Abstract
Protein kinase A (PKA) signaling exemplifies phosphorylation-based signaling as we understand it today. Its catalytic-subunit structure and dynamics continue to advance our understanding of kinase mechanics as the first protein kinase catalytic domain to be identified, sequenced, cloned, and structurally detailed. The PKA holoenzyme elaborates on the role of its regulatory subunits and maintains our understanding of cAMP-dependent cellular signaling. The activation of PKA holoenzymes by cAMP is an example of specialized protein allostery, emphasizing the relevance of protein binding interfaces, unstructured regions, isoform diversity, and dynamics-based allostery. This review provides the most up-to-date overview of PKA structure and function, including a description of the catalytic and regulatory subunits’ structures. In addition, the structure, activation, and allostery of holoenzymes are covered.
1. Introduction
Several cellular signaling and developmental processes are regulated by the counteractions of two sets of enzymes: protein kinases (those that phosphorylate proteins) and protein phosphatases (those that remove these phosphates from proteins) [1,2,3]. Dysregulated protein kinases are associated with inflammatory, neurodegenerative, cardiovascular, and metabolic diseases and various forms of cancer [4,5,6,7,8]. Understandably, protein kinases are critical targets for disease control and therapeutic development strategies [9,10].
The first protein kinase discovered and structurally explored was protein kinase A (or simply PKA) [11,12]. In the 1950s, Edmond H. Fisher and Edwin G. Krebs discovered the activation of glycogen phosphorylase (conversion of inactive phosphorylase b to the active phosphorylase a) to be dependent on a serine phosphorylation by a then unknown “phosphorylase kinase” [13,14]. At the same time, Earl Sutherland and Thomas Rall discovered 3′,5′-cyclic-adenosine monophosphate (cAMP) in liver cells as a ‘second messenger’: a mediator biomolecule that communicated the effect of exogenous hormones into the cell’s interior [15,16]. Eventually, these discoveries converged to uncover a cAMP-dependent protein kinase (now called protein kinase A) that regulated the function of phosphorylase kinase in response to cAMP. These pioneering studies established a connection between hormone action, second messengers, and protein phosphorylation and continue to be the foundational understanding of cellular signaling today. Also, while this was the first evidence of protein kinase–kinase cascades, multisite protein phosphorylation, as suggested by Philip Cohen [17], was quickly discovered to be a signaling norm.
PKA was first purified from rabbit skeletal tissue by D. Walsh [18] and was subsequently reported to be widespread in various mammalian tissues by J.F. Kuo and Paul Greengard [19]. PKA was also the first protein kinase to be sequenced [20], the first to be cloned for purifying in recombinant form [21], and also the first to be explored with X-ray crystallography [22]. PKA is a major target of therapeutic interventions for Alzheimer’s disease, diabetes, and cardiovascular diseases [23,24,25]. PKA and its cAMP-dependent signaling pathway continue to be relevant in furthering our understanding of signaling, protein allostery, and the role of protein kinases in human disease [26]. The cAMP-sensitive PKA holoenzyme is made of two types of subunits: the regulatory (R)-subunit that contains cAMP binding sites and the catalytic (C)-subunit that performs phosphotransfer onto substrate proteins. The C-subunit of PKA serves as the prototype for the entire eukaryotic protein kinase (EPK) superfamily. This review focuses on explaining the structural properties of the PKA subunits, its holoenzyme architecture, and the allosteric underpinnings of cAMP-dependent PKA signaling. We also highlight the amino acid networks that mediate the interactions between the R- and C-subunits of PKA and how these interactions serve as critical allosteric sites for the holoenzyme structures.
2. cAMP-Mediated PKA Signaling
Canonical PKA signaling is initiated in response to hormone binding to GPCRs and the subsequent activation of the adenylate cyclase (AC) enzyme that converts ATP to cAMP (Figure 1) [27,28,29]. As cAMP diffuses into the cell interior, it binds the basal PKA signalosome that typically consists of two R-subunits and two C-subunits in a tetrameric holoenzyme (R2C2) anchored to an A-kinase anchoring protein (AKAP). Both R-subunits bind two molecules of cAMP, each using their cyclic nucleotide binding (CNB) domains to simultaneously release two C-subunit monomers from the holoenzyme. These free C-subunits then perform their biological function by phosphorylating target proteins on their Ser/Thr residues [30]. Substrates of the PKA C-subunit include CREB, RAF, BAD, GSK3, CIP4, and other proteins critical to cell survival [31,32,33]. Finally, termination of PKA signaling is achieved by the action of phosphodiesterase (PDE) enzymes that hydrolyze cAMP and promote the re-formation of the R2C2 holoenzyme complexes [34,35]. Here, the PKA C-subunit participates in maintaining cAMP homeostasis by feedback inhibition of ACs and activation of PDEs to promote a lowering of the levels of available cAMP molecules [36,37].
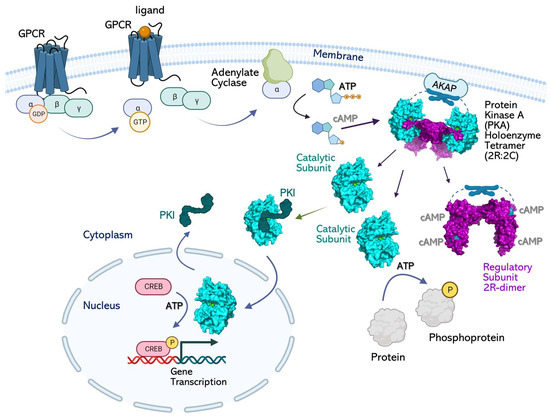
Figure 1.
Signaling pathway of cAMP-dependent protein kinase A (PKA). Basal PKA is a tetrameric holoenzyme that contains two catalytic (C)-subunits and two regulatory (R)-subunits. The PKA holoenzyme is localized to membranes or cell organelles by their interactions with AKAP proteins. Signaling is initiated by ligand binding to heterotrimeric G-proteins and the dissociation of its α-subunit to bind and activate adenylate cyclase (AC). AC converts ATP to the “second messenger” cAMP. As cAMP diffuses in the cytosol, it binds the PKA R-subunits’ CNB domains and dissociates the holoenzyme to free the C-subunit. The “active” free C-subunit phosphorylates its substrate proteins in the cytosol. A subpopulation of C-subunits binds the inhibitory protein PKI and shuttles to the nucleus. After dissociation from PKI, the C-subunit phosphorylates CREB and initiates target gene transcription.
Spatial regulation of PKA holoenzyme signaling is achieved by their binding to various AKAPs in the cell [38]. Approximately 70 AKAPs can target PKA holoenzymes to various compartments in the cell; splice variants of AKAP genes additionally increase their spatial diversity [38,39,40]. For example, AKAPs on the outer mitochondrial membranes are critical for PKA-holoenzyme localization and signaling to prevent apoptosis [41]. In neurons, AKAPs anchor PKA holoenzymes to plasma membranes and the NR1 subunit of NMDA receptors and allow PKA to regulate synaptic plasticity [42,43]. Recently, AKAP79/150-mediated, PKA-dependent regulation of L-type Ca2+ channels was also reported at cellular membranes [44]. AKAPs (like WAVE1) allow for the association of PKA holoenzymes with the cytoskeletal network to regulate basic cellular processes [45]. Muscle-specific AKAP (mAKAP) anchors the PKA holoenzyme to the nuclear envelope and allows for PKA-mediated gene regulation [46]. While the extended diffusion of the free C-subunit from the dissociated holoenzyme remains controversial [47,48,49], free C-subunit has been reported in the nucleus, where it regulates gene transcription by phosphorylating the cAMP-response element binding (CREB) protein (Figure 1) [50,51].
The free PKA C-subunit is also reported to associate with a cAMP-independent inhibitor protein called PKI that allows for the shuttling of the C-subunit into and out of the nucleus by using its nuclear export sequence (NES) [52,53]. This NES is unmasked when PKI attaches to the PKA C-subunit. PKI is a heat-stable inhibitor of the C-subunit that is widely distributed in mammalian tissues, including the brain, heart, liver, testes, muscles, etc. [53,54]. PKI is approximately 75 amino acids long and is mostly unstructured. Endogenous PKI includes three isoforms, PKIα, β, and γ, and is localized in the cell cytoplasm and nuclei [53]. PKIα and PKIγ are quite abundant and are expressed in the brain, liver, heart, pancreas, kidney, and colon, whereas PKIβ is expressed in the testes [55]. The N-terminus of PKIα includes residues (1–25) that strongly bind to and inhibit the C-subunit (Kd = 2 nM) [53,56]. This segment, called IP20, is routinely used in biochemical experiments to study the C-subunit and is allied interactions with the R-subunits [56,57,58,59] (Figure 2D). The PKIβ isoform binds the C-subunit with a Kd = 7.1 nM. PKIγ uses a unique Cys13 residue to bind the C-subunit at Kd = 0.4 nM. All three isoforms use their pseudosubstrate site to bind the C-subunit active site and to inhibit its ATP-bound “active” conformation.
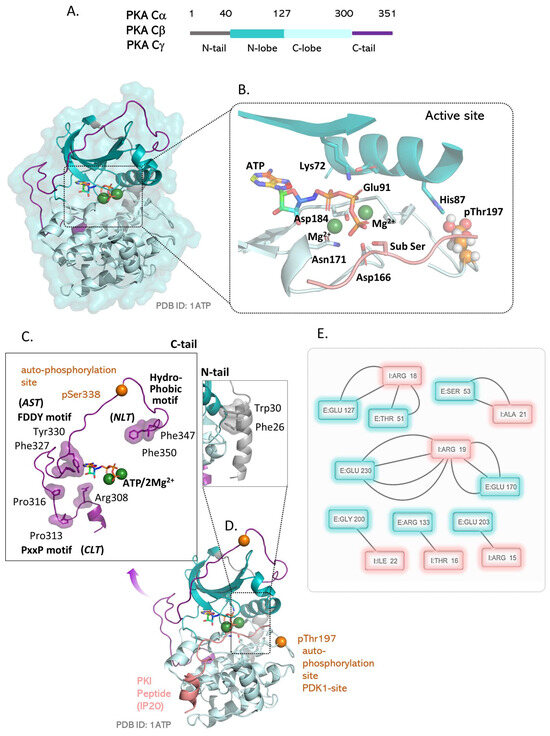
Figure 2.
Structure and properties of the C-subunit of PKA. (A) The C-subunit contains a kinase core flanked by two Nt and Ct tails. The kinase core is a bi-lobal structure that encloses an ATP-binding pocket between the two lobes. (B) The active site of the C-subunit binds ATP with two divalent metal ions that participate in the transition state formation. Lys72 from β3 contacts the Glu91 from the αC helix and also contacts the phosphates of ATP. His87 of the αC helix engages the activation loop phosphorylation site pThr197 and maintains the kinase active conformation. The catalytic residues Asp166, Asp184, and Asn171 coordinate catalysis at the active site. (C) The N-terminal and C-terminal tails tether to the catalytic subunit at specific locations. The N tail docks to the back of the kinase domain using Phe26 and Trp30 aromatic residues. The C-terminal tail uses three tether points: the N-lobe tether (NLT) includes the hydrophobic motif residues Phe347 and Phe350; the active-site tether (AST) includes the FDDY motif residues Phe327 and Tyr330; the C-lobe tether (CLT) includes the PxxP residues Pro313, Pro316, and Arg308. (D) Two phosphorylations are critical to the kinase functioning of the C-subunit. The pThr197 phosphorylation on the activation loop triggers its catalytic activity. The pSer338 phosphorylation allows for tethering of the Ct tail to the kinase core and stabilizes the C-subunit. (E) Contact maps obtained for the peptide bound conformation of the subunit (PDB:1ATP) highlights residues in the substrate binding cleft that recognize the PKA specific Arg-Arg-X-X-Ser/Thr motif.
3. PKA and Human Disease
Given the critical importance of the PKA subunits in cellular signaling, it is not surprising that mutations or aberrations in the activity of any of its subunits are associated with human diseases. Mutations in either the R- or C-subunits that interfere with their interactions to form holoenzymes lead to unregulated PKA signaling. The diseases arising from these mutations include Cushing disease (mutations on the C-subunit) [60,61,62] and multiple endocrine neoplasia syndrome, known as Carney complex disease (CNC) and acrodysostosis (mutations on the RI-subunit) [63,64,65]. The inhibitory role of the R-subunits is indicative of their role as tumor suppressors in CNC-associated tumors [65,66]. Decreased expression of the R-subunits (30% decrease in RIα and 65% decrease in RIβ) is associated with the idiopathic autoimmune disease called systemic lupus erythematosus (SLE) that is characterized by impaired T-lymphocyte functions [67,68]. PKA is reported to play a critical role in the regulation of metabolism and triglyceride storage, such that it is a lucrative target for therapeutic targeting or obesity and aging [69,70]. Leptin is reported to improve the antiproliferative activity of cAMP-increasing agents in breast cancer cells, where PKA inhibitors (like KT-5720) suppress the antiproliferative effects of leptin plus cAMP elevation [71,72,73,74].
A specific chimera created by the fusion of a chaperonin-binding domain fused to the Cα isoform of the catalytic subunit is observed to cause a rare hepatocarcinoma called fibrolamellar carcinoma (FLC) [75,76]. The Cβ isoform of the catalytic subunit is reported to be a direct transcriptional target of c-MYC and is upregulated in c-MYC associated cell transformation [77]. PKA’s kinase activity regulates actin dynamics by modulating structural proteins, such as integrins, myosin light chain, and VASP, as well as regulatory proteins, such as Rho GTPases, Src kinases, p21-activated kinases, phosphatases, and proteases [78]. PKA’s activity is reported to play a role in hypoxia-mediated epithelial–mesenchymal transition, migration, and invasion in lung cancer cells [79]. PKA activity is associated with cell migration and invasion in cancers of the breast and ovaries [80,81].
Research indicates that PKA type I and type II R-subunits are inversely expressed throughout cell differentiation and ontogeny, where they govern proper cell proliferation and differentiation into nondividing states [82,83]. The RI:RII ratios are substantially greater in normal breast specimens with enhanced proliferation [83]. Antisense repression of RI, which upregulates RII, downregulates a wide variety of genes involved in cell proliferation and transformation while upregulating cell differentiation and reverse transformation genes in prostate cancers [84,85]. Despite the fact that the RI:RII ratio varies widely amongst breast cancers, those with a high RI:RII ratio have a poor prognosis in terms of early disease recurrence and mortality following initial therapy [83].
4. The PKA Catalytic Subunit
Four isoforms of the C-subunit are reported in mammals, including Cα, Cβ, Cγ, and PrKX, as coded by the genes PRKACA, PRKACB, PRKACG, PRKX, and PRKY [21,86,87,88,89,90]. Additionally, multiple splice variants have been reported for the Cα and Cβ isoforms [91,92]. There are two alternative 5′ exons in the PRKACA code for the two variants Cα1 and Cα2, respectively [93,94,95,96]. The Cα1 form is the predominant ubiquitously expressed catalytic subunit [90]. The Cα2 form is reported to be expressed exclusively in mammalian sperm, where it regulates sperm motility and fertilization [97,98,99]. Splicing of the PRKACB gene at exons 2 and 4 creates multiple Cβ isoforms, including Cβ1, Cβ2, Cβ3, Cβ4, Cβ3ab, Cβ3b, Cβ3abc, Cβ4ab, Cβ4b, and Cβ4abc proteins [100,101,102,103]. Cβδ4 is a particular isoform expressed in the brains of higher primates, where it is reported to permanently associate with R-subunits and is insensitive to cAMP-mediated activation [104]. Human Cα1 and Cβ1 are 93% identical, indicating that the two PRKACA, PRKACB genes are a result of gene duplication [90]. The Cγ isoform is expressed exclusively in the testes, but its biological role remains unknown [105]. Biochemical experiments indicate that this isoform strongly associates with R-subunits and requires higher cAMP levels for holoenzyme dissociation and activation [106]. PrKX is a unique isoform encoded on the X chromosome and associates specifically with the RIα isoform of the R-subunit [87]. PrKX is understudied and shares just 56% identity with Cα1. The biological function of PrKX and its associated protein PrKY is unclear [90].
Critical post-translational modifications of the C-subunit include its N-terminal myristylation (that allows for its anchoring to plasma membranes) [107] and two phosphorylation sites, Thr197 and Ser338, that regulate its activation [108,109]. Both Thr197 and Ser338 are auto-phosphorylation sites [108,110] where the activation-loop (Thr197) phosphorylation can also be mediated by other kinases, such as PDK1 (Figure 2C) [111].
The structure of the C-subunit continues to serve as the exemplar for the EPK kinase domain architecture ever since its initial detailing [11,22]. It contains 351 amino acids, where residues 40–300 are the core of every EPK kinase domain. This kinase core contains two lobes: the N lobe that has the five-stranded β-sheet (β1–β5) and the functional αC helix and the C lobe that is predominantly helical and houses the β6–β7 β-sheet (Figure 2A). By convention, all helices are named alphabetically, while all strands are numbered in order of their positioning from the N-terminus of the C-subunit. This nomenclature is applied to all EPK structures, where the αC helix of the N lobe is still called the αC, even in the absence of the αA/αB helices in many cases. The kinase active site is sandwiched in the cleft separating the N and C lobes. The C lobe has a surface grove that binds peptide substrates. The N lobe contains residues for ATP binding that are situated under a glycine-rich loop that connects the β1 and β2 strands. A conserved Lys72 [112] from the β3 strand engages the divalent ions (Mg2+ or Mn2+) that stabilize ATP in the charged cleft while simultaneously making a salt bridge with a conserved Glu91 of the αC helix [113]. A short linker, called the ‘hinge’, connects the two lobes and contacts the adenine ring of ATP via hydrogen bonds. The activation loop phosphorylation pThr197 arranges the activation segment (from residues 184–208, DGF-APE motif) appropriately to place the catalytic loop (containing the HRD motif) in a catalytically suitable orientation. A critical Cys199 at the +2 position to the Thr197 phosphorylation site makes the C-subunit resistant to inactivation by protein phosphatases [114].
The N-terminus of the αC helix contains a pH-sensitive histidine (H87) [115] that makes a salt bridge with the phosphorylated pThr197 of the activation loop (Figure 2B). Residues from the magnesium binding loop (DFG motif) and Lys168 coordinate the ATP and metal ions to facilitate phosphotransfer to the incoming substrate Ser/Thr. At the PKA active site, Asp184 and Asn171 bind the “primary” Mg2+ ion (M1) [116] that helps position the terminal gamma phosphate of ATP for nucleophilic attack. A transition state is formed between the substrate Ser/Thr, two Mg2+ ions, and ATP with the terminal phosphate stabilized by hydrogen bonds. QM/MM studies show that Asp166 of the C-subunit functions as a catalytic base and accepts a proton as delivered by the substrate Ser/Thr [117]. The catalytic cycle concludes with the dissociation of a Mg2+-bound ADP from the PKA active site. This last step is the rate limiting step in the C-subunit’s steady-state kinetics and accounts for its kcat = 20 s−1 [118].
The N- and C-terminal regions outside the kinase core are called the C-subunit Nt and Ct tails, respectively (Figure 2A) [119,120]. The Nt and Ct tails are both tethered to the kinase core and play a key role in controlling the C-subunit’s interaction with other proteins [121,122]. The Nt tail is the large, amphipathic, αA helix that contains the C-subunit myristylation site Asn2. The residues Phe26 and Trp30 anchor the αA helix to the hydrophobic kinase core (Figure 2C). The Ct tail is a conserved structural feature of all AGC kinases [122] and contains three tethering regions. The N-lobe tether (NLT) binds at the N-terminus of the αC helix and contains the residues Phe347 and Phe350 that are required to recruit PDK1 for activation-loop phosphorylation [119]. Phosphorylation of Ser338 allows for stable binding of NLT to the kinase core and promotes kinase function [120]. The active site tether (AST) includes the residues Phe327 and Tyr330 in the FDDY motif that participate in ATP binding and in allosterically maintaining the “closed” catalytically competent conformation of the C-subunit. The C-lobe tether (CLT) uses Arg308 to bind Phe100 and Phe102 at the αC-β4 loop of the C-subunit. This interaction maintains the monomeric conformation of PKA (and other AGC kinases) as it engages the αC-β4 loop that can serve as a back-to-back dimerization motif [123] (Figure 2C). CLT also contains a Pro-X-X-Pro motif (Pro 313 and Pro316) that binds SH3 domains in interacting proteins.
A comparison of PKA structures in the inactive/dephosphorylated vs. the active/phosphorylated forms (Figure 3A,B) highlights the local spatial pattern (LSP) alignment-derived kinase “spines” [124]. These are non-linear motifs in the kinase core that illustrate its switching between kinase conformations and how activation affects internal restructuring of the kinase domain. An assembled regulatory (R)-spine is a signature of the active C-subunit conformation. This motif is formed by four residues, including Leu95 from the αC helix, Leu106 of the β4 strand, Phe185 of the magnesium positioning loop DFG motif, and Tyr184 of the catalytic loop HRD motif. In the active conformation that contains a phosphorylated pThr197, the R-spine is dynamically aligned at the core of the C-subunit. In the dephosphorylated C-subunit, electrostatic contacts between the pThr197 and corresponding His87, Lys189, Thr195, and Arg165 are removed, such that its activation segment becomes unstructured and the αC helix swings outwards. In this inactive conformation, the R-spine is misaligned (Figure 3B). Another hydrophobic motif revealed with LSP alignment is the catalytic (C)-spine that highlights the catalytically competent conformation of PKA. The C-spine includes the residues Val57; Ala70 from the N lobe; and the residues Leu171, Leu172, and Ile173 from the C-subunit that bind the adenine ring of ATP and orient it suitably in the PKA active site. Other C-spine residues, including Met127, Leu227, and Met231, integrate entropy-driven dynamic information through the large lobe of the C-subunit [56,125,126].
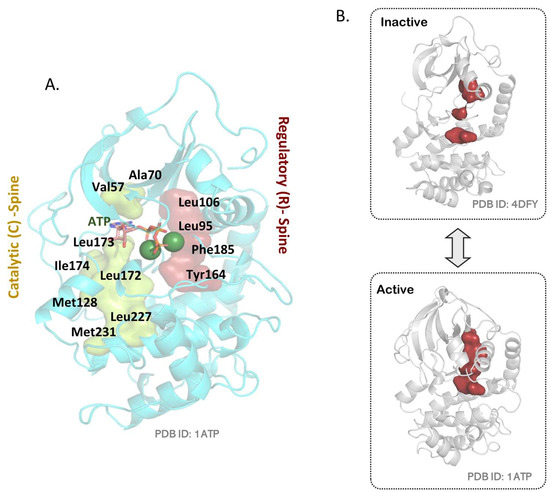
Figure 3.
Kinase “spines” in the C-subunit. (A) The regulatory (R)-spine and catalytic (C)-spine as observed in the C-subunit. (B) An assembled/aligned R-spine is the signature of an active kinase conformation (PDB:1ATP). In the absence of Thr197 phosphorylation, the C-subunit is inactive (PDB:4DFY) and shows a disconnected R-spine.
An activated/free C-subunit is an efficient Ser/Thr phosphotransferase that identifies substrate sites in two general recognition motifs: Arg–Arg–X–Ser/Thr–Hyd and Arg–X–X–Arg–X–X–Ser/Thr–Hyd, (where Hyd is a hydrophobic residue, and X denotes any residue) [119]. The P-2/P-3 Arg residues of the substrate engage with Glu127, Glu170, and Glu230 on the C-subunit’s active-site surface. A contact network obtained from the crystal structure of the C-subunit bound to ATP/Mg2+ and IP20 (peptide derived from the N-terminus of PKI) also shows residues Thr51 and Ser53 to make hydrogen bonds with the peptide (Figure 2D, Supplementary Table S1). PKA’s turnover rate (kcat) averages 20 per second for small peptide substrates, like kemptide, and is limited by the last step of ADP release from its active site [127]. While the steady-state kinetics are constrained by protein movements and dynamic conformations through successive catalytic cycles to produce a slower kcat, pre-steady-state kinetics have shown the rate of phosphotransfer at its active site to be >500 per second [128]. The C-subunit binds ATP and ADP with micromolar affinity (Kd = 20–25 μM) [56] and peptide substrates with a Km of approximately 10–20 μM [129]. However, inhibitory peptides, like IP20, bind the C-subunit cooperatively with ATP with a high nanomolar affinity (Kd = 2 nM) [56,130].
Somatic mutations in the C-subunit have been reported to be associated with cortisol-secreting adrenocortical adenomas responsible for Cushing’s syndrome [131]. The most abundant mutation, as observed in 65% of patients with Cushing’s syndrome, changes Leu206 to arginine [61,132,133,134]. The location of the L206R mutation interferes with the C-subunit’s association with PKI and the R-subunits, such that the mutant is constitutively active to cause Cushing’s syndrome [61]. Biochemical experiments comparing the L206R mutant to the wild-type protein show that this mutation causes dynamic changes in the enzyme’s intramolecular allosteric network, resulting in nucleotide/pseudo-substrate binding cooperativity losses [60]. Recently, an in-frame fusion of the PRKACA gene with the heat-shock DNAJB1 gene has been reported in the fibrolamellar variant of hepatocellular carcinomas [75,135]. The resultant chimeric enzyme contains the J domain of the chaperonin-binding domain of the heat-shock protein 40 or DnaJ (the amino-terminal 69 residues), fused to the carboxyl-terminal 336 residues of the catalytic subunit [62]. Unlike the Cushing’s syndrome mutations, this tumorigenic chimera does not show any alterations in associating with the R-subunits and in being sensitive to cAMP signaling [62]. Here, approximately a ten-fold increase in chimeric transcript levels (under the control of the DNAJB1 gene promoter) results in increased kinase activity and upregulation of PKA signaling to cause tumor pathogenesis [136].
5. The PKA Regulatory Subunit
In mammals, four isoforms of the PKA R-subunit can organize four kinds of PKA holoenzymes: two each of the type-I and type-II R-subunits, viz., RI (RIα2C2, RIβ2C2) and RII (RIIα2C2, RIIβ2C2) [137]. These RI and RII isoforms differ in their affinities to binding cAMP, cellular localization, and affinity to AKAPs. RIα is the most abundant and widely distributed isoform that maintains the RIα2C2 holoenzymes in the cytosol [138]. RIα is also the only embryonic lethal isoform [138], and various mutations in RIα lead to Carney complex disease (CNC) and acrodysostosis [139,140]. Mutations and loss of expression of RIβ lead to neurodegenerative disorders [141]. RIIβ-knockout mice exhibit a lean phenotype and show resistance to diet-induced obesity [142].
All four isoforms share a common domain organization, where the N-terminal dimerization domain (D/D domain) is connected to the two tandem CNBA and CNBB domains by a long unstructured linker (Figure 4A). The D/D domain serves as a docking site for binding AKAPs and recruiting the PKA holoenzyme to various cellular locations and is the most divergent in sequence of the four isoforms. Deletion of the D/D domain does not impair R- and C-subunit binding, and deletion mutants have been used to study the R–C heterodimer [143,144]. The linker connecting the CNB domains to the D/D domains is an intrinsically disordered region (IDR) that is highly dynamic and functions to mediate protein–protein interactions by recruiting interacting binding proteins [11]. It contains the inhibitor site with the pseudosubstrate Arg-Arg-X-X motif to bind the C-subunit’s active site cleft [145]. Here, the biggest difference between the RI and RII isoforms is the presence of C-subunit-specific substrate sites in the RII subunit that make the RIIα2C2, RIIβ2C2 holoenzymes sensitive to phosphorylation-based control [50,146]. The RII subunit is both a substrate and an inhibitor of the C subunit; the phosphorylated RII subunit cannot dissociate from the holoenzyme without binding cAMP. The RI subunit functions as a true inhibitor and uses its pseudosubstrate region to bind the C-subunit with high affinity [145].
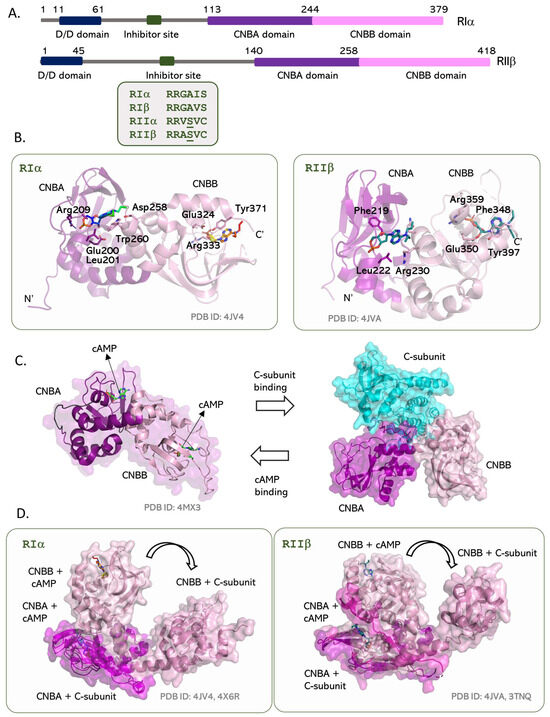
Figure 4.
The type-I and type-II R-subunits of PKA. (A) Domain arrangement of the two types of R-subunits is similar, where both contain a dimerization domain connected to their cAMP binding domains by a long linker. This linker includes the pseudosubstrate/inhibitor site that binds the active site of the C-subunit in the holoenzyme. The type-II R-subunits contain a Ser residue in this site that can be phosphorylated by the C-subunit. (B) Tandem cAMP binding CNBA and CNBB domains in RIα and RIIβ. Structures of the cAMP-bound state show isotype differences in the orientation of the CNBB domain versus the CNBA domain. (C) R-subunits toggle between an active (cAMP bound) and inactive (C-subunit bound) conformation. (D) Binding of the C-subunit to the R-subunit isotypes requires a huge conformational change at the CNBA:CNBB interface that creates an extended R:C interaction surface.
While each CNB domain is highly conserved, the relative orientations of the two domains are distinct in the type-I and type-II R-subunits (Figure 4B) [147]. The two CNB domains are eight-stranded β-barrels with an N-terminal N3A motif in a helix-turn-helix motif preceding the β1 strand and the αB/C helix following the β8 element. These contain an evolutionarily conserved phosphate-binding cassette (PBC) that binds cAMP molecules [147]. The PBC cassette includes an Arg residue (Arg209 in CNBA (RIα)/Arg230 in CNBA (RIIβ) and Arg333 in CNBB (RIα)/Arg359 in CNBA (RIIβ)) that form hydrogen bonds with the phosphates of cAMP. Mutating the PBC arginine to lysine creates cAMP-resistant R-subunit constructs that have been utilized to crystallize the holoenzyme structures [146,148,149]. The cAMP bound and unbound conformations of the CNB domain are distinct. In the cAMP-bound conformation, PBC moves towards the phosphate group of cAMP, and the αB/C helix moves ‘in’ to push the N3A motif ‘out’. The reverse happens when cAMP dissociates from the CNB domain [150]. The bound cAMP is stabilized using hydrophobic interactions with a “capping” residue that comes from outside the CNB β-barrel and is isoform specific [147]. Differences in the capping residues also exemplify the difference between the tandem domain constructs of type-I and type-II R-subunits. In RIα, Trp260 at the CNBA–CNBB interface caps cAMP in the CNBA site, and Tyr371 caps cAMP in the CNBB site. Here, the N3A motif of the CNBB merges with the αB/C element of CNBA to create an interdomain interface with the capping residue for the CNBA site. The W260A mutation in RIα is sufficient to uncouple allosteric interactions between the tandem CNBA and CNBB domains [151,152]. In RIIβ, cAMP in the CNBA site is capped by the long side chain of Arg381 from the αB of the CNBB domain, while cAMP in the CNBB site is stabilized by Tyr397.
The CNBB domain binds cAMP with higher affinity and slower off-rates when compared to the CNBA domain in solution. Dynamics data obtained from NMR, H/D exchange, and MD simulations hint towards a unidirectional flow of allosteric signals from the CNBA to the CNBB domain [152,153]. Mutations of the PBC arginine in the CNBA domain of RIα (R209K) cause internal dynamics to change in its β2–β3 loop, but a corresponding change in the CNBB domain (R333K) does not alter its chemical shifts. The R209K mutation shows a global reduction in protection factors across the tandem CNBA:CNBB length, including the β2–β3, PBC, and B/C helix of CNBA and N3A, β2–β3, and αB–αC of CNBB. However, in the R33K mutation, loss of protection is limited to PBC and αB–αC of CNBB alone. In RIIβ, a PBC arginine mutation of the CNBA domain (R230K) decreases the affinity (increase in activation coefficient) for cAMP binding to the holoenzyme from 584 nM for the WT to 12.9μM in the mutant [149]. In contrast, a mutation of the corresponding PBC arginine in the CNBB domain slightly increases the affinity (decrease in activation coefficient) for cAMP binding to 490 nM. Taken together, these observations suggest that cAMP binding to the CNBA domain is allosterically coupled to dynamic changes in the CNBB domain but not vice versa. Molecular dynamics simulations explain how the lower affinity the cAMP-binding CNBA domain impedes cAMP release from the CNBB domain, even allowing C-subunit binding to the R-subunit in the presence of cAMP at the CNBB site [152]. This unidirectional allosteric communication is lost in the W260A mutation as mentioned above.
6. The PKA Holoenzymes and Contact Networks of R:C Complexes
The structures of the CNBA domains of type-I and type-II R-subunits with the C-subunit explain the mode of inhibition observed in the heterodimers (Figure 5 and Table 1) [154,155]. In both types, the inhibitor sequence of the R-subunit is positioned in the peptide-binding cleft of the C-subunit and engages its Glu127, Glu170, Glu203, and Glu230 residues via the two arginine residues in the Arg-Arg-X-X motif. Additionally, in the ATP-bound, ‘closed’ conformation of the C-subunit, residues from the glycine-rich loop, including Thr51 and Ser53, make hydrogen bonds with the inhibitor site residues of the R-subunit (Supplementary Tables S2 and S3). Isotype specific contacts include residues Arg95, Arg230, His138, and Tyr205 on the RIα CNBA domain that make contacts with Lys168, Trp196, Lys213, and Tyr247 on the C-subunit, respectively. Correspondingly, the RIIβ CNBA domain specifically uses Tyr226, Arg110, Ala111, and Ala259 to engage Tyr247, Lys168, Thr201, and Arg194 on the C-subunit, respectively. The CNBA domain of RIIβ uses its Tyr118 to hydrogen bond with the activation loop phosphorylation site pThr197 of the C-subunit in a specific interaction.
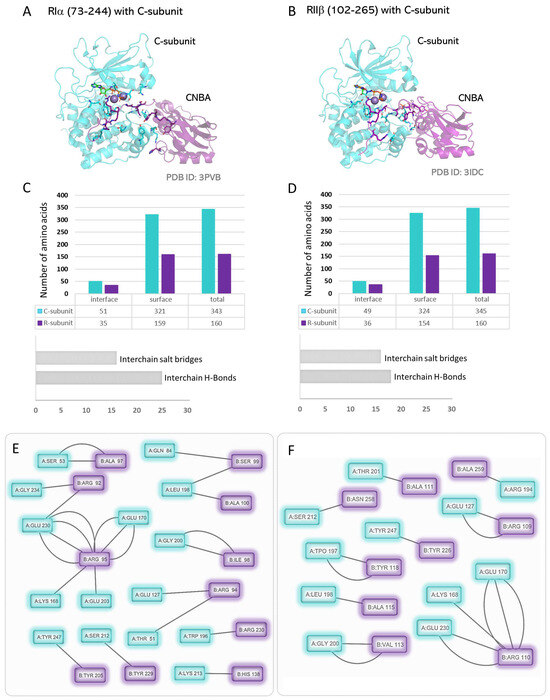
Figure 5.
The smallest R:C heterodimers: (A) Structure of the CNBA and inhibitor segment of RIα bound to the C-subunit. (B) Structure of the CNBA and inhibitor segment of RIIβ bound to the C-subunit. (C,D) R:C interaction interface parameters for the C-subunit and the CNBA domains of the type-I and type-II R-subunits. (E,F) Contact network at the R:C interaction interface as computed from the structures of the C-subunit with the CNBA domains of the type-I and type-II R-subunits.

Table 1.
Protein kinase A holoenzyme structures as available from the PDB.
The relative orientation of the two CNBA and CNB domains in the R-subunit is observed to be significantly different in the cAMP-bound (active) and C-subunit (inactive) conformations (Figure 4C,D). In the C-subunit-bound conformation, the C-terminal αB–αC helices of CNBA and the N-terminal αC′:A helix of CNBB merge to form the B/C helix that creates space to bind the C-subunit to the tandem domains. The binding of the C-subunit shields the PBC of CNBA and makes it inaccessible to cAMP [144]. The cAMP-binding to the two tandem CNB domains kinks their connecting B/C helix (residues Arg226–Ser249 in RIα) to fold the two domains over each other at Tyr244 (RIα). This αC/αC′ helix of the CNBA domain bends between residues Tyr244-Glu245 (RIα). The binding of the R-subunit linker and the CNBA domain is retained in a high-affinity interaction in the absence of the CNBB domain [143]. While the conformations of the type-I and type-II R-subunits in the cAMP-bound state are significantly different, both types bind the C-subunit in a similar orientation to create near identical R:C heterodimers (Figure 6).
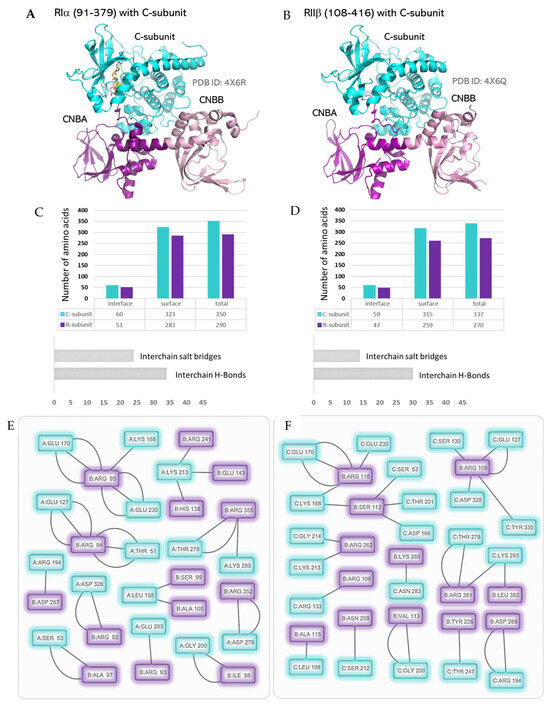
Figure 6.
The R:C heterodimers: (A) Structure of RIα (including the tandem CNBA/CNBB domains and the inhibitory segment while excluding the dimerization domain) bound to the C-subunit. (B) Structure of RIIβ (excluding the dimerization domain and including the tandem CNBA/CNBB domains with the inhibitory segment) bound to the C-subunit. (C,D) R:C interaction interface parameters for the C-subunit and the tandem CNBA/CNBB domains of the type-I and type-II R-subunits. (E,F): Contact network at the R:C interaction interface as computed from the structures of the C-subunit with the type-I and type-II R-subunits.
The structures of the tandem CNBA and CNBB domains with the C-subunit (R:C heterodimers where the R-subunit lacks the D/D domain) reveal important R:C interacting residues (Figure 6) [156]. Alongside the common interactions observed in the CNBA:C-subunit structure, specific isoform interactions are also observed in these structures. The CNBB domain of RIα uses the residues Arg230, Arg241, Arg352, Asp67, and Arg355 to engage with the C-subunit residues Lys213, Asp276, Thr278, Arg194, and Lys285, respectively (Supplementary Tables S4 and S5). Also, in these tandem constructs, the inhibitor site Arg92 of RIα engages with the FDDY Asp328 positions in the active-site tether of the Ct tail of the C-subunit. The corresponding Arg106 in the tandem domain construct of RIIβ does not engage with the C-subunit’s Ct tail and makes hydrogen bonds with Arg133 instead. The CNBB domain of RIIβ uses Asn258, Arg262, Lys285, and Arg281 to engage the residues Ser212, Lys213, Asn283, and Thr278, respectively, on the C-subunit surface. Asp288 of RIIβ makes a strong salt bridge with the activation loop Arg194 of the C-subunit.
The isoform-specific contact residues on the C-subunit are highlighted in Figure 7. These include the residues Asn84, Ser212, and Asp276 that interact specifically with the RIα isoform and the residues Asp166, pThr197, Gly214, Asn283, and Asp 241 that specifically engage RIIβ. Overall, the C-subunit makes 12 more interactions (salt bridges and hydrogen bonds included) with the tandem CNBA:CNBB domains of RIα compared to RIIβ. Surprisingly, these interactions do not correlate with the activation parameters of the RIα2C2 (Ka = 101 nM cAMP, Hill Coefficient = 1.7) [149] or RIIβ2C2 (Ka = 584 nM cAMP, Hill Coefficient = 1.8) [149] and indicate an allosteric role of the regions outside of the tandem CNB domain regions. Combined with small angle X-ray scattering (SAXS) data showing different solution structures for the type-I and type-II holoenzymes [149], it becomes important to analyze the full-length R2C2 holoenzymes rather than the R:C heterodimers. The N linker that connects the D/D domain to the inhibitory site is unstructured in the heterodimers. In the R2C2 holoenzymes, the N linker contributes to isoform-specific organization of the R- and C-subunits.
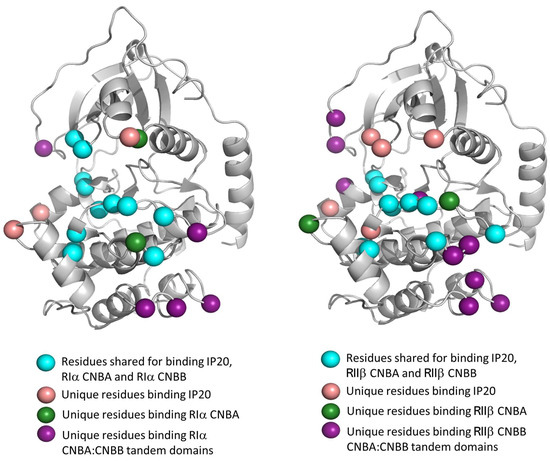
Figure 7.
C-subunit footprints in the R:C heterodimers. Contact maps reveal the specific residues on the C-subunit that interact with RIα (left) or RIIβ (right) CNBA or tandem CNBA/CNBB domains. Residues where identified by analyzing PDB structures in PDBePISA [157] and comparing the type-I and type-II holoenzyme complexes. Detailed residue list, including hydrogen bonding and salt bridge profiles, is provided in Supplementary Tables S1–S5.
In the RIα2C2 holoenzymes, the complex is Y-shaped, where the N linker of one heterodimer stabilizes the other heterodimer to assemble an elongated holoenzyme (Figure 8) [145,158]. In the two conformations observed in the same crystal [158], the R-subunits are in the center with the CNBA and CNBB domains sitting in a head-to-toe orientation, while the C-subunit is on the outside. In one conformation, the N3A motifs of the two R-subunit CNBA domains nucleate the interaction interface. This RIα2C2 holoenzyme captures the ‘closed’ conformation of the C-subunit with an ATP/2Mg2+ bound at its active site. This extended RIα2C2 conformation is stabilized by ATP and requires higher cAMP levels for activation (EC50 ∼2600 nM). The second conformation in the RIα2C2 holoenzyme includes RIα dimers bound to the ‘open’ apo conformation of the C-subunit and use the αN helices of the CBNA domains to dimerize. This is the more compact of the two conformations and is sensitive to activation by cAMP (EC50 ∼450 nM). Size exclusion and fluorescence polarization (FP) experiments show that the RIα2C2 holoenzyme can switch between these two conformations and, in fact, uses these as a sensory switch to capture the ATP-bound C-subunit for inactivation [158].
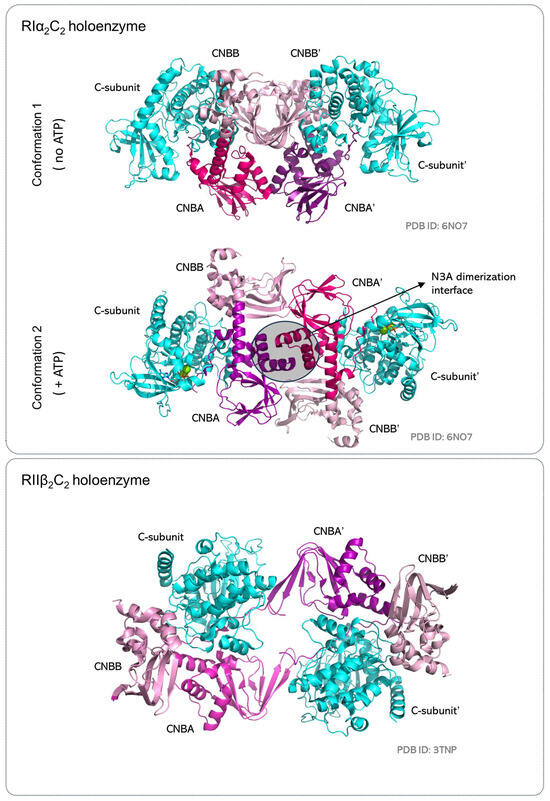
Figure 8.
The R2C2 holoenzymes. Two R:C heterodimers assemble in a 2-fold symmetry to organize the R2C2 holoenzymes. The RIα2C2 holoenzyme is elongated and dimerizes with the R-subunits in the center and the C-subunits at the periphery. It also toggles between two conformations as it functions as an ATP sensor to capture the catalytically competent C-subunit. The RIIβ2C2 holoenzyme is compact and uses the C-subunit and the CNBA domains of the R-subunits to form the dimerization interface. Conformation of the RIIβ2C2 holoenzyme does not vary in the presence or absence of ATP at the C-subunit active site.
In contrast, the RIIβ2C2 holoenzyme is a compact, dumbbell-shaped structure that uses the C-subunit and the CNBA domain of the R-subunit as the dimerization interface. Also, unlike the RIα2C2 holoenzyme, the RIIβ2C2 holoenzyme captures the C-subunit in the ‘closed’ conformation even in the absence of ATP at its active site. Experimental data confirm the RIIβ2C2 holoenzyme to be insensitive to ATP concentration in solution [158]. Here, RIIβ shows a higher preference to bind the apo and ADP-bound C-subunit (EC50 ∼5 nM) compared to the ATP-bound active kinase domain (EC50 = 10 nM). As the inhibitor site of RIIβ includes a C-subunit phosphorylation site, RIIβ2C2 holoenzyme appears to be coupled to RIIβ phosphorylation rather than ATP levels in the cellular milieu. Moreover, RIIβ2C2 holoenzyme activation by cAMP is easier for the ATP-bound C-subunit (EC50 = 94 nM) when compared to the apo or ADP-bound C-subunit in the RIIβ2C2 complex (EC50 ∼200 nM) [158].
In conclusion, the varied quaternary structures of the type-I and type-II holoenzyme are reflective of their biological function. The RIα2C2 holoenzymes are localized to the mitochondria, where they regulate stress-related PKA signaling [159]. The extended quaternary assembly of this holoenzyme makes it an effective ATP sensor that allows PKA activation in ATP-depleted conditions [159,160]. In contrast, the RIIβ2C2 holoenzyme localizes to plasma membranes, where its activation is rendered insensitive to ATP levels.
7. Conclusions and Future Directions
PKA holoenzyme diversity and allostery continues to be a relevant topic for understanding cAMP-dependent signaling. The recent structures of the full-length holoenzymes have been successful in accomplishing quaternary structure diversity, but many questions remain unanswered. How does the C-subunit choose its R-subunit for holoenzyme assembly? Do the R-subunits dimerize first, or do the R:C homodimers come together to form the holoenzymes? We still do not understand how holoenzymes are activated. Do both the R:C homodimers dissociate together or one after another? What are the isoform-specific dynamics-based parameters for holoenzyme activation? Does interaction of AKAPs affect holoenzyme activation? While many milestones in PKA allostery have been achieved, the complete mechanistic picture of PKA allostery continues to be an ongoing and persuasive study.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/kinasesphosphatases1040016/s1, Table S1: Interface parameters C-subunit and IP20 peptide (PDB ID: 1ATP); Table S2; Interface parameters C-subunit and CNBA(73-244)of RIA (PDB ID: 3PVB); Table S3: Interface parameters C-subunit and CNBA(102-265)of RIIB (PDB ID: 3IDC); Table S4: Interface parameters C-subunit and CNBA + CNBB (91-379) of RIA (PDB ID: 4X6R); Table S5: Interface parameters C-subunit and CNBA + CNBB (108-416) of RIIB (PDB ID: 4X6Q).
Author Contributions
C.L.W., A.E.C. and L.K.M. wrote, edited, and revised the manuscript. LKM developed, visualized, and oversaw the manuscript writing process, which included studying current literature and analyzing crystal structures. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by the SC COBRE in Antioxidants and Redox Signaling of the National Institute of General Medical Sciences (NIGMS) (Grant number: 1P30GM140964) and SCTR NIH/NCATS (Grant Number: UL1TR001450) to LKM.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This manuscript does not generate any new data. All interaction networks have been deduced from available crystal structures as shown in Table 1. All interactions (hydrogen bonds and salt bridges) as obtained from structural analysis have been reported in Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wiśniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Smoly, I.; Shemesh, N.; Ziv-Ukelson, M.; Ben-Zvi, A.; Yeger-Lotem, E. An Asymmetrically Balanced Organization of Kinases versus Phosphatases across Eukaryotes Determines Their Distinct Impacts. PLoS Comput. Biol. 2017, 13, e1005221. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, I.; Leiva, M.; Sabio, G. The role of stress kinases in metabolic disease. Nat. Rev. Endocrinol. 2020, 16, 697–716. [Google Scholar] [CrossRef]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase mutations in human disease: Interpreting genotype–phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef]
- Kumar, A.; Cookson, M.R. Role of LRRK2 kinase dysfunction in Parkinson disease. Expert. Rev. Mol. Med. 2011, 13, e20. [Google Scholar] [CrossRef]
- Castelo-Soccio, L.; Kim, H.; Gadina, M.; Schwartzberg, P.L.; Laurence, A.; O’Shea, J.J. Protein kinases: Drug targets for immunological disorders. Nat. Rev. Immunol. 2023, 1–20. [Google Scholar] [CrossRef]
- Stenberg, K.A.; Riikonen, P.T.; Vihinen, M. KinMutBase, a database of human disease-causing protein kinase mutations. Nucleic Acids Res. 2000, 28, 369–371. [Google Scholar] [CrossRef][Green Version]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Taylor, S.S.; Wu, J.; Bruystens, J.G.H.; Del Rio, J.C.; Lu, T.-W.; Kornev, A.P.; Ten Eyck, L.F. From structure to the dynamic regulation of a molecular switch: A journey over 3 decades. J. Biol. Chem. 2021, 296, 100746. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Zhang, P.; Steichen, J.M.; Keshwani, M.M.; Kornev, A.P. PKA: Lessons learned after twenty years. Biochim. Biophys. Acta 2013, 1834, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.H.; Krebs, E.G. Conversion of phosphorylase b to phosphorylase a in muscle extracts. J. Biol. Chem. 1955, 216, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Krebs, E.G.; Kent, A.B.; Fischer, E.H. The muscle phosphorylase b kinase reaction. J. Biol. Chem. 1958, 231, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Rall, T.W.; Sutherland, E.W. Formation of a cyclic adenine ribonucleotide by tissue particles. J. Biol. Chem. 1958, 232, 1065–1076. [Google Scholar] [CrossRef]
- Sutherland, E.W.; Rall, T.W. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 1958, 232, 1077–1091. [Google Scholar] [CrossRef]
- Cohen, P. The regulation of protein function by multisite phosphorylation—A 25 year update. Trends Biochem. Sci. 2000, 25, 596–601. [Google Scholar] [CrossRef]
- Walsh, D.A.; Perkins, J.P.; Krebs, E.G. An adenosine 3’,5’-monophosphate-dependant protein kinase from rabbit skeletal muscle. J. Biol. Chem. 1968, 243, 3763–3765. [Google Scholar] [CrossRef]
- Kuo, J.F.; Greengard, P. Cyclic nucleotide-dependent protein kinases, iv. widespread occurrence of adenosine 3′,5′-monophosphate-dependent protein kinase in various tissues and phyla of the animal kingdom. Proc. Natl. Acad. Sci. USA 1969, 64, 1349–1355. [Google Scholar] [CrossRef]
- Shoji, S.; Ericsson, L.H.; Walsh, K.A.; Fischer, E.H.; Titani, K. Amino acid sequence of the catalytic subunit of bovine type II adenosine cyclic 3’,5’-phosphate-dependent protein kinase. Biochemistry 1983, 22, 3702–3709. [Google Scholar] [CrossRef]
- Uhler, M.D.; Carmichael, D.F.; Lee, D.C.; Chrivia, J.C.; Krebs, E.G.; McKnight, G.S. Isolation of cDNA clones coding for the catalytic subunit of mouse cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1986, 83, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.-H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Dema, A.; Perets, E.; Schulz, M.S.; Deák, V.A.; Klussmann, E. Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell. Signal. 2015, 27, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Wild, A.R.; Dell’Acqua, M.L. Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol. Ther. 2018, 185, 99–121. [Google Scholar] [CrossRef]
- Sapio, L.; Gallo, M.; Illiano, M.; Chiosi, E.; Naviglio, D.; Spina, A.; Naviglio, S. The natural cAMP elevating compound forskolin in cancer therapy: Is it time? J. Cell. Physiol. 2017, 232, 922–927. [Google Scholar] [CrossRef]
- Kleppe, R.; Krakstad, C.; Selheim, F.; Kopperud, R.; Ove Doskeland, S. The cAMP-dependent protein kinase pathway as therapeutic target-possibilities and pitfalls. Curr. Top. Med. Chem. 2011, 11, 1393–1405. [Google Scholar] [CrossRef]
- Oldham, W.M.; Hamm, H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef]
- Grisan, F.; Iannucci, L.F.; Surdo, N.C.; Gerbino, A.; Zanin, S.; Di Benedetto, G.; Pozzan, T.; Lefkimmiatis, K. PKA compartmentalization links cAMP signaling and autophagy. Cell Death Differ. 2021, 28, 2436–2449. [Google Scholar] [CrossRef]
- Li, F.; Gangal, M.; Juliano, C.; Gorfain, E.; Taylor, S.S.; Johnson, D.A. Evidence for an internal entropy contribution to phosphoryl transfer: A study of domain closure, backbone flexibility, and the catalytic cycle of cAMP-dependent protein kinase. J. Mol. Biol. 2002, 315, 459–469. [Google Scholar] [CrossRef][Green Version]
- Zhao, L.; Liu, J.; He, C.; Yan, R.; Zhou, K.; Cui, Q.; Meng, X.; Li, X.; Zhang, Y.; Nie, Y. Protein kinase A determines platelet life span and survival by regulating apoptosis. J. Clin. Investig. 2017, 127, 4338–4351. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.; Brennesvik, E.O.; Lai, Y.-C.; Shepherd, P.R. GSK-3β regulation in skeletal muscles by adrenaline and insulin: Evidence that PKA and PKB regulate different pools of GSK-3. Cell. Signal. 2007, 19, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Tonucci, F.M.; Almada, E.; Borini-Etichetti, C.; Pariani, A.; Hidalgo, F.; Rico, M.J.; Girardini, J.; Favre, C.; Goldenring, J.R.; Menacho-Marquez, M. Identification of a CIP4 PKA phosphorylation site involved in the regulation of cancer cell invasiveness and metastasis. Cancer Lett. 2019, 461, 65–77. [Google Scholar] [CrossRef] [PubMed]
- London, E.; Bloyd, M.; Stratakis, C.A. PKA functions in metabolism and resistance to obesity: Lessons from mouse and human studies. J. Endocrinol. 2020, 246, R51–R64. [Google Scholar] [CrossRef] [PubMed]
- Moleschi, K.; Melacini, G. Signaling at crossroads: The dialogue between PDEs and PKA is spoken in multiple languages. Biophys. J. 2014, 107, 1259–1260. [Google Scholar] [CrossRef]
- Murthy, K.S.; Zhou, H.; Makhlouf, G.M. PKA-dependent activation of PDE3A and PDE4 and inhibition of adenylyl cyclase V/VI in smooth muscle. Am. J. Physiol.-Cell Physiol. 2002, 282, C508–C517. [Google Scholar] [CrossRef]
- Khannpnavar, B.; Mehta, V.; Qi, C.; Korkhov, V. Structure and function of adenylyl cyclases, key enzymes in cellular signaling. Curr. Opin. Struct. Biol. 2020, 63, 34–41. [Google Scholar] [CrossRef]
- Torres-Quesada, O.; Mayrhofer, J.E.; Stefan, E. The many faces of compartmentalized PKA signalosomes. Cell. Signal. 2017, 37, 1–11. [Google Scholar] [CrossRef]
- Zhu, Y.R.; Jiang, X.X.; Zheng, Y.; Xiong, J.; Wei, D.; Zhang, D.M. Cardiac function modulation depends on the A-kinase anchoring protein complex. J. Cell. Mol. Med. 2019, 23, 7170–7179. [Google Scholar] [CrossRef]
- Wong, W.; Scott, J.D. AKAP signalling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef]
- Amer, Y.O.; Hebert-Chatelain, E. Mitochondrial cAMP-PKA signaling: What do we really know? Biochim. et Biophys. Acta (BBA)-Bioenerg. 2018, 1859, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Westphal, R.S.; Tavalin, S.J.; Lin, J.W.; Alto, N.M.; Fraser, I.D.; Langeberg, L.K.; Sheng, M.; Scott, J.D. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science 1999, 285, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Dell’Acqua, M.L.; Smith, K.E.; Gorski, J.A.; Horne, E.A.; Gibson, E.S.; Gomez, L.L. Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur. J. Cell Biol. 2006, 85, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Pallien, T.; Klussmann, E. New aspects in cardiac L-type Ca2+ channel regulation. Biochem. Soc. Trans. 2020, 48, 39–49. [Google Scholar] [CrossRef]
- Scott, J. A-kinase-anchoring proteins and cytoskeletal signalling events. Biochem. Soc. Trans. 2003, 31, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Rababa’h, A.; Singh, S.; Suryavanshi, S.V.; Altarabsheh, S.E.; Deo, S.V.; McConnell, B.K. Compartmentalization role of A-kinase anchoring proteins (AKAPs) in mediating protein kinase A (PKA) signaling and cardiomyocyte hypertrophy. Int. J. Mol. Sci. 2014, 16, 218–229. [Google Scholar] [CrossRef]
- Smith, F.D.; Reichow, S.L.; Esseltine, J.L.; Shi, D.; Langeberg, L.K.; Scott, J.D.; Gonen, T. Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. Elife 2013, 2, e01319. [Google Scholar] [CrossRef]
- Smith, F.D.; Esseltine, J.L.; Nygren, P.J.; Veesler, D.; Byrne, D.P.; Vonderach, M.; Strashnov, I.; Eyers, C.E.; Eyers, P.A.; Langeberg, L.K. Local protein kinase A action proceeds through intact holoenzymes. Science 2017, 356, 1288–1293. [Google Scholar] [CrossRef]
- Walker-Gray, R.; Stengel, F.; Gold, M.G. Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches. Proc. Natl. Acad. Sci. USA 2017, 114, 10414–10419. [Google Scholar] [CrossRef]
- Martin, B.R.; Deerinck, T.J.; Ellisman, M.H.; Taylor, S.S.; Tsien, R.Y. Isoform-specific PKA dynamics revealed by dye-triggered aggregation and DAKAP1α-mediated localization in living cells. Chem. Biol. 2007, 14, 1031–1042. [Google Scholar] [CrossRef]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- De Boer, A.; Letzel, T.; Lingeman, H.; Irth, H. Systematic development of an enzymatic phosphorylation assay compatible with mass spectrometric detection. Anal. Bioanal. Chem. 2005, 381, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Dalton, G.D.; Smith, F.L.; Smith, P.A.; Dewey, W.L. Alterations in brain Protein Kinase A activity and reversal of morphine tolerance by two fragments of native Protein Kinase A inhibitor peptide (PKI). Neuropharmacology 2005, 48, 648–657. [Google Scholar] [CrossRef]
- Liu, C.; Ke, P.; Zhang, J.; Zhang, X.; Chen, X. Protein Kinase Inhibitor Peptide as a Tool to Specifically Inhibit Protein Kinase A. Front. Physiol. 2020, 11, 574030. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Yu, L.; Tu, Q.; Zhang, M.; He, H.; Chen, W.; Gao, J.; Yu, J.; Wu, Q.; Zhao, S. Cloning and mapping of human PKIB and PKIG, and comparison of tissue expression patterns of three members of the protein kinase inhibitor family, including PKIA. Biochem. J. 2000, 349, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, L.G.; Kornev, A.P.; McClendon, C.L.; Veglia, G.; Taylor, S.S. Mutation of a kinase allosteric node uncouples dynamics linked to phosphotransfer. Proc. Natl. Acad. Sci. USA 2017, 114, E931–E940. [Google Scholar] [CrossRef]
- Saldanha, S.A.; Kaler, G.; Cottam, H.B.; Abagyan, R.; Taylor, S.S. Assay principle for modulators of protein-protein interactions and its application to non-ATP-competitive ligands targeting protein kinase A. Anal. Chem. 2006, 78, 8265–8272. [Google Scholar] [CrossRef][Green Version]
- Haushalter, K.J.; Casteel, D.E.; Raffeiner, A.; Stefan, E.; Patel, H.H.; Taylor, S.S. Phosphorylation of protein kinase A (PKA) regulatory subunit RIalpha by protein kinase G (PKG) primes PKA for catalytic activity in cells. J. Biol. Chem. 2018, 293, 4411–4421. [Google Scholar] [CrossRef]
- Bruystens, J.G.H.; Wu, J.; Fortezzo, A.; Kornev, A.P.; Blumenthal, D.K.; Taylor, S.S. PKA RIα Homodimer Structure Reveals an Intermolecular Interface with Implications for Cooperative cAMP Binding and Carney Complex Disease. Structure 2014, 22, 59–69. [Google Scholar] [CrossRef]
- Walker, C.; Wang, Y.; Olivieri, C.; Karamafrooz, A.; Casby, J.; Bathon, K.; Calebiro, D.; Gao, J.; Bernlohr, D.A.; Taylor, S.S.; et al. Cushing’s syndrome driver mutation disrupts protein kinase A allosteric network, altering both regulation and substrate specificity. Sci. Adv. 2019, 5, eaaw9298. [Google Scholar] [CrossRef]
- Beuschlein, F.; Fassnacht, M.; Assié, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.L.; Wieland, T.; Sbiera, S.; Faucz, F.R. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N. Engl. J. Med. 2014, 370, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Ginter, C.; Cassidy, M.; Franklin, M.C.; Rudolph, M.J.; Robine, N.; Darnell, R.B.; Hendrickson, W.A. Structural insights into mis-regulation of protein kinase A in human tumors. Proc. Natl. Acad. Sci. USA 2015, 112, 1374–1379. [Google Scholar] [CrossRef] [PubMed]
- Bertherat, J.; Horvath, A.; Groussin, L.; Grabar, S.; Boikos, S.; Cazabat, L.; Libe, R.; René-Corail, F.; Stergiopoulos, S.; Bourdeau, I. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): Phenotype analysis in 353 patients and 80 different genotypes. J. Clin. Endocrinol. Metab. 2009, 94, 2085–2091. [Google Scholar] [CrossRef]
- Bossis, I.; Stratakis, C.A. Minireview: PRKAR1A: Normal and abnormal functions. Endocrinology 2004, 145, 5452–5458. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, L.S.; Sandrini, F.; Monbo, J.; Lin, J.P.; Carney, J.A.; Stratakis, C.A. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum. Mol. Genet. 2000, 9, 3037–3046. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Kirschner, L.S.; Carney, J.A. Clinical and molecular features of the Carney complex: Diagnostic criteria and recommendations for patient evaluation. J. Clin. Endocrinol. Metab. 2001, 86, 4041–4046. [Google Scholar] [CrossRef]
- Laxminarayana, D.; Kammer, G.M. mRNA mutations of type I protein kinase A regulatory subunit α in T lymphocytes of a subject with systemic lupus erythematosus. Int. Immunol. 2000, 12, 1521–1529. [Google Scholar] [CrossRef]
- Pérez-Lorenzo, M.J.; Galindo, M.; García-González, A.J.; Criado, G. Increased expression of A-kinase anchoring proteins in T cells from systemic lupus erythematosus patients. J. Transl. Med. 2010, 8, P49. [Google Scholar] [CrossRef]
- Enns, L.C.; Ladiges, W. Protein kinase A signaling as an anti-aging target. Ageing Res. Rev. 2010, 9, 269–272. [Google Scholar] [CrossRef]
- Enns, L.C.; Morton, J.F.; Mangalindan, R.S.; McKnight, G.S.; Schwartz, M.W.; Kaeberlein, M.R.; Kennedy, B.K.; Rabinovitch, P.S.; Ladiges, W.C. Attenuation of age-related metabolic dysfunction in mice with a targeted disruption of the Cβ subunit of protein kinase A. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2009, 64, 1221–1231. [Google Scholar] [CrossRef]
- Gertler, A.; Solomon, G. Leptin-activity blockers: Development and potential use in experimental biology and medicine. Can. J. Physiol. Pharmacol. 2013, 91, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Spina, A.; Di Maiolo, F.; Esposito, A.; D’Auria, R.; Di Gesto, D.; Chiosi, E.; Sorvillo, L.; Naviglio, S. Integrating leptin and cAMP signalling pathways in triple-negative breast cancer cells. Front. Biosci. 2013, 18, 133–144. [Google Scholar] [CrossRef]
- Naviglio, S.; Di Gesto, D.; Romano, M.; Sorrentino, A.; Illiano, F.; Sorvillo, L.; Abbruzzese, A.; Marra, M.; Caraglia, M.; Chiosi, E.; et al. Leptin enhances growth inhibition by cAMP elevating agents through apoptosis of MDA-MB-231 breast cancer cells. Cancer Biol. Ther. 2009, 8, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Naviglio, S.; Di Gesto, D.; Illiano, F.; Chiosi, E.; Giordano, A.; Illiano, G.; Spina, A. Leptin potentiates antiproliferative action of cAMP elevation via protein kinase A down-regulation in breast cancer cells. J. Cell Physiol. 2010, 225, 801–809. [Google Scholar] [CrossRef]
- Graham, R.P.; Jin, L.; Knutson, D.L.; Kloft-Nelson, S.M.; Greipp, P.T.; Waldburger, N.; Roessler, S.; Longerich, T.; Roberts, L.R.; Oliveira, A.M. DNAJB1-PRKACA is specific for fibrolamellar carcinoma. Mod. Pathol. 2015, 28, 822–829. [Google Scholar] [CrossRef]
- Turnham, R.E.; Smith, F.D.; Kenerson, H.L.; Omar, M.H.; Golkowski, M.; Garcia, I.; Bauer, R.; Lau, H.-T.; Sullivan, K.M.; Langeberg, L.K.; et al. An acquired scaffolding function of the DNAJ-PKAc fusion contributes to oncogenic signaling in fibrolamellar carcinoma. eLife 2019, 8, e44187. [Google Scholar] [CrossRef]
- Wu, K.-J.; Mattioli, M.; Morse, H.C.; Dalla-Favera, R. c-MYC activates protein kinase A (PKA) by direct transcriptional activation of the PKA catalytic subunit beta (PKA-Cβ) gene. Oncogene 2002, 21, 7872–7882. [Google Scholar] [CrossRef]
- Howe, A.K. Regulation of actin-based cell migration by cAMP/PKA. Biochim. Biophys. Acta 2004, 1692, 159–174. [Google Scholar] [CrossRef]
- Shaikh, D.; Zhou, Q.; Chen, T.; Ibe, J.C.; Raj, J.U.; Zhou, G. cAMP-dependent protein kinase is essential for hypoxia-mediated epithelial-mesenchymal transition, migration, and invasion in lung cancer cells. Cell Signal. 2012, 24, 2396–2406. [Google Scholar] [CrossRef]
- Jiang, P.; Enomoto, A.; Takahashi, M. Cell biology of the movement of breast cancer cells: Intracellular signalling and the actin cytoskeleton. Cancer Lett. 2009, 284, 122–130. [Google Scholar] [CrossRef]
- McKenzie, A.J.; Campbell, S.L.; Howe, A.K. Protein kinase A activity and anchoring are required for ovarian cancer cell migration and invasion. PLoS ONE 2011, 6, e26552. [Google Scholar] [CrossRef] [PubMed]
- Cho-Chung, Y.S. Protein kinase A-directed antisense blockade of cancer growth: Single gene-based therapeutic approach. Appl. Antisense Oligonucleotide Technol. 1998, 263–281. [Google Scholar]
- Stratakis, C.A.; Cho-Chung, Y.S. Protein kinase A and human disease. Trends Endocrinol. Metab. 2002, 13, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Cho-Chung, Y.S.; Nesterova, M.V. Tumor reversion: Protein kinase A isozyme switching. Ann. N. Y. Acad. Sci. 2005, 1058, 76–86. [Google Scholar] [CrossRef]
- Tortora, G.; Ciardiello, F. Protein kinase A as target for novel integrated strategies of cancer therapy. Ann. N. Y. Acad. Sci. 2002, 968, 139–147. [Google Scholar] [CrossRef]
- Beebe, S.J.; Øyen, O.; Sandberg, M.; Frøysa, A.; Hansson, V.; Jahnsen, T. Molecular cloning of a tissue-specific protein kinase (Cγ) from human testis—Representing a third isoform for the catalytic subunit of cAMP-dependent protein kinase. Mol. Endocrinol. 1990, 4, 465–475. [Google Scholar] [CrossRef]
- Zimmermann, B.; Chiorini, J.A.; Ma, Y.; Kotin, R.M.; Herberg, F.W. PrKX is a novel catalytic subunit of the cAMP-dependent protein kinase regulated by the regulatory subunit type I. J. Biol. Chem. 1999, 274, 5370–5378. [Google Scholar] [CrossRef]
- Skalhegg, B.S.; Tasken, K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front. Biosci.-Landmark 2000, 5, D678–D693. [Google Scholar] [CrossRef]
- Schiebel, K.; Winkelmann, M.; Mertz, A.; Xu, X.; Page, D.C.; Weil, D.; Petit, C.; Rappold, G.A. Abnormal XY interchange between a novel isolated protein kinase gene, PRKY, and its homologue, PRKX, accounts for one third of all (Y+)XX males and (Y-)XY females. Hum. Mol. Genet. 1997, 6, 1985–1989. [Google Scholar] [CrossRef]
- Søberg, K.; Jahnsen, T.; Rognes, T.; Skålhegg, B.S.; Laerdahl, J.K. Evolutionary paths of the cAMP-dependent protein kinase (PKA) catalytic subunits. PLoS ONE 2013, 8, e60935. [Google Scholar] [CrossRef]
- Agustin, J.T.S.; Wilkerson, C.G.; Witman, G.B. The unique catalytic subunit of sperm cAMP-dependent protein kinase is the product of an alternative Cα mRNA expressed specifically in spermatogenic cells. Mol. Biol. Cell 2000, 11, 3031–3044. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guthrie, C.R.; Skålhegg, B.S.; McKnight, G.S. Two novel brain-specific splice variants of the murine Cβ gene of cAMP-dependent protein kinase. J. Biol. Chem. 1997, 272, 29560–29565. [Google Scholar] [CrossRef]
- Showers, M.O.; Maurer, R.A. Cloning of cDNA for the catalytic subunit of cAMP-dependent protein kinase. Methods Enzymol. 1988, 159, 311–318. [Google Scholar] [CrossRef] [PubMed]
- San Agustin, J.T.; Leszyk, J.D.; Nuwaysir, L.M.; Witman, G.B. The catalytic subunit of the cAMP-dependent protein kinase of ovine sperm flagella has a unique amino-terminal sequence. J. Biol. Chem. 1998, 273, 24874–24883. [Google Scholar] [CrossRef]
- San Agustin, J.T.; Witman, G.B. Differential expression of the C(s) and Calpha1 isoforms of the catalytic subunit of cyclic 3’,5’-adenosine monophosphate-dependent protein kinase testicular cells. Biol. Reprod. 2001, 65, 151–164. [Google Scholar] [CrossRef]
- Reinton, N.; Orstavik, S.; Haugen, T.B.; Jahnsen, T.; Taskén, K.; Skålhegg, B.S. A novel isoform of human cyclic 3’,5’-adenosine monophosphate-dependent protein kinase, c alpha-s, localizes to sperm midpiece. Biol. Reprod. 2000, 63, 607–611. [Google Scholar] [CrossRef]
- Nolan, M.A.; Babcock, D.F.; Wennemuth, G.; Brown, W.; Burton, K.A.; McKnight, G.S. Sperm-specific protein kinase A catalytic subunit Calpha2 orchestrates cAMP signaling for male fertility. Proc. Natl. Acad. Sci. USA 2004, 101, 13483–13488. [Google Scholar] [CrossRef]
- Desseyn, J.L.; Burton, K.A.; McKnight, G.S. Expression of a nonmyristylated variant of the catalytic subunit of protein kinase A during male germ-cell development. Proc. Natl. Acad. Sci. USA 2000, 97, 6433–6438. [Google Scholar] [CrossRef]
- Skålhegg, B.S.; Huang, Y.; Su, T.; Idzerda, R.L.; McKnight, G.S.; Burton, K.A. Mutation of the Calpha subunit of PKA leads to growth retardation and sperm dysfunction. Mol. Endocrinol. 2002, 16, 630–639. [Google Scholar] [CrossRef]
- Uhler, M.D.; Chrivia, J.C.; McKnight, G.S. Evidence for a second isoform of the catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem. 1986, 261, 15360–15363. [Google Scholar] [CrossRef]
- Wiemann, S.; Kinzel, V.; Pyerin, W. Isoform C beta 2, an unusual form of the bovine catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem. 1991, 266, 5140–5146. [Google Scholar] [CrossRef] [PubMed]
- Ørstavik, S.; Reinton, N.; Frengen, E.; Langeland, B.T.; Jahnsen, T.; Skålhegg, B.S. Identification of novel splice variants of the human catalytic subunit Cbeta of cAMP-dependent protein kinase. Eur. J. Biochem. 2001, 268, 5066–5073. [Google Scholar] [CrossRef] [PubMed]
- Kvissel, A.K.; Ørstavik, S.; Øistad, P.; Rootwelt, T.; Jahnsen, T.; Skålhegg, B.S. Induction of Cbeta splice variants and formation of novel forms of protein kinase A type II holoenzymes during retinoic acid-induced differentiation of human NT2 cells. Cell. Signal. 2004, 16, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.C.; Kvissel, A.K.; Hafte, T.T.; Avellan, C.I.; Eikvar, S.; Rootwelt, T.; Ørstavik, S.; Skålhegg, B.S. Inactive forms of the catalytic subunit of protein kinase A are expressed in the brain of higher primates. FEBS J. 2008, 275, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Beebe, S.J.; Salomonsky, P.; Jahnsen, T.; Li, Y. The C gamma subunit is a unique isozyme of the cAMP-dependent protein kinase. J. Biol. Chem. 1992, 267, 25505–25512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Morris, G.Z.; Beebe, S.J. Characterization of the cAMP-dependent protein kinase catalytic subunit Cgamma expressed and purified from sf9 cells. Protein Expr. Purif. 2004, 35, 156–169. [Google Scholar] [CrossRef]
- Carr, S.A.; Biemann, K.; Shoji, S.; Parmelee, D.C.; Titani, K. n-Tetradecanoyl is the NH2-terminal blocking group of the catalytic subunit of cyclic AMP-dependent protein kinase from bovine cardiac muscle. Proc. Natl. Acad. Sci. USA 1982, 79, 6128–6131. [Google Scholar] [CrossRef]
- Adams, J.A.; McGlone, M.L.; Gibson, R.; Taylor, S.S. Phosphorylation modulates catalytic function and regulation in the cAMP-dependent protein kinase. Biochemistry 1995, 34, 2447–2454. [Google Scholar] [CrossRef]
- Taylor, S.S.; Radzio-Andzelm, E. Chapter 179—cAMP-Dependent Protein Kinase. In Handbook of Cell Signaling, 2nd ed.; Bradshaw, R.A., Dennis, E.A., Eds.; Academic Press: San Diego, CA, USA, 2010; pp. 1461–1469. [Google Scholar]
- Keshwani, M.M.; Klammt, C.; von Daake, S.; Ma, Y.; Kornev, A.P.; Choe, S.; Insel, P.A.; Taylor, S.S. Cotranslational cis-phosphorylation of the COOH-terminal tail is a key priming step in the maturation of cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 2012, 109, E1221–E1229. [Google Scholar] [CrossRef]
- Moore, M.J.; Kanter, J.R.; Jones, K.C.; Taylor, S.S. Phosphorylation of the Catalytic Subunit of Protein Kinase A: AUTOPHOSPHORYLATION VERSUS PHOSPHORYLATION BY PHOSPHOINOSITIDE-DEPENDENT KINASE-1. J. Biol. Chem. 2002, 277, 47878–47884. [Google Scholar] [CrossRef]
- Carrera, A.C.; Alexandrov, K.; Roberts, T.M. The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc. Natl. Acad. Sci. USA 1993, 90, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Meharena, H.S.; Fan, X.; Ahuja, L.G.; Keshwani, M.M.; McClendon, C.L.; Chen, A.M.; Adams, J.A.; Taylor, S.S. Decoding the Interactions Regulating the Active State Mechanics of Eukaryotic Protein Kinases. PLoS Biol. 2016, 14, e2000127. [Google Scholar] [CrossRef] [PubMed]
- Humphries, K.M.; Deal, M.S.; Taylor, S.S. Enhanced dephosphorylation of cAMP-dependent protein kinase by oxidation and thiol modification. J. Biol. Chem. 2005, 280, 2750–2758. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.; Taylor, S.S. Kinetic analysis of cAMP-dependent protein kinase: Mutations at histidine 87 affect peptide binding and pH dependence. Biochemistry 1995, 34, 16203–16209. [Google Scholar] [CrossRef] [PubMed]
- Khavrutskii, I.V.; Grant, B.; Taylor, S.S.; McCammon, J.A. A transition path ensemble study reveals a linchpin role for Mg(2+) during rate-limiting ADP release from protein kinase A. Biochemistry 2009, 48, 11532–11545. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheng, Y.; Zhang, Y.; McCammon, J.A. How Does the cAMP-Dependent Protein Kinase Catalyze the Phosphorylation Reaction: An ab Initio QM/MM Study. J. Am. Chem. Soc. 2005, 127, 1553–1562. [Google Scholar] [CrossRef]
- Adams, J.A. Kinetic and catalytic mechanisms of protein kinases. Chem. Rev. 2001, 101, 2271–2290. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kim, C.; Cheng, C.Y.; Brown, S.H.; Wu, J.; Kannan, N. Signaling through cAMP and cAMP-dependent protein kinase: Diverse strategies for drug design. Biochim. et Biophys. Acta (BBA)-Proteins Proteom. 2008, 1784, 16–26. [Google Scholar] [CrossRef]
- Taylor, S.S.; Søberg, K.; Kobori, E.; Wu, J.; Pautz, S.; Herberg, F.W.; Skålhegg, B.S. The Tails of Protein Kinase A. Mol. Pharmacol. 2022, 101, 219–225. [Google Scholar] [CrossRef]
- Sastri, M.; Barraclough, D.M.; Carmichael, P.T.; Taylor, S.S. A-kinase-interacting protein localizes protein kinase A in the nucleus. Proc. Natl. Acad. Sci. USA 2005, 102, 349–354. [Google Scholar] [CrossRef]
- Kannan, N.; Haste, N.; Taylor, S.S.; Neuwald, A.F. The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc. Natl. Acad. Sci. USA 2007, 104, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.S.; Kornev, A.P.; Hu, J.; Ahuja, L.G.; Taylor, S.S. Kinases and pseudokinases: Lessons from RAF. Mol. Cell. Biol. 2014, 34, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Kornev, A.P.; Haste, N.M.; Taylor, S.S.; Ten Eyck, L.F. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 17783–17788. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, L.G.; Aoto, P.C.; Kornev, A.P.; Veglia, G.; Taylor, S.S. Dynamic allostery-based molecular workings of kinase:peptide complexes. Proc. Natl. Acad. Sci. USA 2019, 116, 15052–15061. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Manus, V.S.; Kim, J.; Li, G.; Ahuja, L.G.; Aoto, P.; Taylor, S.S.; Veglia, G. Globally correlated conformational entropy underlies positive and negative cooperativity in a kinase’s enzymatic cycle. Nat. Commun. 2019, 10, 799. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.A.; Taylor, S.S. Energetic limits of phosphotransfer in the catalytic subunit of cAMP-dependent protein kinase as measured by viscosity experiments. Biochemistry 1992, 31, 8516–8522. [Google Scholar] [CrossRef]
- Lew, J.; Taylor, S.S.; Adams, J.A. Identification of a partially rate-determining step in the catalytic mechanism of cAMP-dependent protein kinase: A transient kinetic study using stopped-flow fluorescence spectroscopy. Biochemistry 1997, 36, 6717–6724. [Google Scholar] [CrossRef]
- Adams, J.A.; Taylor, S.S. Phosphorylation of peptide substrates for the catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem. 1993, 268, 7747–7752. [Google Scholar] [CrossRef]
- Masterson, L.R.; Cembran, A.; Shi, L.; Veglia, G. Allostery and binding cooperativity of the catalytic subunit of protein kinase A by NMR spectroscopy and molecular dynamics simulations. Adv. Protein Chem. Struct. Biol. 2012, 87, 363–389. [Google Scholar] [CrossRef]
- Lodish, M.; Stratakis, C.A. A genetic and molecular update on adrenocortical causes of Cushing syndrome. Nat. Rev. Endocrinol. 2016, 12, 255–262. [Google Scholar] [CrossRef]
- Cao, Y.; He, M.; Gao, Z.; Peng, Y.; Li, Y.; Li, L.; Zhou, W.; Li, X.; Zhong, X.; Lei, Y. Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science 2014, 344, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Maekawa, S.; Ishii, R.; Sanada, M.; Morikawa, T.; Shiraishi, Y.; Yoshida, K.; Nagata, Y.; Sato-Otsubo, A.; Yoshizato, T. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 2014, 344, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.; Scholl, U.I.; Healy, J.M.; Choi, M.; Prasad, M.L.; Nelson-Williams, C.; Kunstman, J.W.; Korah, R.; Suttorp, A.-C.; Dietrich, D. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat. Genet. 2014, 46, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Honeyman, J.N.; Simon, E.P.; Robine, N.; Chiaroni-Clarke, R.; Darcy, D.G.; Lim, I.I.P.; Gleason, C.E.; Murphy, J.M.; Rosenberg, B.R.; Teegan, L. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 2014, 343, 1010–1014. [Google Scholar] [CrossRef]
- Xu, L.; Hazard, F.K.; Zmoos, A.-F.; Jahchan, N.; Chaib, H.; Garfin, P.M.; Rangaswami, A.; Snyder, M.P.; Sage, J. Genomic analysis of fibrolamellar hepatocellular carcinoma. Hum. Mol. Genet. 2015, 24, 50–63. [Google Scholar] [CrossRef]
- Tasken, K.; Skålhegg, B.; Solberg, R.; Andersson, K.; Taylor, S.; Lea, T.; Blomhoff, H.; Jahnsen, T.; Hansson, V. Novel isozymes of cAMP-dependent protein kinase exist in human cells due to formation of RI alpha-RI beta heterodimeric complexes. J. Biol. Chem. 1993, 268, 21276–21283. [Google Scholar] [CrossRef]
- Amieux, P.S.; McKnight, G.S. The essential role of RIα in the maintenance of regulated PKA activity. Ann. N. Y. Acad. Sci. 2002, 968, 75–95. [Google Scholar] [CrossRef]
- Horvath, A.; Bertherat, J.; Groussin, L.; Guillaud-Bataille, M.; Tsang, K.; Cazabat, L.; Libe, R.; Remmers, E.; René-Corail, F.; Faucz, F.R. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): An update. Hum. Mutat. 2010, 31, 369–379. [Google Scholar] [CrossRef]
- Bruystens, J.G.; Wu, J.; Fortezzo, A.; Del Rio, J.; Nielsen, C.; Blumenthal, D.K.; Rock, R.; Stefan, E.; Taylor, S.S. Structure of a PKA RIalpha Recurrent Acrodysostosis Mutant Explains Defective cAMP-Dependent Activation. J. Mol. Biol. 2016, 428, 4890–4904. [Google Scholar] [CrossRef]
- Wong, T.H.; Chiu, W.Z.; Breedveld, G.J.; Li, K.W.; Verkerk, A.J.; Hondius, D.; Hukema, R.K.; Seelaar, H.; Frick, P.; Severijnen, L.-A. PRKAR1B mutation associated with a new neurodegenerative disorder with unique pathology. Brain 2014, 137, 1361–1373. [Google Scholar] [CrossRef]
- Schreyer, S.A.; Cummings, D.E.; McKnight, G.S.; LeBoeuf, R.e.C. Mutation of the RIIβ subunit of protein kinase A prevents diet-induced insulin resistance and dyslipidemia in mice. Diabetes 2001, 50, 2555–2562. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ringheim, G.E.; Taylor, S.S. Dissecting the domain structure of the regulatory subunit of cAMP-dependent protein kinase I and elucidating the role of MgATP. J. Biol. Chem. 1990, 265, 4800–4808. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Cheng, C.Y.; Saldanha, S.A.; Taylor, S.S. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 2007, 130, 1032–1043. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Ilouz, R.; Zhang, P.; Kornev, A.P. Assembly of allosteric macromolecular switches: Lessons from PKA. Nat. Rev. Mol. Cell Biol. 2012, 13, 646–658. [Google Scholar] [CrossRef]
- Zhang, P.; Knape, M.J.; Ahuja, L.G.; Keshwani, M.M.; King, C.C.; Sastri, M.; Herberg, F.W.; Taylor, S.S. Single Turnover Autophosphorylation Cycle of the PKA RIIβ Holoenzyme. PLoS Biol. 2015, 13, e1002192. [Google Scholar] [CrossRef]
- Berman, H.M.; Ten Eyck, L.F.; Goodsell, D.S.; Haste, N.M.; Kornev, A.; Taylor, S.S. The cAMP binding domain: An ancient signaling module. Proc. Natl. Acad. Sci. USA 2005, 102, 45–50. [Google Scholar] [CrossRef]
- Herberg, F.W.; Taylor, S.S.; Dostmann, W.R. Active site mutations define the pathway for the cooperative activation of cAMP-dependent protein kinase. Biochemistry 1996, 35, 2934–2942. [Google Scholar] [CrossRef]
- Zhang, P.; Smith-Nguyen, E.V.; Keshwani, M.M.; Deal, M.S.; Kornev, A.P.; Taylor, S.S. Structure and allostery of the PKA RIIbeta tetrameric holoenzyme. Science 2012, 335, 712–716. [Google Scholar] [CrossRef]
- Kornev, A.P.; Taylor, S.S.; Ten Eyck, L.F. A generalized allosteric mechanism for cis-regulated cyclic nucleotide binding domains. PLoS Comput. Biol. 2008, 4, e1000056. [Google Scholar] [CrossRef]
- Akimoto, M.; McNicholl, E.T.; Ramkissoon, A.; Moleschi, K.; Taylor, S.S.; Melacini, G. Mapping the free energy landscape of PKA inhibition and activation: A double-conformational selection model for the tandem cAMP-binding domains of PKA RIα. PLoS Biol. 2015, 13, e1002305. [Google Scholar] [CrossRef]
- Guo, C.; Zhou, H.-X. Unidirectional allostery in the regulatory subunit RIα facilitates efficient deactivation of protein kinase A. Proc. Natl. Acad. Sci. USA 2016, 113, E6776–E6785. [Google Scholar] [CrossRef] [PubMed]
- McNicholl, E.T.; Das, R.; SilDas, S.; Taylor, S.S.; Melacini, G. Communication between tandem cAMP binding domains in the regulatory subunit of protein kinase A-Iα as revealed by domain-silencing mutations. J. Biol. Chem. 2010, 285, 15523–15537. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, A.J.; Wu, J.; Kim, C.; Yang, J.; Bruystens, J.; Cheung, N.; Pennypacker, J.K.; Blumenthal, D.A.; Kornev, A.P.; Taylor, S.S. Realizing the allosteric potential of the tetrameric protein kinase A RIalpha holoenzyme. Structure 2011, 19, 265–276. [Google Scholar] [CrossRef]
- Brown, S.H.; Wu, J.; Kim, C.; Alberto, K.; Taylor, S.S. Novel isoform-specific interfaces revealed by PKA RIIbeta holoenzyme structures. J. Mol. Biol. 2009, 393, 1070–1082. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Ye, F.; Bastidas, A.C.; Kornev, A.P.; Wu, J.; Ginsberg, M.H.; Taylor, S.S. An Isoform-Specific Myristylation Switch Targets Type II PKA Holoenzymes to Membranes. Structure 2015, 23, 1563–1572. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krissinel, E. and Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Lu, T.W.; Wu, J.; Aoto, P.C.; Weng, J.H.; Ahuja, L.G.; Sun, N.; Cheng, C.Y.; Zhang, P.; Taylor, S.S. Two PKA RIalpha holoenzyme states define ATP as an isoform-specific orthosteric inhibitor that competes with the allosteric activator, cAMP. Proc. Natl. Acad. Sci. USA 2019, 116, 16347–16356. [Google Scholar] [CrossRef]
- Day, M.E.; Gaietta, G.M.; Sastri, M.; Koller, A.; Mackey, M.R.; Scott, J.D.; Perkins, G.A.; Ellisman, M.H.; Taylor, S.S. Isoform-specific targeting of PKA to multivesicular bodies. J. Cell Biol. 2011, 193, 347–363. [Google Scholar] [CrossRef]
- Ferretti, A.C.; Tonucci, F.M.; Hidalgo, F.; Almada, E.; Larocca, M.C.; Favre, C. AMPK and PKA interaction in the regulation of survival of liver cancer cells subjected to glucose starvation. Oncotarget 2016, 7, 17815–17828. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).