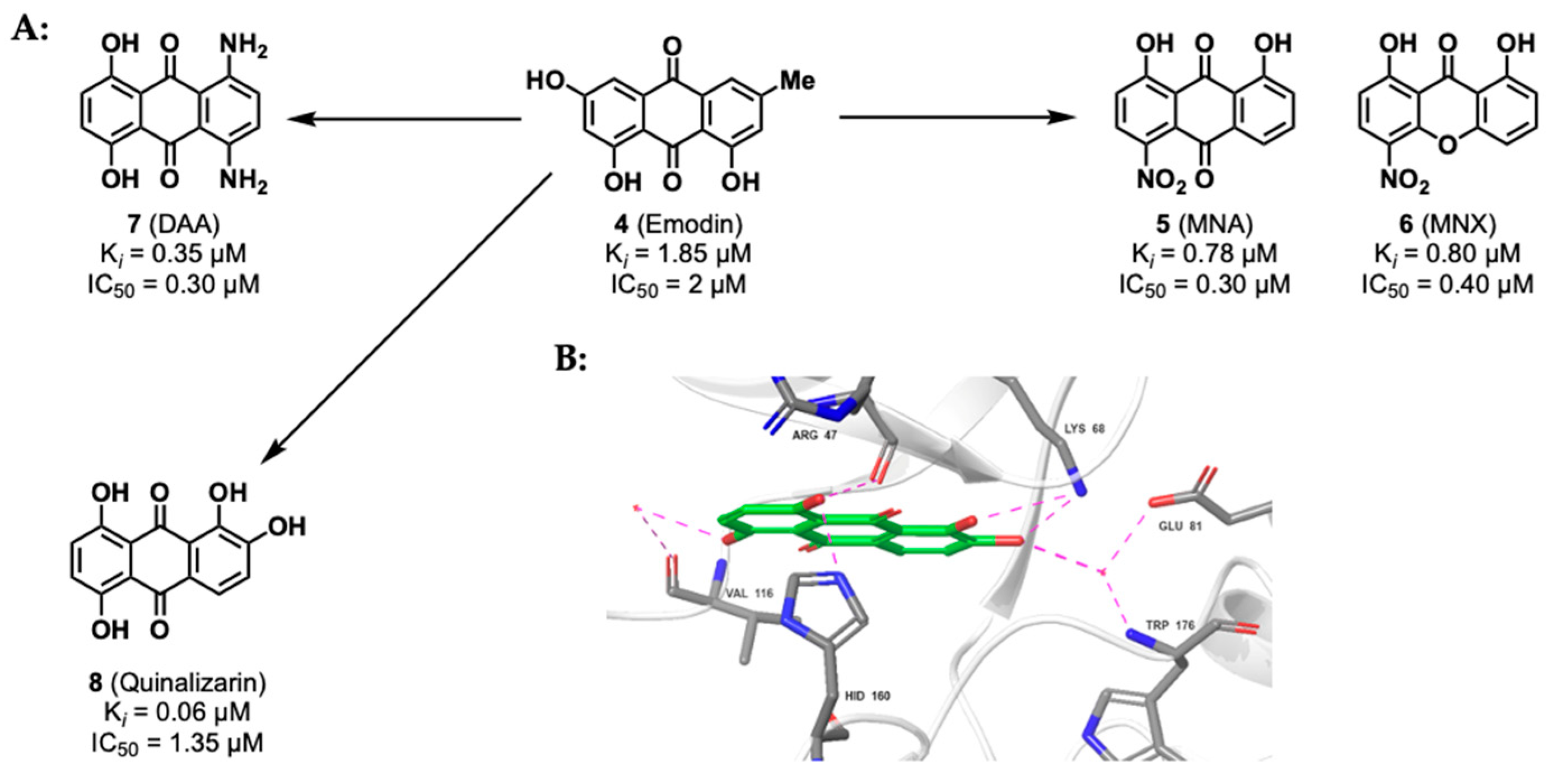
2.1.2. Anthraquinone Derivatives
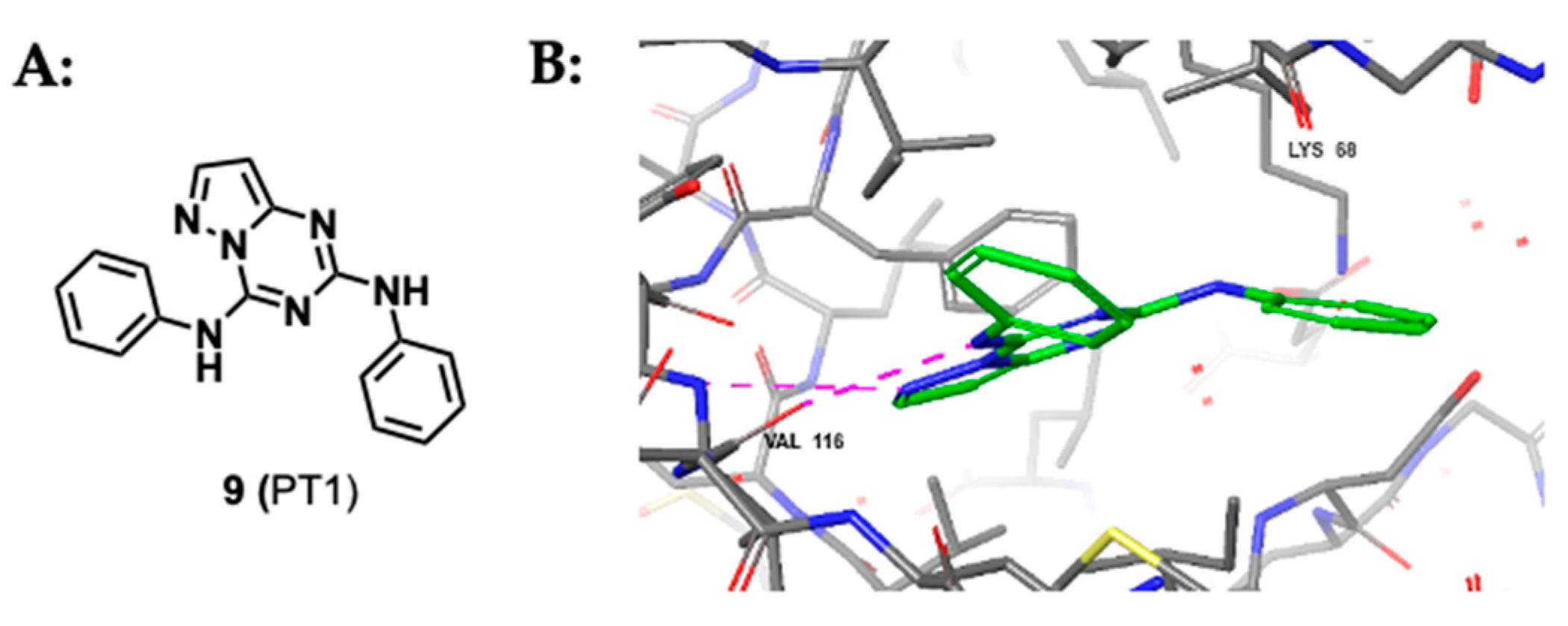
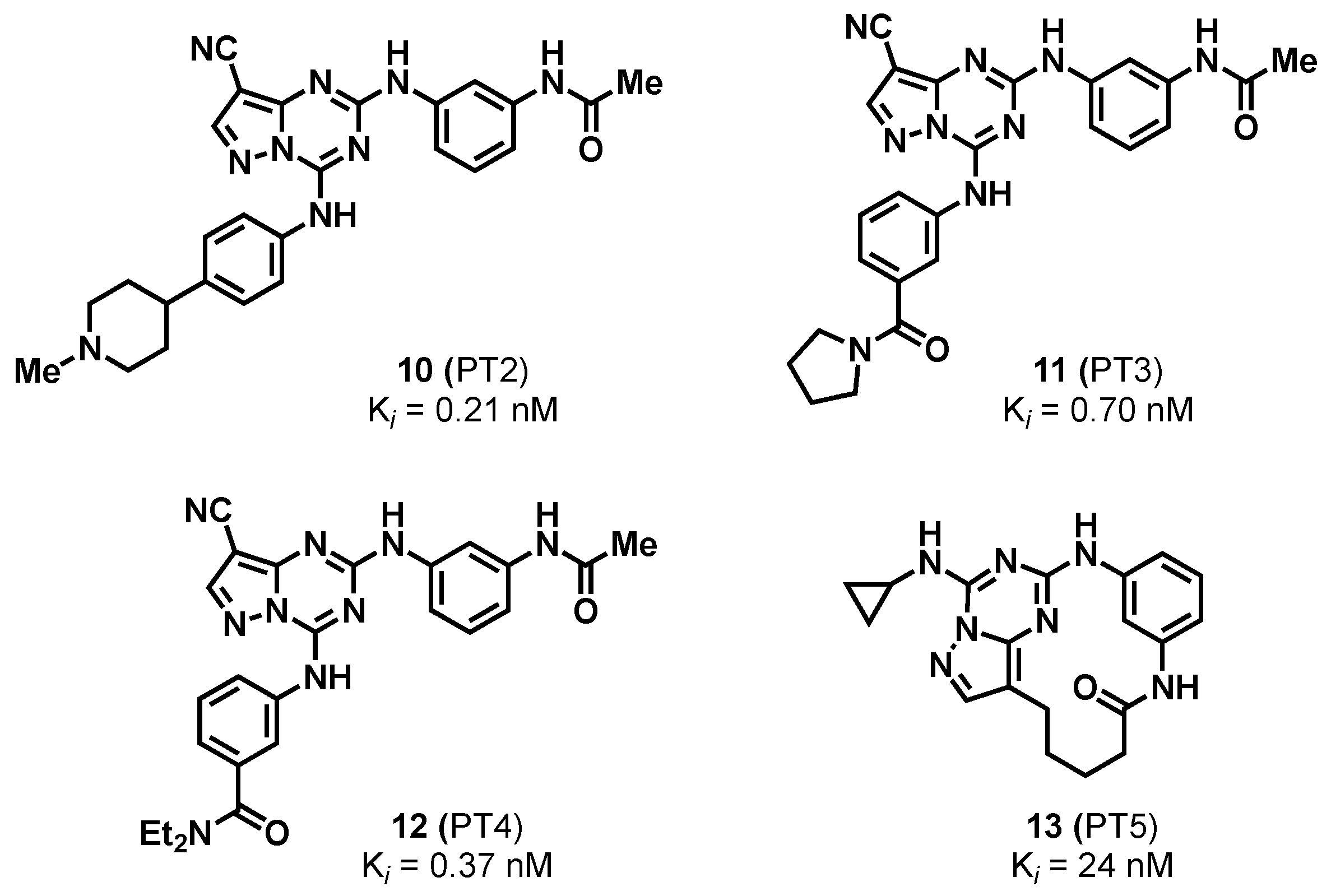
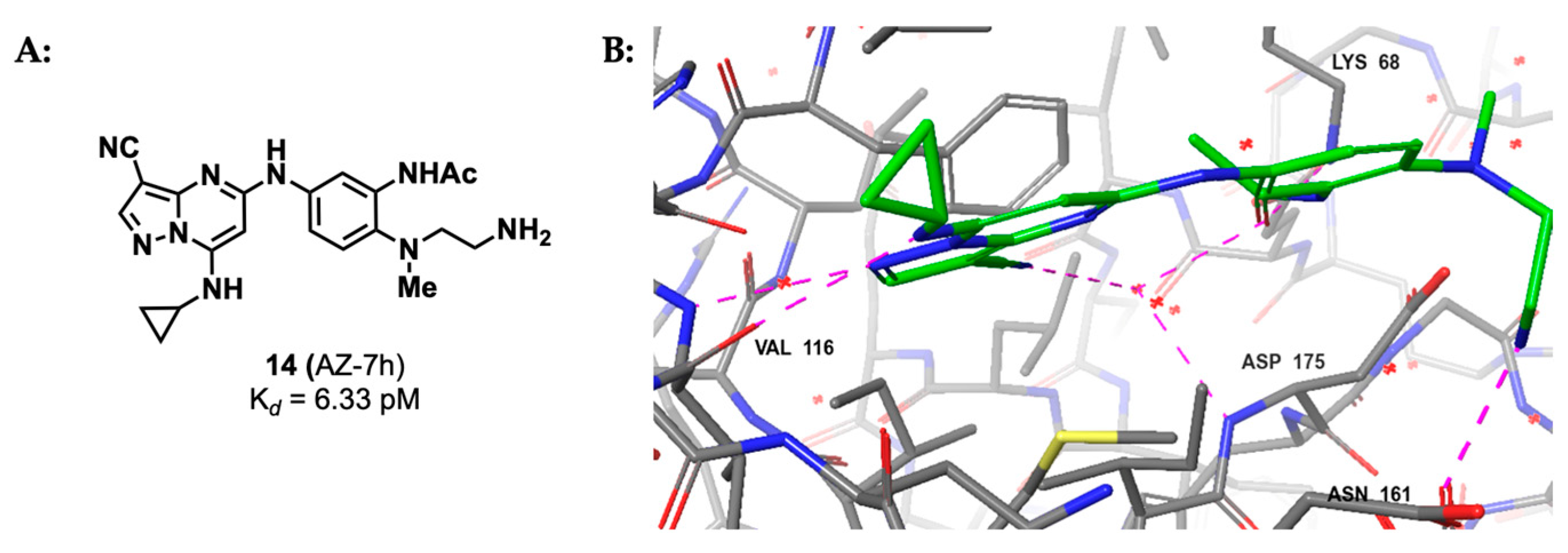
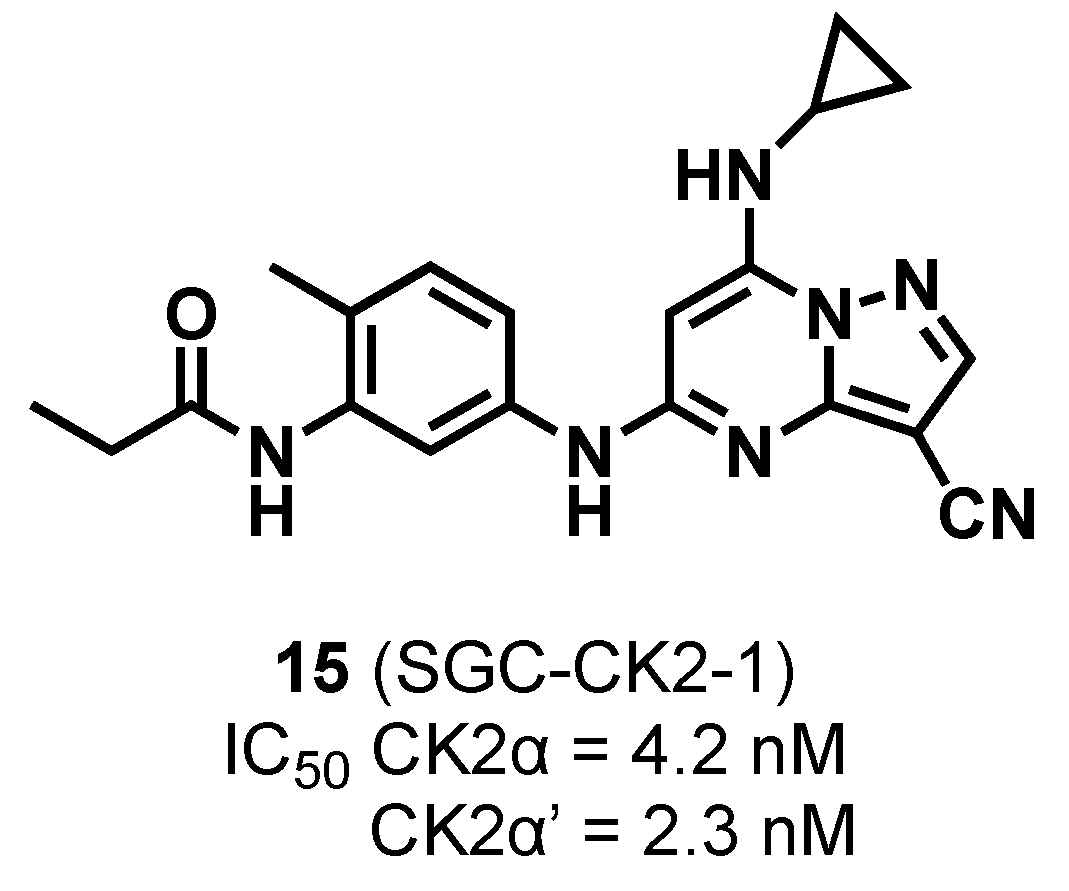
2.1.3. Pyrazolo-Triazines and Pyrazolo-Pyrimidines
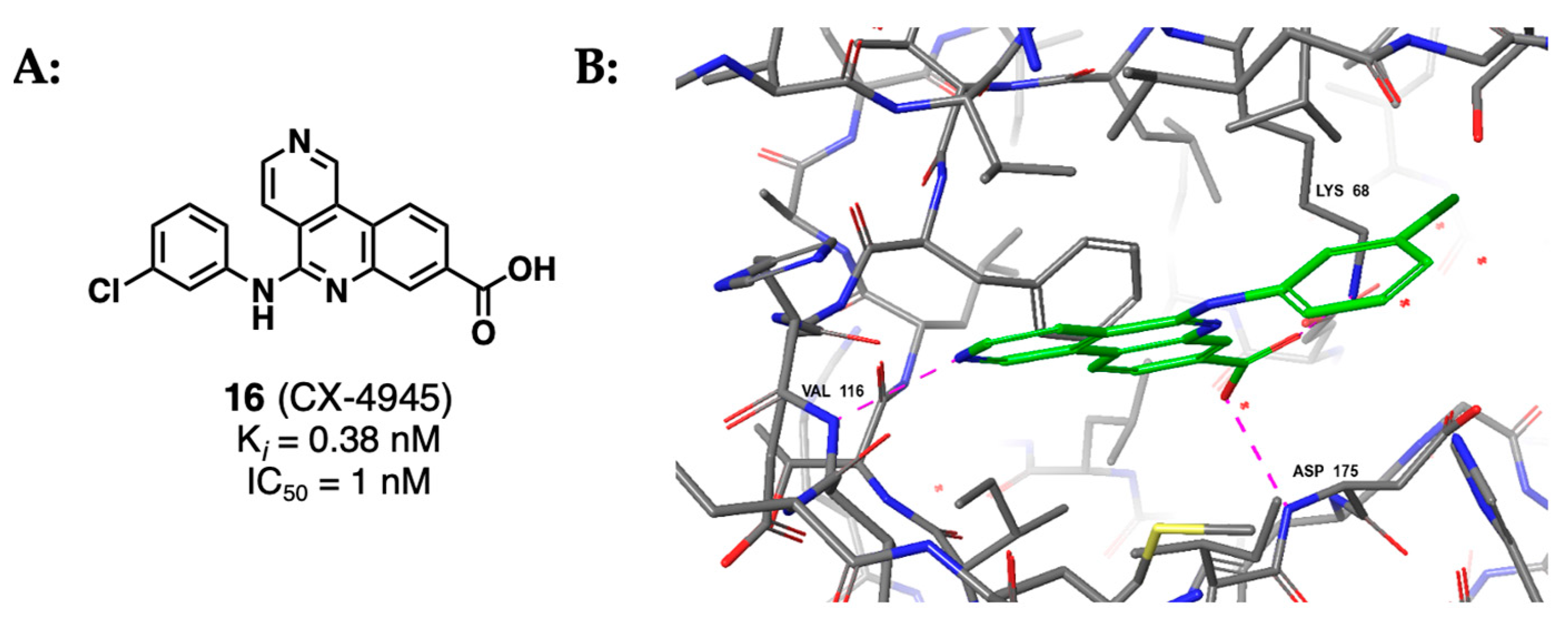
2.1.4. Indoloquinazolines Such as CX-4945
3. Extending beyond the ATP Site
3.1. Dual-Binding Ligands
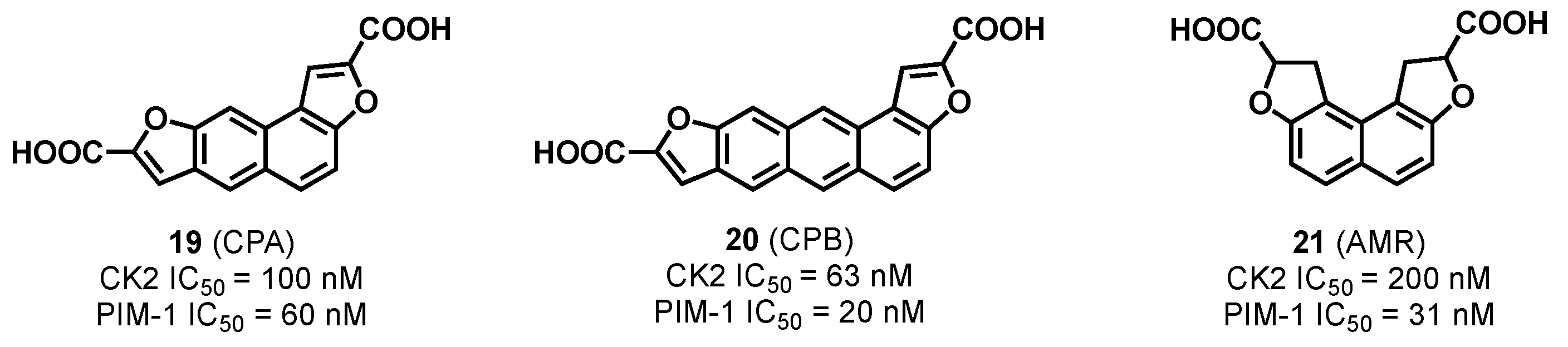
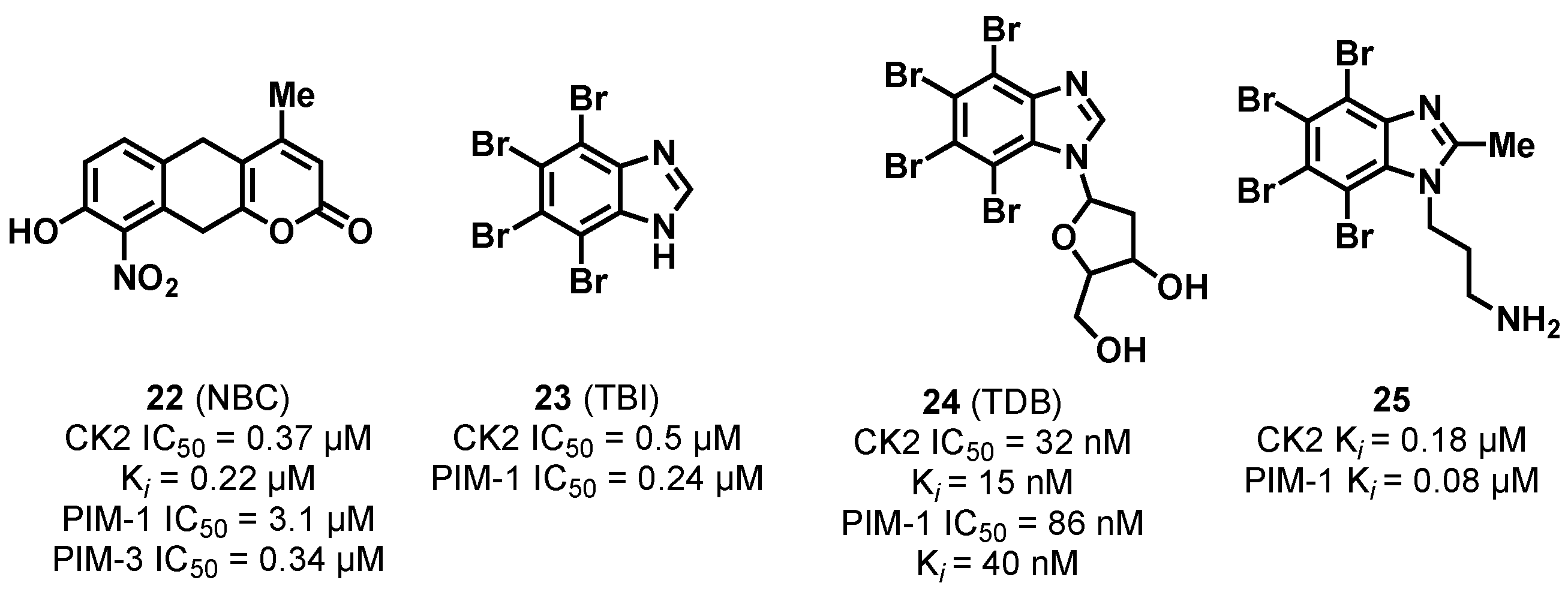
3.1.1. CK2/PIM Dual-Binding Ligands
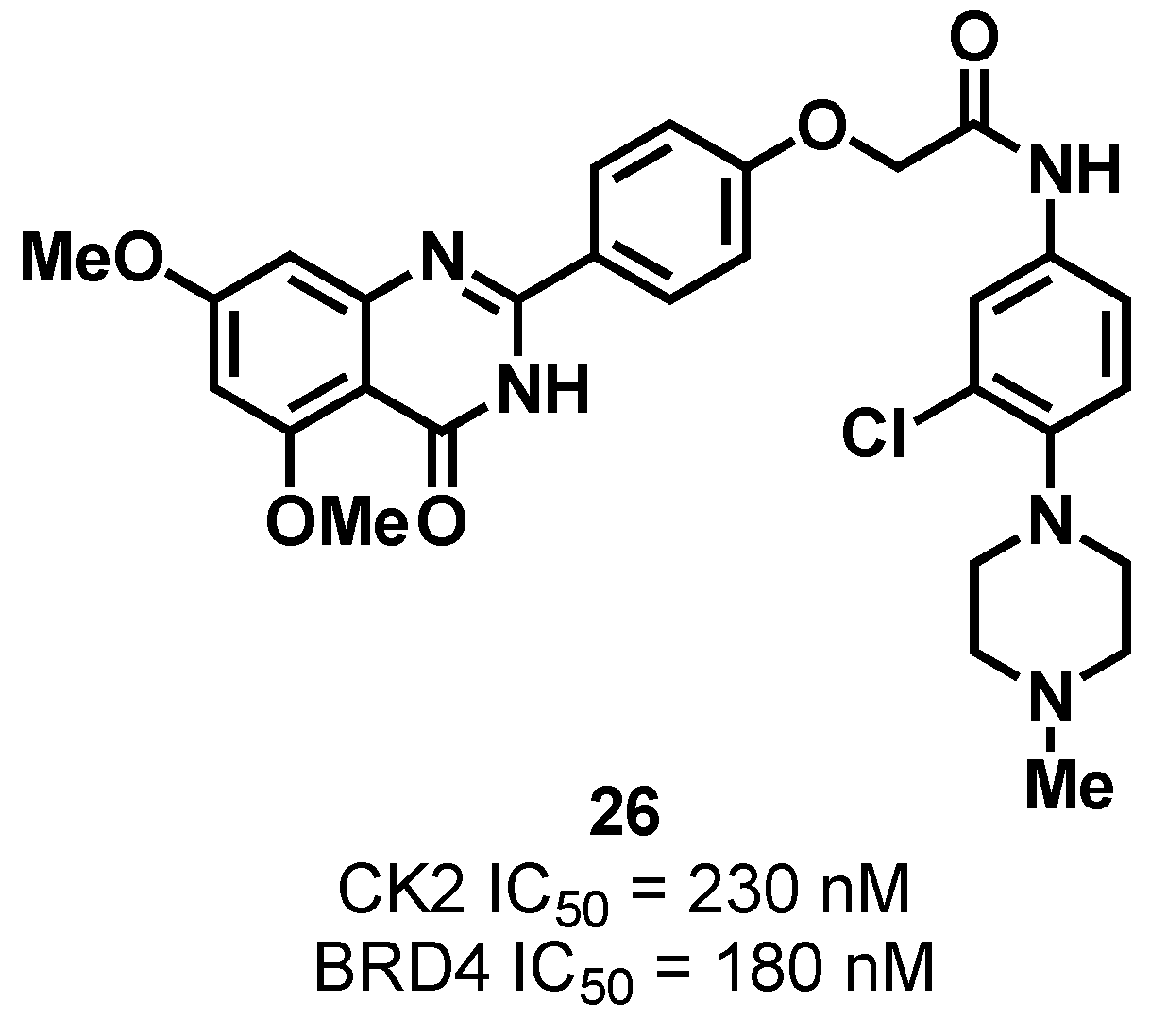
3.1.2. CK2/BRD4 Dual-Binding Ligands
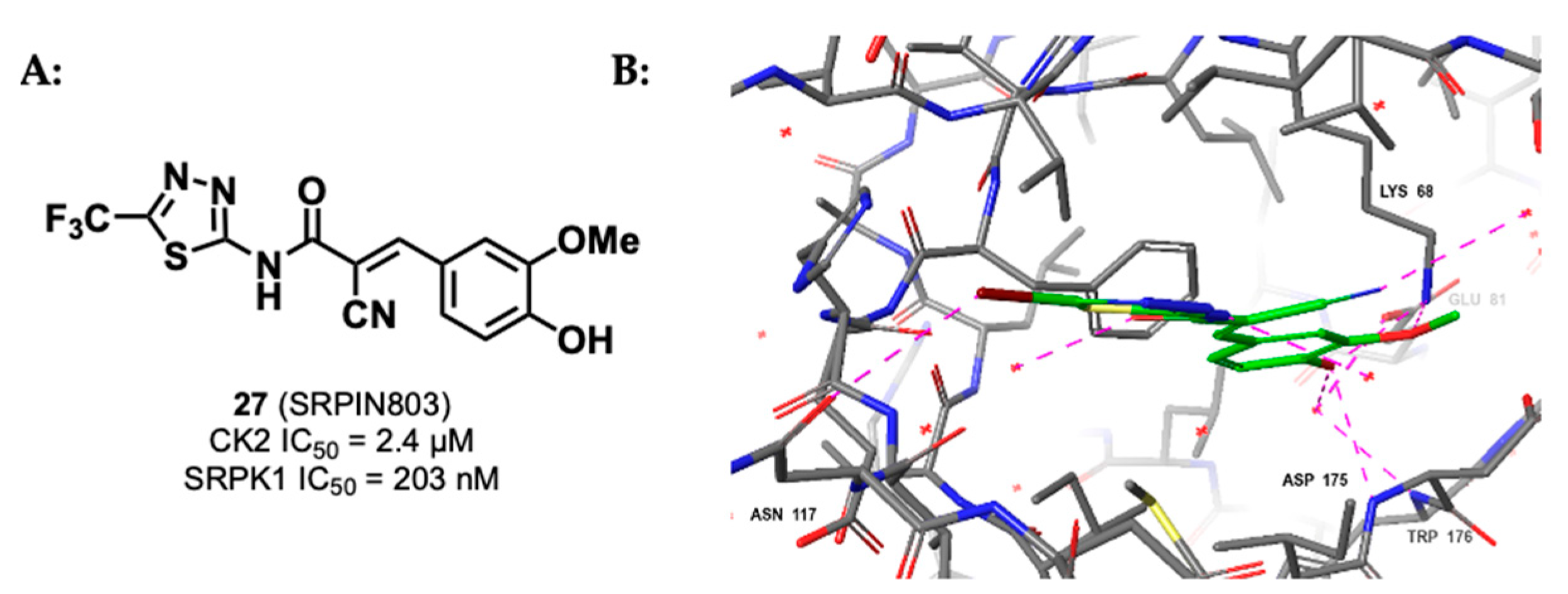
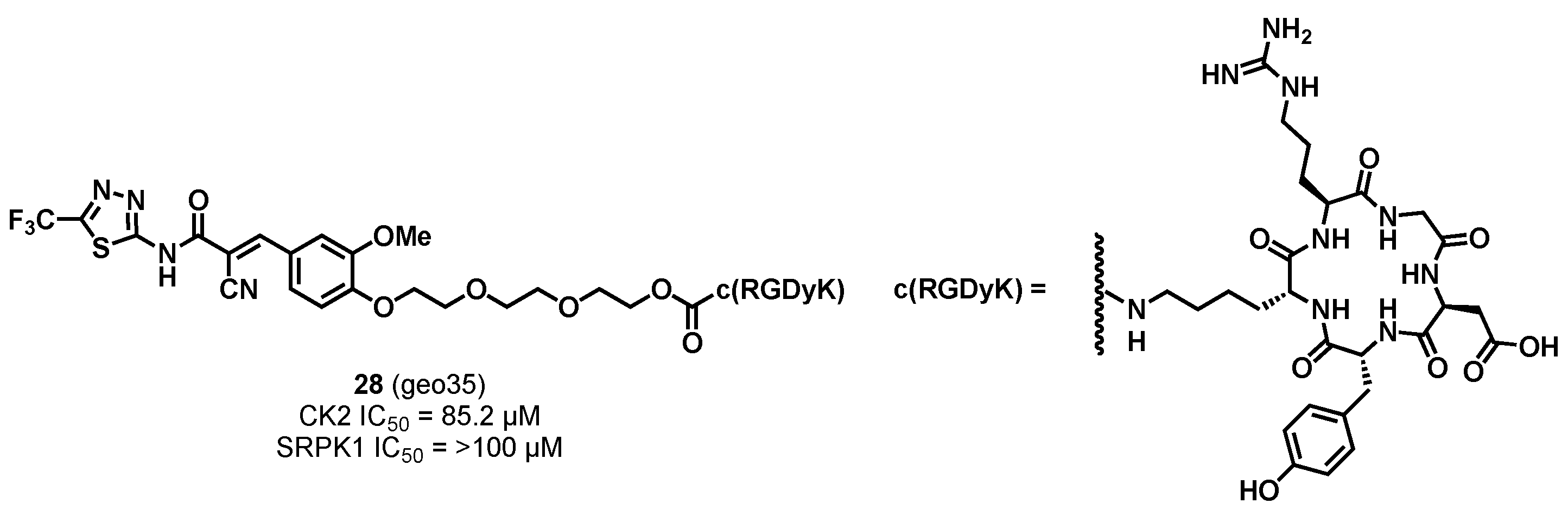
3.1.3. CK2/SRPK1 Dual Inhibitor
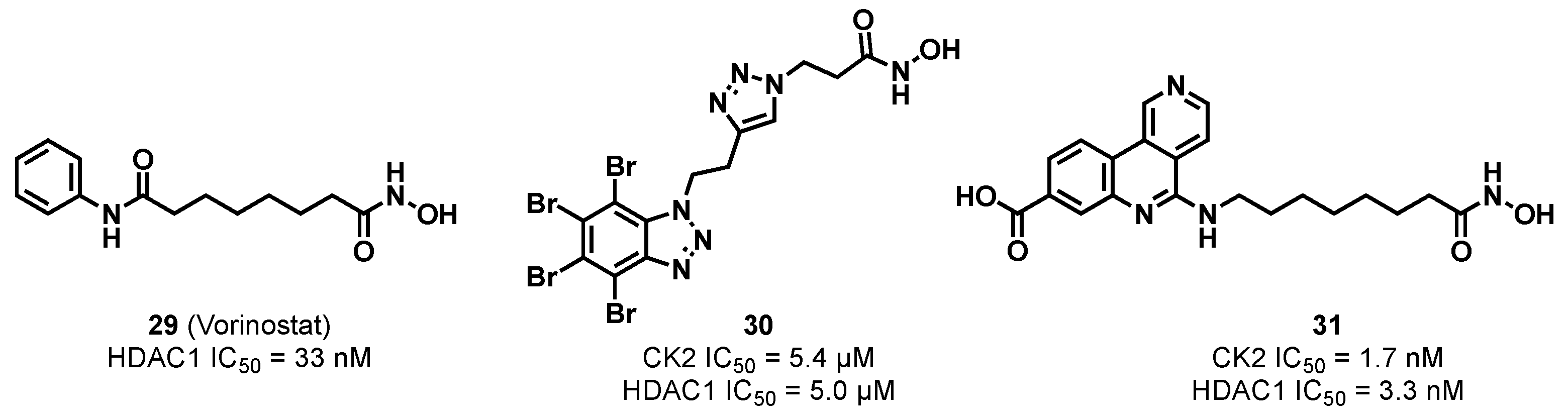
3.1.4. CK2/HDAC1 Dual Inhibitor
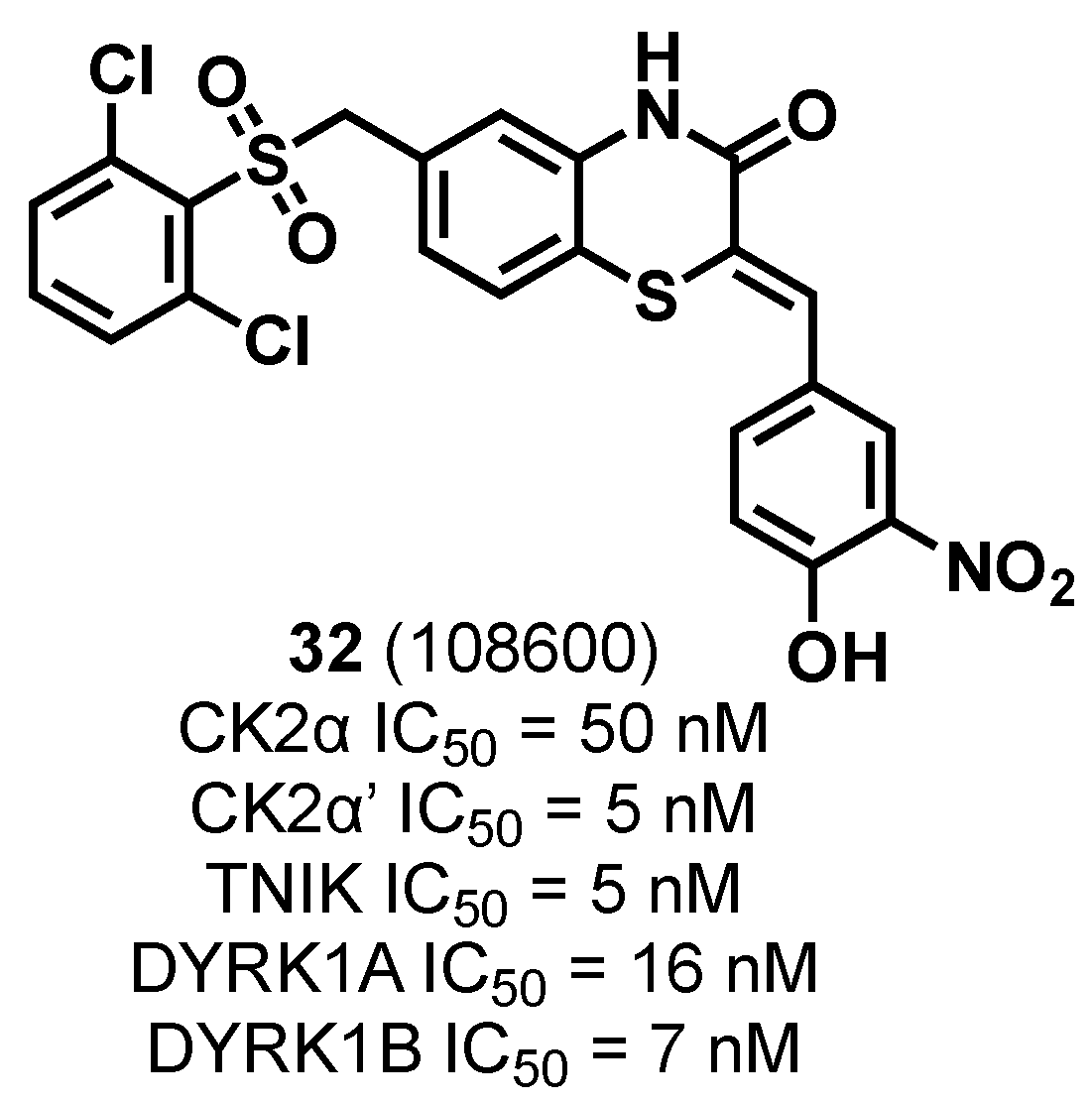
3.1.5. CK2/TNIK/DYRK1 Multiple Inhibitor
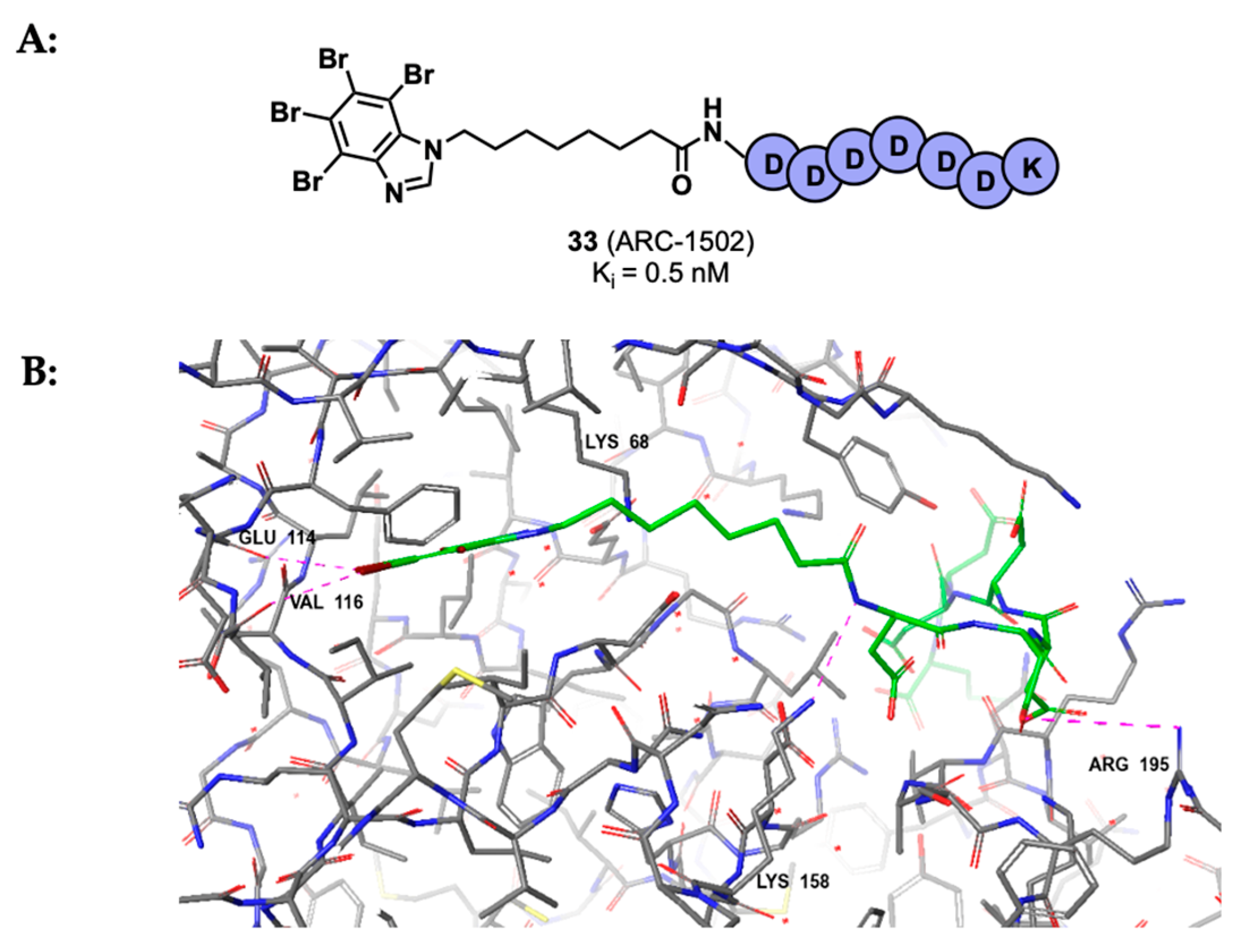
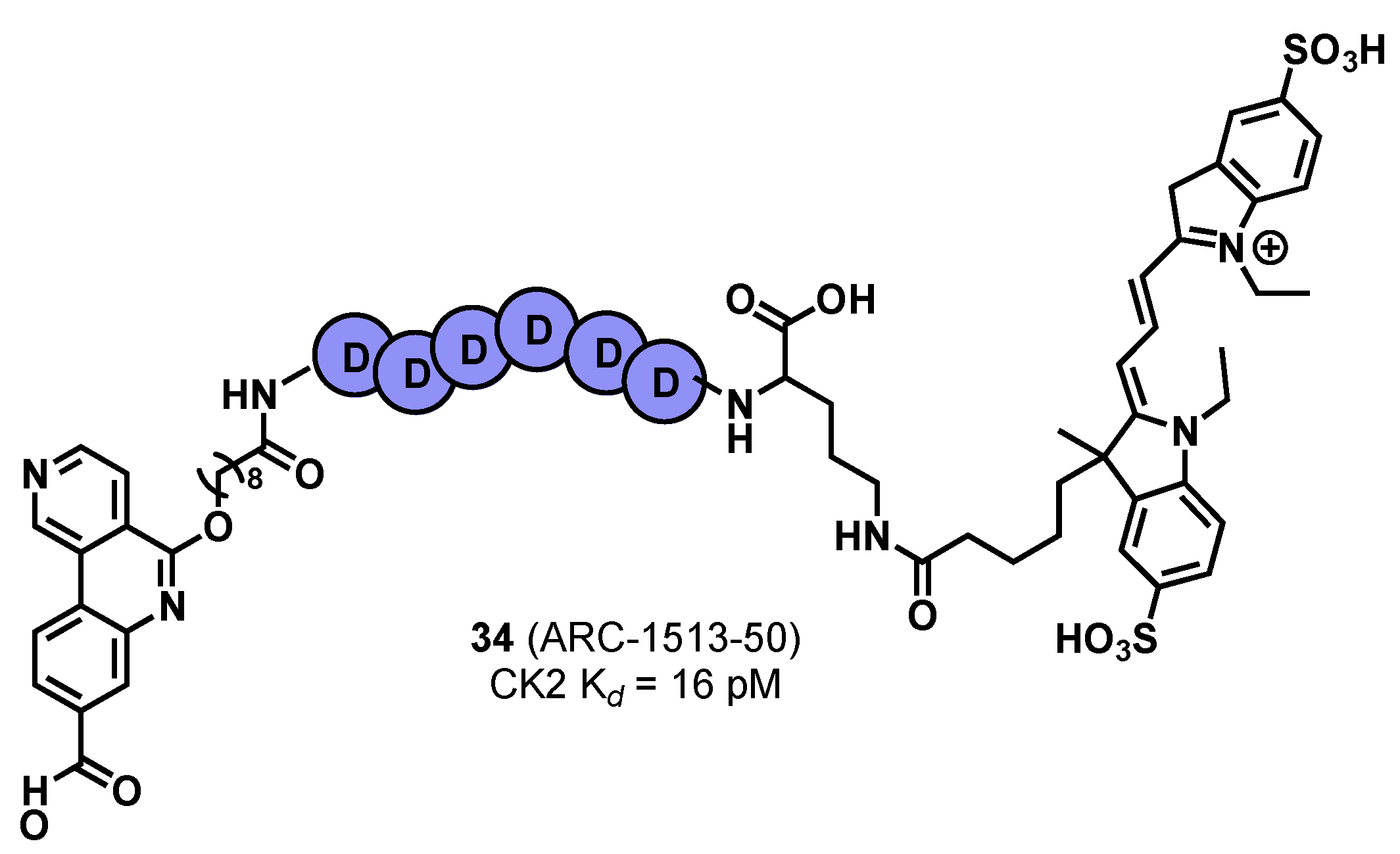
3.2. Substrate Binding Site Inhibition
3.3. Bi-Specific ATP/Substrate Competitive Inhibition
3.4. Inhibitors Acting in the αD Site
3.5. Holoenzyme Assembly Inhibition
3.6. CK2 Proteolysis-Targeting Chimeras
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hou, Z.; Liu, H. Mapping the Protein Kinome: Current Strategy and Future Direction. Cells 2023, 12, 925. [Google Scholar] [CrossRef]
- Obsilova, V.; Obsil, T. The 14-3-3 Proteins as Important Allosteric Regulators of Protein Kinases. Int. J. Mol. Sci. 2020, 21, 8824. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Small Molecule Protein Kinase Inhibitors Approved by Regulatory Agencies Outside of the United States. Pharmacol. Res. 2023, 194, 106847. [Google Scholar] [CrossRef] [PubMed]
- Montenarh, M. Cellular Regulators of Protein Kinase CK2. Cell Tissue Res. 2010, 342, 139. [Google Scholar] [CrossRef]
- Kannan, N.; Neuwald, A.F. Evolutionary Constraints Associated with Functional Specificity of the CMGC Protein Kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2α. Protein Sci. 2004, 13, 2059. [Google Scholar] [CrossRef]
- Burnett, G.; Kennedy, E.P. The Enzymatic Phosphorylation of Proteins. J. Biol. Chem. 1954, 211, 969. [Google Scholar] [CrossRef]
- Venerando, A.; Ruzzene, M.; Pinna, L.A. Casein Kinase: The Triple Meaning of a Misnomer. Biochem. J. 2014, 460, 141. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Guerra, B.; Ermakowa, I.; Issinger, O.G. Crystal structure of human protein kinase CK2: Insights into basic properties of the CK2 holoenzyme. EMBO J. 2001, 20, 5320. [Google Scholar] [CrossRef]
- Pinna, L.A. Protein Kinase CK2: A Challenge to Canons. J. Cell Sci. 2002, 115, 3873. [Google Scholar] [CrossRef]
- Roffey, S.E.; Litchfield, D.W. CK2 Regulation: Perspectives in 2021. Biomedicines 2021, 9, 1361. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Wang, J.; Zhou, Z.; Cao, S.; Zhang, J. Strategies of Targeting CK2 in Drug Discovery: Challenges, Opportunities, and Emerging Prospects. J. Med. Chem. 2023, 66, 2257. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Ghisellini, P.; Pinna, L.A. Unique activation mechanism of protein kinase CK2. J. Biol. Chem. 2002, 277, 22509. [Google Scholar] [CrossRef] [PubMed]
- Núñez de Villavicencio-Díaz, T.; Mazola, Y.; Perera Negrín, Y.; Cruz García, Y.; Guirola Cruz, O.; Perea Rodríguez, S.E. Predicting CK2 Beta-Dependent Substrates Using Linear Patterns. Biochem. Biophys. Rep. 2015, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Gottardo, M.F.; Capobianco, C.S.; Sidabra, J.E.; Garona, J.; Perera, Y.; Perea, S.E.; Alonso, D.F.; Farina, H.G. Preclinical Efficacy of CIGB-300, an Anti-CK2 Peptide, on Breast Cancer Metastasic Colonization. Sci. Rep. 2020, 10, 14689. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B. Protein Kinase CK2 Subunits Are Positive Regulators of AKT Kinase. Int. J. Oncol. 2006, 28, 685. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yeh, J.; Van Waes, C. Protein Kinase CK2 Mediates Inhibitor-Kappa B Kinase and Aberrant Nuclear Factor-κB Activation by Serum Factor(s) in Head and Neck Squamous Carcinoma Cells. Cancer Res. 2006, 66, 6722. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qin, H.; Frank, S.J.; Deng, L.; Litchfield, D.W.; Tefferi, A.; Pardanani, A.; Lin, F.-T.; Li, J.; Sha, B.; et al. A CK2-Dependent Mechanism for Activation of the JAK-STAT Signaling Pathway. Blood 2011, 118, 156. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, H. Casein Kinase 2 Is Activated and Essential for Wnt/β-Catenin Signaling. J. Biol. Chem. 2006, 281, 18394. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Liu, Y.; Xia, R.; Tong, C.; Yue, T.; Jiang, J.; Jia, J. Casein Kinase 2 Promotes Hedgehog Signaling by Regulating Both Smoothened and Cubitus Interruptus. J. Biol. Chem. 2010, 285, 37218. [Google Scholar] [CrossRef]
- Zhang, S.; Long, H.; Yang, Y.-L.; Wang, Y.; Hsieh, D.; Li, W.; Au, A.; Stoppler, H.J.; Xu, Z.; Jablons, D.M.; et al. Inhibition of CK2α Down-Regulates Notch1 Signalling in Lung Cancer Cells. J. Cell. Mol. Med. 2013, 17, 854. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Sig. Transduct. Target Ther. 2021, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Pinna, L.A. Addiction to Protein Kinase CK2: A Common Denominator of Diverse Cancer Cells? Biochim. Biophys. Acta BBA—Proteins Proteom. 2010, 1804, 499. [Google Scholar] [CrossRef]
- Faust, M.; Montenarch, M. Subcellular localisation of protein kinase CK2. A key to its function? Cell Tissue Res. 2000, 301, 329. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Li, B.; Kren, B.T.; Gravely, A.A.; Caicedo-Granados, E.; Klein, M.A.; Ahmed, K. CX-4945 and siRNA-Mediated Knockdown of CK2 Improves Cisplatin Response in HPV(+) and HPV(−) HNSCC Cell Lines. Biomedicines 2021, 9, 571. [Google Scholar] [CrossRef] [PubMed]
- Meza, C.P.Q.; Ruzeene, M. Protein Kinase CK2 and SARS-CoV-2: An Expected Interplay Story. Kinases Phosphatases 2023, 1, 141. [Google Scholar] [CrossRef]
- Breen, M.E.; Soellner, M.B. Small Molecule Substrate Phosphorylation Site Inhibitors of Protein Kinases: Approaches and Challenges. ACS Chem. Biol. 2015, 10, 175. [Google Scholar] [CrossRef]
- Grygier, P.; Pustelny, K.; Nowak, J.; Golik, P.; Popowicz, G.M.; Plettenburg, O.; Dubin, G.; Menezes, F.; Czarna, A. Silmitasertib (CX-4945), a Clinically Used CK2-Kinase Inhibitor with Additional Effects on GSK3β and DYRK1A Kinases: A Structural Perspective. J. Med. Chem. 2023, 66, 4009. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Yun, J.-S.; Kim, W.-K.; Chun, H.-S.; Jin, H.; Cho, S.; Chang, J.H. Structural Basis for the Selective Inhibition of Cdc2-Like Kinases by CX-4945. BioMed Res. Int. 2019, 2019, 6125068. [Google Scholar] [CrossRef]
- Iegre, J.; Atkinson, E.L.; Brear, P.D.; Cooper, B.M.; Hyvönen, M.; Spring, D.R. Chemical Probes Targeting the Kinase CK2: A Journey Outside the Catalytic Box. Org. Biomol. Chem. 2021, 19, 4380. [Google Scholar] [CrossRef]
- Schwartz, P.A.; Murray, B.W. Protein Kinase Biochemistry and Drug Discovery. Bioorg. Chem. 2011, 39, 192. [Google Scholar] [CrossRef]
- Nuñez de Villavicencio-Diaz, T.; Rabalski, A.J.; Litchfield, D.W. Protein Kinase CK2: Intricate Relationships within Regulatory Cellular Networks. Pharmaceuticals 2017, 10, 27. [Google Scholar] [CrossRef] [PubMed]
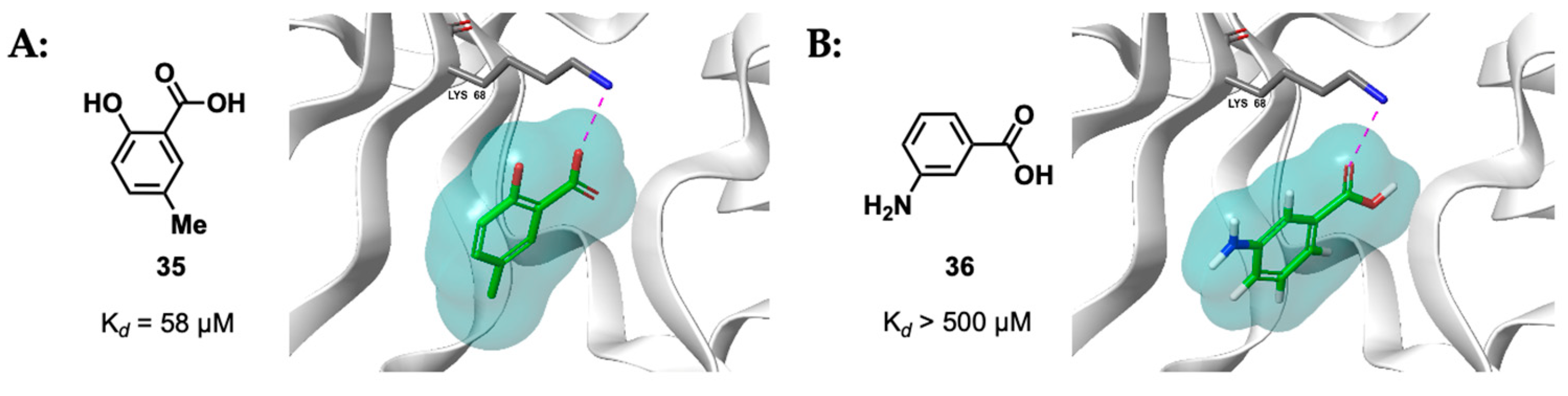
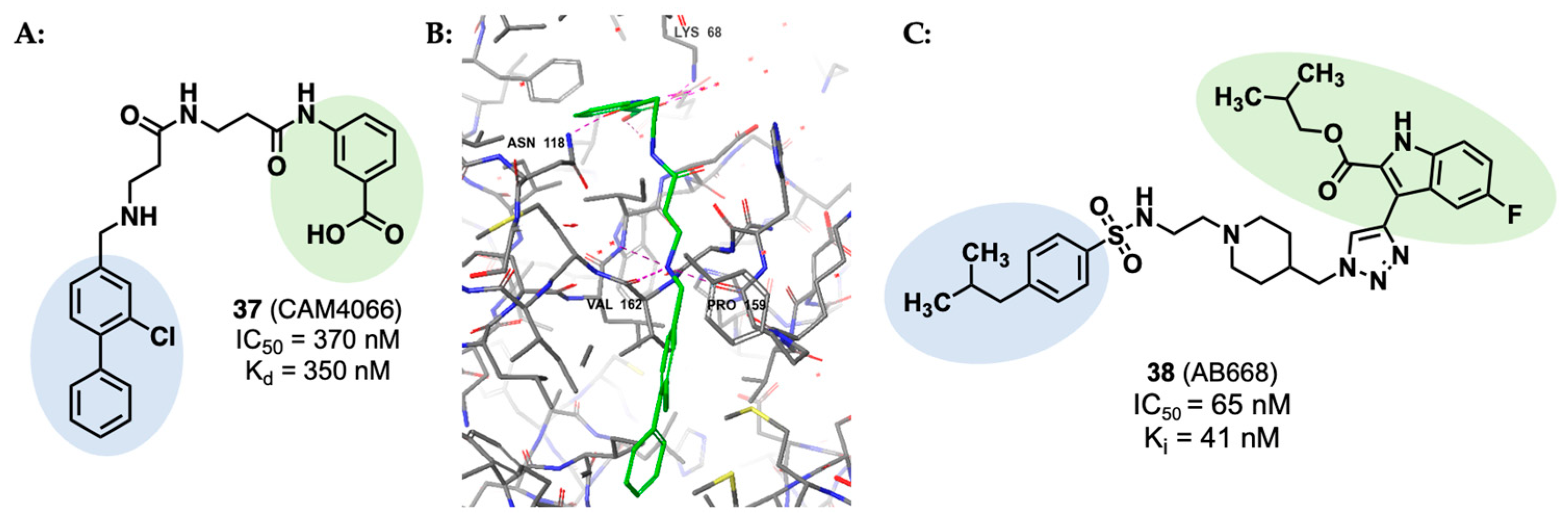
- De Fusco, C.; Brear, P.; Iegre, J.; Georgiou, K.H.; Sore, H.F.; Hyvönen, M.; Spring, D.R. A Fragment-Based Approach Leading to the Discovery of a Novel Binding Site and the Selective CK2 Inhibitor CAM4066. Bioorg. Med. Chem. 2017, 25, 3471. [Google Scholar] [CrossRef]
- Niefind, K.; Guerra, B.; Pinna, L.A.; Issinger, O.-G.; Schomburg, D. Crystal Structure of the Catalytic Subunit of Protein Kinase CK2 from Zea Mays at 2.1 Å Resolution. EMBO J. 1998, 17, 2451. [Google Scholar] [CrossRef] [PubMed]
- Papinutto, E.; Ranchio, A.; Lolli, G.; Pinna, L.A.; Battistutta, R. Structural and Functional Analysis of the Flexible Regions of the Catalytic α-Subunit of Protein Kinase CK2. J. Struct. Biol. 2012, 177, 382. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Raaf, J.; Issinger, O.-G. Protein kinase CK2 in health and disease: Protein kinase CK2: From structures to insights. Cell Mol. Life Sci. 2009, 66, 1800. [Google Scholar] [CrossRef] [PubMed]
- Lettieri, A.; Borgo, C.; Zanieri, L.; D’Amore, C.; Oleari, R.; Paganoni, A.; Pinna, L.A.; Cariboni, A.; Salvi, M. Protein Kinase CK2 Subunits Differentially Perturb the Adhesion and Migration of GN11 Cells: A Model of Immature Migrating Neurons. Int. J. Mol. Sci. 2019, 20, 5951. [Google Scholar] [CrossRef] [PubMed]
- Bibby, A.C.; Litchfield, D.W. The Multiple Personalities of the Regulatory Subunit of Protein Kinase CK2: CK2 Dependent and CK2 Independent Roles Reveal a Secret Identity for CK2beta. Int. J. Biol. Sci. 2005, 1, 67. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation, and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–5. [Google Scholar] [CrossRef]
- Filhol, O.; Martiel, J.-L.; Cochet, C. Protein Kinase CK2: A New View of an Old Molecular Complex. EMBO Rep. 2004, 5, 351. [Google Scholar] [CrossRef]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein Kinase CK2 in Health and Disease. Cell. Mol. Life Sci. 2009, 66, 1858. [Google Scholar] [CrossRef]
- Chua, M.M.J.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Kren, B.T.; Afzal, M.; Scaria, G.A.; Klein, M.A.; Ahmed, K. Protein Kinase CK2—Diverse Roles in Cancer Cell Biology and Therapeutic Promise. Mol. Cell. Biochem. 2023, 478, 899. [Google Scholar] [CrossRef]
- Faust, R.A.; Gapany, M.; Tristani, P.; Davis, A.; Adams, G.L.; Ahmed, K. Elevated Protein Kinase CK2 Activity in Chromatin of Head and Neck Tumors: Association with Malignant Transformation. Cancer Lett. 1996, 101, 31. [Google Scholar] [CrossRef]
- Yenice, S.; Davis, A.T.; Goueli, S.A.; Akdas, A.; Limas, C.; Ahmed, K. Nuclear casein kinase 2 (CK-2) activity in human normal, benign hyperplastic, and cancerous prostate. Prostate 1994, 24, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Mandato, E.; Manni, S.; Zaffino, F.; Semenzato, G.; Piazza, F. Targeting CK2-Driven Non-Oncogene Addiction in B-Cell Tumors. Oncogene 2016, 35, 6045. [Google Scholar] [CrossRef]
- Zou, J.; Luo, H.; Zeng, Q.; Dong, Z.; Wu, D.; Liu, L. Protein Kinase CK2α Is Overexpressed in Colorectal Cancer and Modulates Cell Proliferation and Invasion via Regulating EMT-Related Genes. J. Transl. Med. 2011, 9, 97. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Jiang, S.-S.; Zhang, X.-F.; Zhou, Z.-Q.; Pan, Q.-Z.; Chen, C.-L.; Zhao, J.-J.; Tang, Y.; Xia, J.-C.; Weng, D.-S. Protein Kinase CK2α Catalytic Subunit Is Overexpressed and Serves as an Unfavorable Prognostic Marker in Primary Hepatocellular Carcinoma. Oncotarget 2015, 6, 34800. [Google Scholar] [CrossRef]
- Turowec, J.P.; Vilk, G.; Gabriel, M.; Litchfield, D.W. Characterizing the Convergence of Protein Kinase CK2 and Caspase-3 Reveals Isoform-Specific Phosphorylation of Caspase-3 by CK2α’: Implications for Pathological Roles of CK2 in Promoting Cancer Cell Survival. Oncotarget 2013, 4, 560. [Google Scholar] [CrossRef]
- Zhou, B.; Ritt, D.A.; Morrison, D.K.; Der, C.J.; Cox, A.D. Protein Kinase CK2α Maintains Extracellular Signal-Regulated Kinase (ERK) Activity in a CK2α Kinase-Independent Manner to Promote Resistance to Inhibitors of RAF and MEK but Not ERK in BRAF Mutant Melanoma. J. Biol. Chem. 2016, 291, 17804. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Setoguchi, T.; Tsuru, A. Inhibition of casein kinase 2 prevents growth of human osteosarcoma. Oncol. Rep. 2017, 37, 1141. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, E.L.; Iegre, J.; Brear, P.D.; Zhabina, E.A.; Hyvönen, M.; Spring, D.R. Downfalls of Chemical Probes Acting at the Kinase ATP-Site: CK2 as a Case Study. Molecules 2021, 26, 1977. [Google Scholar] [CrossRef] [PubMed]
- Tamm, I.; Folkers, K.; Shunk, C.H.; Horsfall, F.L. Inhibition of Influenza Virus Multiplication by N-Glycosides of Benzimidazoles. J. Exp. Med. 1954, 99, 227. [Google Scholar] [CrossRef] [PubMed]
- Zandomeni, R.; Zandomeni, M.C.; Shugar, D.; Weinmann, R. Casein Kinase Type II Is Involved in the Inhibition by 5,6-Dichloro-1-Beta-D-Ribofuranosylbenzimidazole of Specific RNA Polymerase II Transcription. J. Biol. Chem. 1986, 261, 3414. [Google Scholar] [CrossRef]
- Szyszka, R.; Grankowski, N.; Felczak, K.; Shugar, D. Halogenated Benzimidazoles and Benzotriazoles as Selective Inhibitors of Protein Kinases CK-I and CK-II from Saccharomyces Cerevisiae and Other Sources. Biochem. Biophys. Res. Commun. 1995, 208, 418. [Google Scholar] [CrossRef]
- Wiley, R.H.; Hussung, K.F. Halogenated Benzotriazoles. J. Am. Chem. Soc. 1957, 79, 4395. [Google Scholar] [CrossRef]
- Pagano, M.A.; Meggio, F.; Ruzzene, M.; Andrzejewska, M.; Kazimierczuk, Z.; Pinna, L.A. 2-Dimethylamino-4,5,6,7-Tetrabromo-1H-Benzimidazole: A Novel Powerful and Selective Inhibitor of Protein Kinase CK2. Biochem. Biophys. Res. Commun. 2004, 321, 1040. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Bain, J.; Kazimierczuk, Z.; Sarno, S.; Ruzzene, M.; Di Maira, G.; Elliott, M.; Orzeszko, A.; Cozza, G.; Meggio, F.; et al. The Selectivity of Inhibitors of Protein Kinase CK2: An Update. Biochem. J. 2008, 415, 353. [Google Scholar] [CrossRef]
- Mishra, S.; Pertz, V.; Zhang, B.; Kaur, P.; Shimada, H.; Groffen, J.; Kazimierczuk, Z.; Pinna, L.A.; Heisterkamp, N. Treatment of P190 Bcr/Abl Lymphoblastic Leukemia Cells with Inhibitors of the Serine/Threonine Kinase CK2. Leukemia 2007, 21, 178. [Google Scholar] [CrossRef]
- Siddiqui, Y.H.; Kershaw, R.M.; Humphreys, E.H.; Assis Junior, E.M.; Chaudhri, S.; Jayaraman, P.-S.; Gaston, K. CK2 Abrogates the Inhibitory Effects of PRH/HHEX on Prostate Cancer Cell Migration and Invasion and Acts through PRH to Control Cell Proliferation. Oncogenesis 2017, 6, e293. [Google Scholar] [CrossRef]
- Ulges, A.; Witsch, E.J.; Pramanik, G.; Klein, M.; Birkner, K.; Bühler, U.; Wasser, B.; Luessi, F.; Stergiou, N.; Dietzen, S.; et al. Protein Kinase CK2 Governs the Molecular Decision between Encephalitogenic TH17 Cell and Treg Cell Development. Proc. Natl. Acad. Sci. USA 2016, 113, 10145. [Google Scholar] [CrossRef]
- Gartman, J.A.; Tambar, U.K. Recent Total Syntheses of Anthraquinone-Based Natural Products. Tetrahedron 2022, 105, 132501. [Google Scholar] [CrossRef] [PubMed]
- Janeczko, M.; Masłyk, M.; Kubiński, K.; Golczyk, H. Emodin, a Natural Inhibitor of Protein Kinase CK2, Suppresses Growth, Hyphal Development, and Biofilm Formation of Candida Albicans. Yeast 2017, 34, 253. [Google Scholar] [CrossRef]
- Meggio, F.; Pagano, M.A.; Moro, S.; Zagotto, G.; Ruzzene, M.; Sarno, S.; Cozza, G.; Bain, J.; Elliott, M.; Deana, A.D.; et al. Inhibition of Protein Kinase CK2 by Condensed Polyphenolic Derivatives: An In Vitro and In Vivo Study. Biochemistry 2004, 43, 12931. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G. The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals 2017, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- De Moliner, E.; Moro, S.; Sarno, S.; Zagotto, G.; Zanotti, G.; Pinna, L.A.; Battistutta, R. Inhibition of Protein Kinase CK2 by Anthraquinone-Related Compounds: A Structural Insight. J. Biol. Chem. 2003, 278, 1831. [Google Scholar] [CrossRef]
- Cozza, G.; Mazzorana, M.; Papinutto, E.; Bain, J.; Elliott, M.; di Maira, G.; Gianoncelli, A.; Pagano, M.A.; Sarno, S.; Ruzzene, M.; et al. Quinalizarin as a Potent, Selective and Cell-Permeable Inhibitor of Protein Kinase CK2. Biochem. J. 2009, 421, 387. [Google Scholar] [CrossRef]
- Cozza, G.; Venerando, A.; Sarno, S.; Pinna, L.A. The Selectivity of CK2 Inhibitor Quinalizarin: A Reevaluation. BioMed Res. Int. 2015, 2015, e734127. [Google Scholar] [CrossRef]
- Nie, Z.; Perretta, C.; Erickson, P.; Margosiak, S.; Almassy, R.; Lu, J.; Averill, A.; Yager, K.M.; Chu, S. Structure-Based Design, Synthesis, and Study of Pyrazolo[1,5-a][1,3,5]Triazine Derivatives as Potent Inhibitors of Protein Kinase CK2. Bioorg. Med. Chem. Lett. 2007, 17, 4191. [Google Scholar] [CrossRef]
- Nie, Z.; Perretta, C.; Erickson, P.; Margosiak, S.; Lu, J.; Averill, A.; Almassy, R.; Chu, S. Structure-Based Design and Synthesis of Novel Macrocyclic Pyrazolo[1,5-a] [1,3,5]Triazine Compounds as Potent Inhibitors of Protein Kinase CK2 and Their Anticancer Activities. Bioorg. Med. Chem. Lett. 2008, 18, 619. [Google Scholar] [CrossRef]
- Dowling, J.E.; Chuaqui, C.; Pontz, T.W.; Lyne, P.D.; Larsen, N.A.; Block, M.H.; Chen, H.; Su, N.; Wu, A.; Russell, D.; et al. Potent and Selective Inhibitors of CK2 Kinase Identified through Structure-Guided Hybridization. ACS Med. Chem. Lett. 2012, 3, 278. [Google Scholar] [CrossRef]
- Dowling, J.E.; Alimzhanov, M.; Bao, L.; Chuaqui, C.; Denz, C.R.; Jenkins, E.; Larsen, N.A.; Lyne, P.D.; Pontz, T.; Ye, Q.; et al. Potent and Selective CK2 Kinase Inhibitors with Effects on Wnt Pathway Signaling in Vivo. ACS Med. Chem. Lett. 2016, 7, 300. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.I.; Drewry, D.H.; Pickett, J.E.; Tjaden, A.; Krämer, A.; Müller, S.; Gyenis, L.; Menyhart, D.; Litchfield, D.W.; Knapp, S.; et al. Development of a Potent and Selective Chemical Probe for the Pleiotropic Kinase CK2. Cell Chem. Biol. 2021, 28, 546.e10. [Google Scholar] [CrossRef] [PubMed]
- Licciardello, M.P.; Workman, P. A New Chemical Probe Challenges the Broad Cancer Essentiality of CK2. Trends Pharmacol. Sci. 2021, 42, 313. [Google Scholar] [CrossRef]
- Salvi, M.; Borgo, C.; Pinna, L.A.; Ruzzene, M. Targeting CK2 in Cancer: A Valuable Strategy or a Waste of Time? Cell Death Discov. 2021, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.W.; Drewry, D.H.; Axtman, A.D. CK2 Chemical Probes: Past, Present, and Future. Kinases Phosphatases 2023, 1, 288. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288. [Google Scholar] [CrossRef]
- Senhwa Biosciences CX-4945 Granted Orphan Drug Designation by the US FDA in Cholangiocarcinoma. News Release. Senhwa Biosciences, Inc. January 4 2017. Available online: https://www.prnewswire.com/news-releases/senhwa-biosciences-cx-4945-granted-orphan-drug-designation-by-the-us-fda-in-cholangiocarcinoma-300385278.html (accessed on 14 February 2024).
- D’Amore, C.; Borgo, C.; Sarno, S.; Salvi, M. Role of CK2 Inhibitor CX-4945 in Anti-Cancer Combination Therapy—Potential Clinical Relevance. Cell. Oncol. Dordr. 2020, 43, 1003. [Google Scholar] [CrossRef]
- Chon, H.J.; Bae, K.J.; Lee, Y.; Kim, J. The Casein Kinase 2 Inhibitor, CX-4945, as an Anti-Cancer Drug in Treatment of Human Hematological Malignancies. Front. Pharmacol. 2015, 6, 70. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; Bottegoni, G. Multi-Kinase Inhibitors. Curr. Med. Chem. 2015, 22, 695. [Google Scholar] [CrossRef]
- Menyhart, D.; Gyenis, L.; Jurcic, K.; Roffey, S.E.; Puri, A.; Jovanovic, P.; Szkop, K.J.; Pittock, P.; Lajoie, G.; Axtman, A.D.; et al. Comparison of CX-4945 and SGC-CK2-1 as Inhibitors of CSNK2 Using Quantitative Phosphoproteomics: Triple SILAC in Combination with Inhibitor-Resistant CSNK2. Curr. Res. Chem. Biol. 2023, 3, 100041. [Google Scholar] [CrossRef]
- Battistutta, R.; Cozza, G.; Pierre, F.; Papinutto, E.; Lolli, G.; Sarno, S.; O’Brien, S.E.; Siddiqui-Jain, A.; Haddach, M.; Anderes, K.; et al. Unprecedented Selectivity and Structural Determinants of a New Class of Protein Kinase CK2 Inhibitors in Clinical Trials for the Treatment of Cancer. Biochemistry 2011, 50, 8478. [Google Scholar] [CrossRef]
- Ursu, A.; Childs-Disney, J.L.; Angelbello, A.J.; Costales, M.G.; Meyer, S.M.; Disney, M.D. Gini Coefficients as a Single Value Metric to Define Chemical Probe Selectivity. ACS Chem. Biol. 2020, 15, 2031. [Google Scholar] [CrossRef]
- Ember, S.W.; Lambert, Q.T.; Berndt, N.; Gunawan, S.; Ayaz, M.; Tauro, M.; Zhu, J.-Y.; Cranfill, P.J.; Greninger, P.; Lynch, C.C.; et al. Potent Dual BET Bromodomain-Kinase Inhibitors as Value Added Multi-Targeted Chemical Probes and Cancer Therapeutics. Mol. Cancer Ther. 2017, 16, 1054. [Google Scholar] [CrossRef]
- Ciceri, P.; Müller, S.; O’Mahony, A.; Fedorov, O.; Filippakopoulos, P.; Hunt, J.P.; Lasater, E.A.; Pallares, G.; Picaud, S.; Wells, C.; et al. Dual Kinase-Bromodomain Inhibitors for Rationally Designed Polypharmacology. Nat. Chem. Biol. 2014, 10, 305. [Google Scholar] [CrossRef] [PubMed]
- Karim, R.M.; Bikowitz, M.J.; Chan, A.; Zhu, J.-Y.; Grassie, D.; Becker, A.; Berndt, N.; Gunawan, S.; Lawrence, N.J.; Schönbrunn, E. Differential BET Bromodomain Inhibition by Dihydropteridinone and Pyrimidodiazepinone Kinase Inhibitors. J. Med. Chem. 2021, 64, 15772. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Li, X.; Magnuson, N.S. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 2008, 27, 4809. [Google Scholar] [CrossRef] [PubMed]
- Panchal, N.K.; Sabina, E.P. A Serine/Threonine Protein PIM Kinase as a Biomarker of Cancer and a Target for Anti-Tumor Therapy. Life Sci. 2020, 255, 117866. [Google Scholar] [CrossRef] [PubMed]
- Decker, S.; Finter, J.; Forde, A.J.; Kissel, S.; Schwaller, J.; Mack, T.S.; Kuhn, A.; Gray, N.; Follo, M.; Jumaa, H.; et al. PIM Kinases Are Essential for Chronic Lymphocytic Leukemia Cell Survival (PIM2/3) and CXCR4-Mediated Microenvironmental Interactions (PIM1). Mol. Cancer Ther. 2014, 13, 1231. [Google Scholar] [CrossRef]
- Luszczak, S.; Kumar, C.; Sathyadevan, V.K.; Simpson, B.S.; Gately, K.A.; Whitaker, H.C.; Heavey, S. PIM Kinase Inhibition: Co-Targeted Therapeutic Approaches in Prostate Cancer. Signal Transduct. Target. Ther. 2020, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Aparicio, C.; Carnero, A. Pim Kinases in Cancer: Diagnostic, Prognostic and Treatment Opportunities. Biochem. Pharmacol. 2013, 85, 629. [Google Scholar] [CrossRef] [PubMed]
- Arrouchi, H.; Lakhlili, W.; Ibrahimi, A. A Review on PIM Kinases in Tumors. Bioinformation 2019, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- López-Ramos, M.; Prudent, R.; Moucadel, V.; Sautel, C.; Barette, C.; Lafanechère, L.; Mouawad, L.; Grierson, D.; Schmidt, F.; Florent, J.; et al. New Potent Dual Inhibitors of CK2 and Pim Kinases: Discovery and Structural Insights. FASEB J. 2010, 24, 3171. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Mazzorana, M.; Traynor, R.; Ruzzene, M.; Cozza, G.; Pagano, M.A.; Meggio, F.; Zagotto, G.; Battistutta, R.; Pinna, L.A. Structural Features Underlying the Selectivity of the Kinase Inhibitors NBC and dNBC: Role of a Nitro Group That Discriminates between CK2 and DYRK1A. Cell. Mol. Life Sci. 2012, 69, 449. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Sarno, S.; Ruzzene, M.; Girardi, C.; Orzeszko, A.; Kazimierczuk, Z.; Zagotto, G.; Bonaiuto, E.; Di Paolo, M.L.; Pinna, L.A. Exploiting the Repertoire of CK2 Inhibitors to Target DYRK and PIM Kinases. Biochim. Biophys. Acta BBA—Proteins Proteom. 2013, 1834, 1402. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Girardi, C.; Ranchio, A.; Lolli, G.; Sarno, S.; Orzeszko, A.; Kazimierczuk, Z.; Battistutta, R.; Ruzzene, M.; Pinna, L.A. Cell-Permeable Dual Inhibitors of Protein Kinases CK2 and PIM-1: Structural Features and Pharmacological Potential. Cell. Mol. Life Sci. 2014, 71, 3173. [Google Scholar] [CrossRef]
- Girardi, C.; Ottaviani, D.; Pinna, L.A.; Ruzzene, M. Different Persistence of the Cellular Effects Promoted by Protein Kinase CK2 Inhibitors CX-4945 and TDB. BioMed Res. Int. 2015, 2015, 185736. [Google Scholar] [CrossRef]
- Chojnacki, K.; Wińska, P.; Wielechowska, M.; Łukowska-Chojnacka, E.; Tölzer, C.; Niefind, K.; Bretner, M. Biological Properties and Structural Study of New Aminoalkyl Derivatives of Benzimidazole and Benzotriazole, Dual Inhibitors of CK2 and PIM1 Kinases. Bioorg. Chem. 2018, 80, 266. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond Transcriptional Regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Di Micco, R.; Fontanals-Cirera, B.; Low, V.; Ntziachristos, P.; Yuen, S.K.; Lovell, C.D.; Dolgalev, I.; Yonekubo, Y.; Zhang, G.; Rusinova, E.; et al. Control of Embryonic Stem Cell Identity by BRD4-Dependent Transcriptional Elongation of Super-Enhancer-Associated Pluripotency Genes. Cell Rep. 2014, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Lee, A.-Y.; Lai, H.-T.; Zhang, H.; Chiang, C.-M. Phospho Switch Triggers BRD4 Chromatin Binding and Activator Recruitment for Gene-Specific Targeting. Mol. Cell 2013, 49, 843. [Google Scholar] [CrossRef]
- Malvezzi, F.; Stubbs, C.J.; Jowitt, T.A.; Dale, I.L.; Guo, X.; DeGnore, J.P.; Degliesposti, G.; Skehel, J.M.; Bannister, A.J.; McAlister, M.S. Phosphorylation-Dependent BRD4 Dimerization and Implications for Therapeutic Inhibition of BET Family Proteins. Commun. Biol. 2021, 4, 1. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Lee, C.-F.; Lai, H.-T.; Yu, C.-T.; Lee, J.-E.; Zuo, H.; Tsai, S.Y.; Tsai, M.-J.; Ge, K.; Wan, Y.; et al. Opposing Functions of BRD4 Isoforms in Breast Cancer. Mol. Cell 2020, 78, 1114.e10. [Google Scholar] [CrossRef]
- Sanz-Alvarez, M.; Cristobal, I.; Luque, M.; Santos, A.; Zazo, S.; Madoz-Gurpide, J.; Carames, C.; Chiang, C.M.; Garcia-Foncillas, J.; Eroles, P.; et al. Expression of Phosphorylated BRD4 is Markedly Associated with the Activation Status of the PP2A Pathway and Shows a Strong Prognostic Value in Triple Negative Breast Cancer Patients. Cancers 2021, 31, 1246. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, P.; Zou, L.; Zhang, J.; Chen, J.; Yang, C.; He, G.; Liu, B.; Liu, J.; Chiang, C.-M.; et al. Discovery of Novel Dual-Target Inhibitor of Bromodomain-Containing Protein 4/Casein Kinase 2 Inducing Apoptosis and Autophagy-Associated Cell Death for Triple-Negative Breast Cancer Therapy. J. Med. Chem. 2021, 64, 18025. [Google Scholar] [CrossRef]
- Huang, J.-Q.; Duan, L.-X.; Liu, Q.-Y.; Li, H.-F.; Hu, A.-P.; Song, J.-W.; Lin, C.; Huang, B.; Yao, D.; Peng, B.; et al. Serine-Arginine Protein Kinase 1 (SRPK1) Promotes EGFR-TKI Resistance by Enhancing GSK3β Ser9 Autophosphorylation Independent of Its Kinase Activity in Non-Small-Cell Lung Cancer. Oncogene 2023, 42, 1233. [Google Scholar] [CrossRef] [PubMed]
- Nikas, I.P.; Themistocleous, S.C.; Paschou, S.A.; Tsamis, K.I.; Ryu, H.S. Serine-Arginine Protein Kinase 1 (SRPK1) as a Prognostic Factor and Potential Therapeutic Target in Cancer: Current Evidence and Future Perspectives. Cells 2019, 9, 19. [Google Scholar] [CrossRef]
- Yi, N.; Xiao, M.; Jiang, F.; Liu, Z.; Ni, W.; Lu, C.; Ni, R.; Chen, W. SRPK1 Is a Poor Prognostic Indicator and a Novel Potential Therapeutic Target for Human Colorectal Cancer. OncoTargets Ther. 2018, 11, 5359. [Google Scholar] [CrossRef]
- Mylonis, I.; Giannakouros, T. Protein kinase CK2 phosphorylates and activates the SR protein-specific kinase 1. Biochem. Biophys. Res. Commun. 2003, 301, 650. [Google Scholar] [CrossRef]
- Morooka, S.; Hoshina, M.; Kii, I.; Okabe, T.; Kojima, H.; Inoue, N.; Okuno, Y.; Denawa, M.; Yoshida, S.; Fukuhara, J.; et al. Identification of a Dual Inhibitor of SRPK1 and CK2 That Attenuates Pathological Angiogenesis of Macular Degeneration in Mice. Mol. Pharmacol. 2015, 88, 316. [Google Scholar] [CrossRef] [PubMed]
- Leonidis, G.; Dalezis, P.; Trafalis, D.; Beis, D.; Giardoglou, P.; Koukiali, A.; Sigala, I.; Nikolakaki, E.; Sarli, V. Synthesis and Biological Evaluation of a c(RGDyK) Peptide Conjugate of SRPIN803. ACS Omega 2021, 6, 28379. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.C.; Seto, E. Regulation of histone deacetylase 2 by protein kinase CK2. J. Biol. Chem. 2002, 277, 31826. [Google Scholar] [CrossRef] [PubMed]
- Bubna, A.K. Vorinostat—An Overview. Indian J. Dermatol. 2015, 60, 419. [Google Scholar] [CrossRef]
- Ruzzene, M.; Penzo, D.; Pinna, L.A. Protein Kinase CK2 Inhibitor 4,5,6,7-Tetrabromobenzotriazole (TBB) Induces Apoptosis and Caspase-Dependent Degradation of Haematopoietic Lineage Cell-Specific Protein 1 (HS1) in Jurkat Cells. Biochem. J. 2002, 364, 41. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.C.; Hessenauer, A.; Götz, C.; Montenarh, M. DMAT, an Inhibitor of Protein Kinase CK2 Induces Reactive Oxygen Species and DNA Double Strand Breaks. Oncol. Rep. 2009, 21, 1593. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Purwin, M.; Hernández-Toribio, J.; Coderch, C.; Panchuk, R.; Skorokhyd, N.; Filipiak, K.; de Pascual-Teresa, B.; Ramos, A. Design and Synthesis of Novel Dual-Target Agents for HDAC1 and CK2 Inhibition. RSC Adv. 2016, 6, 66595. [Google Scholar] [CrossRef]
- Rangasamy, L.; Ortín, I.; Zapico, J.M.; Coderch, C.; Ramos, A.; de Pascual-Teresa, B. New Dual CK2/HDAC1 Inhibitors with Nanomolar Inhibitory Activity against Both Enzymes. ACS Med. Chem. Lett. 2020, 11, 713. [Google Scholar] [CrossRef]
- Deboever, E.; Fistrovich, A.; Hulme, C.; Dunckley, T. The Omnipresence of DYRK1A in Human Diseases. Int. J. Mol. Sci. 2022, 23, 9355. [Google Scholar] [CrossRef]
- Di Vona, C.; Daniela Bezdan, D.; Islam, A.; Salichs, E.; López-Bigas, N.; Ossowski, S.; de la Luna, S. Chromatin-wide Profiling of DYRK1A Reveals a Role as a Gene-Specific RNA Polymerase II CTD Kinase. Mol. Cell 2015, 57, 506. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, T.; Li, V.S.; Ng, S.S.; Taouatas, N.; Vries, R.G.; Mohammed, S.; Heck, A.J.; Clevers, H. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 2009, 28, 3329. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Padgaonkar, A.A.; Baker, S.J.; Cosenza, S.C.; Rechkoblit, O.; Subbaiah, D.V.; Domingo-Domenech, J.; Bartkowski, A.; Port, E.R.; Aggarwal, A.K.; et al. Simultaneous CK2/TNIK/DYRK1 inhibition by 108600 suppresses triple negative breast cancer stem cells and chemotherapy-resistant disease. Nat. Commun. 2021, 12, 4671. [Google Scholar] [CrossRef]
- Sato, K.; Baker, S.J.; Reddy, E.P.; Irie, H.Y. Abstract 308: Novel Cancer Stem Cell Inhibitor 108600 Modulates Tumor Immunomicroenvironment of Triple Negative Breast Cancer (TNBC). Cancer Res. 2022, 82, PD3-08. [Google Scholar] [CrossRef]
- Meggio, F.; Pinna, L.A.; Marchiori, F.; Borin, G. Polyglutamyl Peptides: A New Class of Inhibitors of Type-2 Casein Kinases. FEBS Lett. 1983, 162, 235. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Reyes, O.; Puchades, Y.; Mendoza, O.; Vispo, N.S.; Torrens, I.; Santos, A.; Silva, R.; Acevedo, B.; López, E.; et al. Antitumor Effect of a Novel Proapoptotic Peptide that Impairs the Phosphorylation by the Protein Kinase 2 (Casein Kinase 2). Cancer Res. 2004, 64, 7127. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Baladron, I.; Garcia, Y.; Perera, Y.; Lopez, A.; Soriano, J.L.; Batista, N.; Palau, A.; Hernández, I.; Farina, H.; et al. CIGB-300, a Synthetic Peptide-Based Drug That Targets the CK2 Phosphoaceptor Domain. Translational and Clinical Research. Mol. Cell. Biochem. 2011, 356, 45. [Google Scholar] [CrossRef]
- Cruz, L.R.; Baladrón, I.; Rittoles, A.; Díaz, P.A.; Valenzuela, C.; Santana, R.; Vázquez, M.M.; García, A.; Chacón, D.; Thompson, D.; et al. Treatment with an Anti-CK2 Synthetic Peptide Improves Clinical Response in COVID-19 Patients with Pneumonia. A Randomized and Controlled Clinical Trial. ACS Pharmacol. Transl. Sci. 2020, 4, 206. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Zanin, S.; Sarno, S.; Costa, E.; Girardi, C.; Ribaudo, G.; Salvi, M.; Zagotto, G.; Ruzzene, M.; Pinna, L.A. Design, Validation and Efficacy of Bisubstrate Inhibitors Specifically Affecting Ecto-CK2 Kinase Activity. Biochem. J. 2015, 471, 415. [Google Scholar] [CrossRef]
- Enkvist, E.; Viht, K.; Bischoff, N.; Vahter, J.; Saaver, S.; Raidaru, G.; Issinger, O.-G.; Niefind, K.; Uri, A. A Subnanomolar Fluorescent Probe for Protein Kinase CK2 Interaction Studies. Org. Biomol. Chem. 2012, 10, 8645. [Google Scholar] [CrossRef]
- Viht, K.; Saaver, S.; Vahter, J.; Enkvist, E.; Lavogina, D.; Sinijärv, H.; Raidaru, G.; Guerra, B.; Issinger, O.-G.; Uri, A. Acetoxymethyl Ester of Tetrabromobenzimidazole-Peptoid Conjugate for Inhibition of Protein Kinase CK2 in Living Cells. Bioconjug. Chem. 2015, 26, 2324. [Google Scholar] [CrossRef]
- Vahter, J.; Viht, K.; Uri, A.; Enkvist, E. Oligo-Aspartic Acid Conjugates with Benzo[c][2,6]Naphthyridine-8-Carboxylic Acid Scaffold as Picomolar Inhibitors of CK2. Bioorg. Med. Chem. 2017, 25, 2277. [Google Scholar] [CrossRef] [PubMed]
- Brear, P.; De Fusco, C.; Hadje Georgiou, K.; Francis-Newton, N.J.; Stubbs, C.J.; Sore, H.F.; Venkitaraman, A.R.; Abell, C.; Spring, D.R.; Hyvönen, M. Specific Inhibition of CK2α from an Anchor Outside the Active Site. Chem. Sci. 2016, 7, 6839. [Google Scholar] [CrossRef]
- Li, C.; Zhang, X.; Zhang, N.; Zhou, Y.; Sun, G.; Zhao, L.; Zhong, R. Identification and Biological Evaluation of CK2 Allosteric Fragments through Structure-Based Virtual Screening. Molecules 2020, 25, 237. [Google Scholar] [CrossRef]
- Bancet, A.; Frem, R.; Jeanneret, F.; Mularoni, A.; Bazelle, P.; Roelants, C.; Delcros, J.G.; Guichou, J.F.; Pillet, C.; Coste, I.; et al. Cancer Selective Cell Death Induction by a Bivalent CK2 Inhibitor Targeting the ATP Site and the Allosteric αD Pocket. iScience. 2024, 27, 108903. [Google Scholar] [CrossRef] [PubMed]
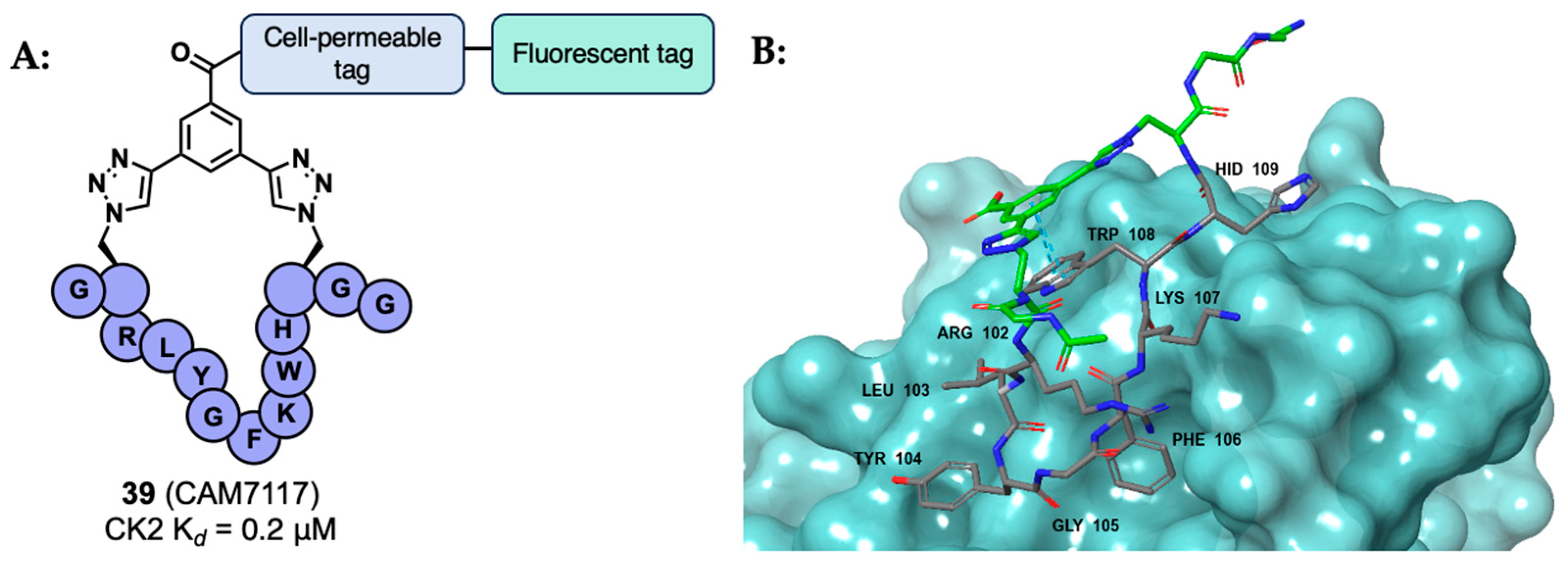
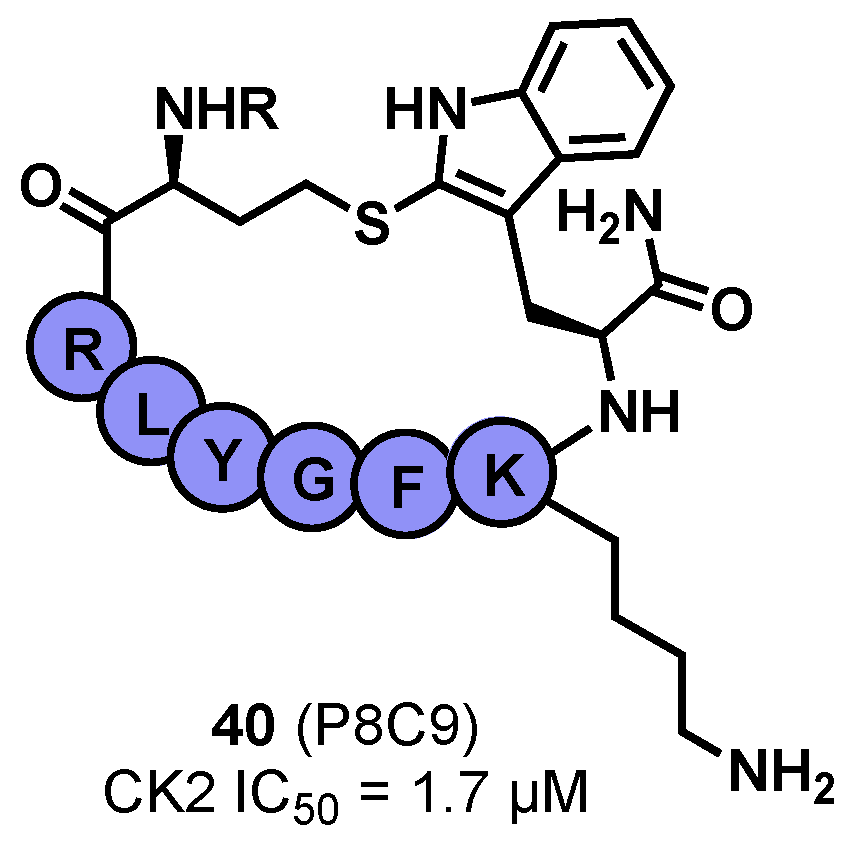
- Iegre, J.; Brear, P.; Baker, D.J.; Tan, Y.S.; Atkinson, E.L.; Sore, H.F.; Donovan, D.H.O.; Verma, C.S.; Hyvönen, M.; Spring, D.R. Efficient Development of Stable and Highly Functionalised Peptides Targeting the CK2α/CK2β Protein–Protein Interaction. Chem. Sci. 2019, 10, 5056. [Google Scholar] [CrossRef]
- Laudet, B.; Barette, C.; Dulery, V.; Renaudet, O.; Dumy, P.; Metz, A.; Prudent, R.; Deshiere, A.; Dideberg, O.; Filhol, O.; et al. Structure-Based Design of Small Peptide Inhibitors of Protein Kinase CK2 Subunit Interaction. Biochem. J. 2007, 408, 363. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, E.; Iegre, J.; D’Amore, C.; Brear, P.; Salvi, M.; Hyvonen, M.; Spring, D. Development of Small Cyclic Peptides Targeting the CK2α/β Interface. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Kufareva, I.; Bestgen, B.; Brear, P.; Prudent, R.; Laudet, B.; Moucadel, V.; Ettaoussi, M.; Sautel, C.F.; Krimm, I.; Engel, M.; et al. Discovery of Holoenzyme-Disrupting Chemicals as Substrate-Selective CK2 Inhibitors. Sci. Rep. 2019, 9, 15893. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181. [Google Scholar] [CrossRef]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great Opportunities for Academia and Industry. Signal Transduct. Target. Ther. 2019, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Schwartz, A.L. The Ubiquitin-Proteasome Pathway: The Complexity and Myriad Functions of Proteins Death. Proc. Natl. Acad. Sci. USA 1998, 95, 2727. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, F.; Liu, N.; Wang, X.; Gou, S. Chemically Induced Degradation of CK2 by Proteolysis Targeting Chimeras Based on a Ubiquitin–Proteasome Pathway. Bioorg. Chem. 2018, 81, 536. [Google Scholar] [CrossRef] [PubMed]



























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Day-Riley, S.; West, R.M.; Brear, P.D.; Hyvönen, M.; Spring, D.R. CK2 Inhibitors Targeting Inside and Outside the Catalytic Box. Kinases Phosphatases 2024, 2, 110-135. https://doi.org/10.3390/kinasesphosphatases2020007
Day-Riley S, West RM, Brear PD, Hyvönen M, Spring DR. CK2 Inhibitors Targeting Inside and Outside the Catalytic Box. Kinases and Phosphatases. 2024; 2(2):110-135. https://doi.org/10.3390/kinasesphosphatases2020007
Chicago/Turabian StyleDay-Riley, Sophie, Rebekah M. West, Paul D. Brear, Marko Hyvönen, and David R. Spring. 2024. "CK2 Inhibitors Targeting Inside and Outside the Catalytic Box" Kinases and Phosphatases 2, no. 2: 110-135. https://doi.org/10.3390/kinasesphosphatases2020007
APA StyleDay-Riley, S., West, R. M., Brear, P. D., Hyvönen, M., & Spring, D. R. (2024). CK2 Inhibitors Targeting Inside and Outside the Catalytic Box. Kinases and Phosphatases, 2(2), 110-135. https://doi.org/10.3390/kinasesphosphatases2020007