
The Role of Mitochondria in Mediation of Skeletal Muscle Repair


Abstract
1. Introduction
2. Muscle Injury and Regeneration
2.1. Inflammatory Response to Muscle Injury
2.2. Myogenic Responses to Muscle Injury
3. Mitochondrial Regulation of Regeneration after Muscle Injury
3.1. Mitochondria-Derived Signaling Pathways Controlling Inflammation
3.1.1. Mitochondrial Dynamics
3.1.2. Mitophagy and Inflammation
3.2. Metabolism in MSCs
3.2.1. Mitochondria in MSC Proliferation and Differentiation
3.2.2. Immunometabolism in Muscle Injury and Repair
Exopher-Macrophage Regulation of Dysfunctional Mitochondria in Injury
Extracellular Vesicle Regulation of Mitochondria in Physiological Injury and Repair
3.3. Mitochondrially-Induced Oxidative Stress in Muscle Injury
3.3.1. Mitochondria Associated Oxidative Stress as a Negative Regulator of Regeneration Following Muscle Injury
3.3.2. Mitochondrial Reactive Oxygen/Nitrogen Species for Protecting Muscle against Injury and Improving Recovery
Hydrogen Peroxide
Molecular Hydrogen
Mitogen-Activated Protein Kinases (MAPK)
Peroxisome Proliferator-Activated Receptor Gamma (PPAR-γ)
Nuclear Factor-κB (NF-κB)
Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2)
Sestrin2
3.4. Alterations in Mitochondrial Genes during Injury and Repair
3.4.1. Molecular Alterations in Mitochondrially Related Genes with Injury
3.4.2. Molecular Alterations in Mitochondrially Related Genes in Repair/Myogenesis
3.5. Mitochondrial Dysfunction and Cellular Regeneration in Myopathies
3.5.1. Oxidative Enzyme Loss and Mitochondrial Dysfunction in DMD Muscle Degeneration
3.5.2. Mitochondrial Molecular Dysfunction in DMD Muscle Degeneration
3.5.3. Disruption of Ion Homeostasis and Mitochondrial Dysfunction in DMD Muscle Degeneration
3.5.4. ROS and Mitochondrial Dysfunction in DMD Muscle Degeneration
3.5.5. Dysregulation of K+ Homeostasis
3.6. Mitochondria in MSC Proliferation
3.7. Mitochondria in MSC Differentiation
3.7.1. Mitochondrial Biogenesis in MSC Differentiation and Regeneration
3.7.2. Mitochondrial Fusion in MSC Differentiation
3.7.3. Mitochondrial Mitophagy in MSC Differentiation
3.7.4. mRNA and microRNA Regulation of Differentiation Activates Mitochondrial Biogenesis
3.7.5. Strategies for Attenuation of Mitochondrial Aging
Resveratrol
Beta-Hydroxy-Beta-Methylbutyrate (HMB)
Epigallocatechin-3-Gallate (EGCg)
Sulforaphane (SFN)
Nicotinamide Riboside (NR)
3.8. Mitochondrial-Nuclear Axis in Physiological Muscle Injury and Repair
3.9. Mitochondrial Transplantation Increases Mitochondria Abundance and Improves Muscle Regeneration
3.9.1. Mitochondrial Regulation of the Extracellular Matrix in Muscle Regeneration
3.9.2. Mitochondria Enhancement Improves Muscle Regeneration
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leduc-Gaudet, J.P.; Hussain, S.N.A.; Barreiro, E.; Gouspillou, G. Mitochondrial Dynamics and Mitophagy in Skeletal Muscle Health and Aging. Int. J. Mol. Sci. 2021, 22, 8179. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.R.; Ryan, M.J.; Hao, Y.; Alway, S.E. Mediation of endogenous antioxidant enzymes and apoptotic signaling by resveratrol following muscle disuse in the gastrocnemius muscles of young and old rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1572–R1581. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.T.; Mohamed, J.S.; Alway, S.E. Effects of resveratrol on the recovery of muscle mass following disuse in the plantaris muscle of aged rats. PLoS ONE 2013, 8, e83518. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gonzalo, R.; McDonnell, A.C.; Simpson, E.J.; Macdonald, I.A.; Rullman, E.; Mekjavic, I.B. Substantial and Reproducible Individual Variability in Skeletal Muscle Outcomes in the Cross-Over Designed Planica Bed Rest Program. Front. Physiol. 2021, 12, 676501. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.; De Martino, E.; Hides, J.; Cable, G.; Elliott, J.M.; Hoggarth, M.; Zange, J.; Lindsay, K.; Debuse, D.; Winnard, A.; et al. Gluteal Muscle Atrophy and Increased Intramuscular Lipid Concentration Are Not Mitigated by Daily Artificial Gravity Following 60-Day Head-Down Tilt Bed Rest. Front. Physiol. 2021, 12, 745811. [Google Scholar] [CrossRef]
- Paez, H.G.; Pitzer, C.R.; Alway, S.E. Age-Related Dysfunction in Proteostasis and Cellular Quality Control in the Development of Sarcopenia. Cells 2023, 12, 249. [Google Scholar] [CrossRef]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Alway, S.E.; Morissette, M.R.; Siu, P.M. Aging and apoptosis in muscle. In Handbook of the Biology of Aging, 7th ed.; Elsevier: Amsterdam, The Netherlands, 2011; pp. 63–118. [Google Scholar]
- Alway, S.E.; Bennett, B.T.; Wilson, J.C.; Edens, N.K.; Pereira, S.L. Epigallocatechin-3-gallate improves plantaris muscle recovery after disuse in aged rats. Exp. Gerontol. 2014, 50, 82–94. [Google Scholar] [CrossRef]
- Pitzer, C.R.; Paez, H.G.; Alway, S.E. The Contribution of Tumor Derived Exosomes to Cancer Cachexia. Cells 2023, 12, 292. [Google Scholar] [CrossRef]
- Counts, B.R.; Halle, J.L.; Carson, J.A. Early-Onset Physical Inactivity and Metabolic Dysfunction in Tumor-bearing Mice Is Associated with Accelerated Cachexia. Med. Sci. Sports Exerc. 2022, 54, 77–88. [Google Scholar] [CrossRef]
- Counts, B.R.; Hardee, J.P.; Fix, D.K.; Vanderveen, B.N.; Montalvo, R.N.; Carson, J.A. Cachexia Disrupts Diurnal Regulation of Activity, Feeding, and Muscle Mechanistic Target of Rapamycin Complex 1 in Mice. Med. Sci. Sports Exerc. 2020, 52, 577–587. [Google Scholar] [CrossRef]
- Siu, P.M.; Alway, S.E. Mitochondria-associated apoptotic signalling in denervated rat skeletal muscle. J. Physiol. 2005, 565, 309–323. [Google Scholar] [CrossRef]
- Shen, Y.; Li, M.; Wang, K.; Qi, G.; Liu, H.; Wang, W.; Ji, Y.; Chang, M.; Deng, C.; Xu, F.; et al. Diabetic Muscular Atrophy: Molecular Mechanisms and Promising Therapies. Front. Endocrinol. 2022, 13, 917113. [Google Scholar] [CrossRef]
- Alway, S.E. Mitochondrial Dysfunction: Linking Type 1 Diabetes and Sarcopenia. Exerc. Sport Sci. Rev. 2019, 47, 63. [Google Scholar] [CrossRef]
- Ferrandi, P.J.; Khan, M.M.; Paez, H.G.; Pitzer, C.R.; Alway, S.E.; Mohamed, J.S. Transcriptome Analysis of Skeletal Muscle Reveals Altered Proteolytic and Neuromuscular Junction Associated Gene Expressions in a Mouse Model of Cerebral Ischemic Stroke. Genes 2020, 11, 726. [Google Scholar] [CrossRef]
- Tuntevski, K.; Hajira, A.; Nichols, A.; Alway, S.E.; Mohamed, J.S. Muscle-specific sirtuin1 gain-of-function ameliorates skeletal muscle atrophy in a pre-clinical mouse model of cerebral ischemic stroke. FASEB Bioadv. 2020, 2, 387–397. [Google Scholar] [CrossRef]
- Guglieri, M.; Bushby, K.; McDermott, M.P.; Hart, K.A.; Tawil, R.; Martens, W.B.; Herr, B.E.; McColl, E.; Speed, C.; Wilkinson, J.; et al. Effect of Different Corticosteroid Dosing Regimens on Clinical Outcomes in Boys With Duchenne Muscular Dystrophy: A Randomized Clinical Trial. JAMA 2022, 327, 1456–1468. [Google Scholar] [CrossRef]
- Garibaldi, M.; Nicoletti, T.; Bucci, E.; Fionda, L.; Leonardi, L.; Morino, S.; Tufano, L.; Alfieri, G.; Lauletta, A.; Merlonghi, G.; et al. Muscle magnetic resonance imaging in myotonic dystrophy type 1 (DM1): Refining muscle involvement and implications for clinical trials. Eur. J. Neurol. 2022, 29, 843–854. [Google Scholar] [CrossRef]
- Hsu, W.C.; Lin, Y.C.; Chuang, H.H.; Yeh, K.Y.; Chan, W.P.; Ro, L.S. A Muscle Biosignature Differentiating Between Limb-Girdle Muscular Dystrophy and Idiopathic Inflammatory Myopathy on Magnetic Resonance Imaging. Front. Neurol. 2021, 12, 783095. [Google Scholar] [CrossRef]
- Rodrigues, F.; Domingos, C.; Monteiro, D.; Morouco, P. A Review on Aging, Sarcopenia, Falls, and Resistance Training in Community-Dwelling Older Adults. Int. J. Environ. Res. Public Health 2022, 19, 874. [Google Scholar] [CrossRef]
- van Gameren, M.; Hoogendijk, E.O.; van Schoor, N.M.; Bossen, D.; Visser, B.; Bosmans, J.E.; Pijnappels, M. Physical activity as a risk or protective factor for falls and fall-related fractures in non-frail and frail older adults: A longitudinal study. BMC Geriatr. 2022, 22, 695. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Volmar, C.H.; Crossland, H.; Phillips, B.E.; Sood, S.; Janczura, K.J.; Tormakangas, T.; Kujala, U.M.; Kraus, W.E.; Atherton, P.J.; et al. Longevity-related molecular pathways are subject to midlife “switch” in humans. Aging Cell 2019, 18, e12970. [Google Scholar] [CrossRef] [PubMed]
- Andres-Mateos, E.; Brinkmeier, H.; Burks, T.N.; Mejias, R.; Files, D.C.; Steinberger, M.; Soleimani, A.; Marx, R.; Simmers, J.L.; Lin, B.; et al. Activation of serum/glucocorticoid-induced kinase 1 (SGK1) is important to maintain skeletal muscle homeostasis and prevent atrophy. EMBO Mol. Med. 2013, 5, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Casado, E.; Khraiwesh, H.; Lopez-Dominguez, J.A.; Montero-Guisado, J.; Lopez-Lluch, G.; Navas, P.; de Cabo, R.; Ramsey, J.J.; Gonzalez-Reyes, J.A.; Villalba, J.M. The Impact of Aging, Calorie Restriction and Dietary Fat on Autophagy Markers and Mitochondrial Ultrastructure and Dynamics in Mouse Skeletal Muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 760–769. [Google Scholar] [CrossRef]
- Jun, J.E.; Lee, S.E.; Lee, Y.B.; Kim, G.; Jin, S.M.; Jee, J.H.; Kim, J.H. Low skeletal muscle mass Accompanied by Abdominal Obesity Additively Increases the Risk of Incident Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2022, dgac662. [Google Scholar] [CrossRef]
- Lepper, C.; Partridge, T.A.; Fan, C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 2011, 138, 3639–3646. [Google Scholar] [CrossRef]
- Sambasivan, R.; Yao, R.; Kissenpfennig, A.; Van, W.L.; Paldi, A.; Gayraud-Morel, B.; Guenou, H.; Malissen, B.; Tajbakhsh, S.; Galy, A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 2011, 138, 3647–3656. [Google Scholar] [CrossRef]
- Dilworth, F.J.; Blais, A. Epigenetic regulation of satellite cell activation during muscle regeneration. Stem Cell Res. Ther. 2011, 2, 18. [Google Scholar] [CrossRef]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar]
- Lukjanenko, L.; Jung, M.J.; Hegde, N.; Perruisseau-Carrier, C.; Migliavacca, E.; Rozo, M.; Karaz, S.; Jacot, G.; Schmidt, M.; Li, L.; et al. Loss of fibronectin from the aged stem cell niche affects the regenerative capacity of skeletal muscle in mice. Nat. Med. 2016, 22, 897–905. [Google Scholar] [CrossRef]
- Talbert, E.E.; Guttridge, D.C. Impaired regeneration: A role for the muscle microenvironment in cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 82–91. [Google Scholar] [CrossRef]
- Arthur, S.T.; Cooley, I.D. The effect of physiological stimuli on sarcopenia; impact of Notch and Wnt signaling on impaired aged skeletal muscle repair. Int. J. Biol. Sci. 2012, 8, 731–760. [Google Scholar] [CrossRef]
- Lee, G.; Espirito Santo, A.I.; Zwingenberger, S.; Cai, L.; Vogl, T.; Feldmann, M.; Horwood, N.J.; Chan, J.K.; Nanchahal, J. Fully reduced HMGB1 accelerates the regeneration of multiple tissues by transitioning stem cells to GAlert. Proc. Natl. Acad. Sci. USA 2018, 115, E4463–E4472. [Google Scholar] [CrossRef]
- Joanisse, S.; Nederveen, J.P.; Snijders, T.; McKay, B.R.; Parise, G. Skeletal Muscle Regeneration, Repair and Remodelling in Aging: The Importance of Muscle Stem Cells and Vascularization. Gerontology 2017, 63, 91–100. [Google Scholar] [CrossRef]
- Alway, S.E.; Bennett, B.T.; Wilson, J.C.; Sperringer, J.; Mohamed, J.S.; Edens, N.K.; Pereira, S.L. Green tea extract attenuates muscle loss and improves muscle function during disuse, but fails to improve muscle recovery following unloading in aged rats. J. Appl. Physiol. 2015, 118, 319–330. [Google Scholar] [CrossRef]
- Globus, R.K.; Morey-Holton, E. Hindlimb unloading: Rodent analog for microgravity. J. Appl. Physiol. 2016, 120, 1196–1206. [Google Scholar] [CrossRef]
- Dziki, J.L.; Giglio, R.M.; Sicari, B.M.; Wang, D.S.; Gandhi, R.M.; Londono, R.; Dearth, C.L.; Badylak, S.F. The Effect of Mechanical Loading Upon Extracellular Matrix Bioscaffold-Mediated Skeletal Muscle Remodeling. Tissue Eng. Part A 2017, 24, 34–46. [Google Scholar] [CrossRef]
- Andrianjafiniony, T.; Dupre-Aucouturier, S.; Letexier, D.; Couchoux, H.; Desplanches, D. Oxidative stress, apoptosis, and proteolysis in skeletal muscle repair after unloading. Am. J. Physiol. Cell Physiol. 2010, 299, C307–C315. [Google Scholar] [CrossRef]
- Jang, Y.C.; Sinha, M.; Cerletti, M.; Dall’Osso, C.; Wagers, A.J. Skeletal muscle stem cells: Effects of aging and metabolism on muscle regenerative function. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 101–111. [Google Scholar] [CrossRef]
- Alway, S.E.; McCrory, J.L.; Kearcher, K.; Vickers, A.; Frear, B.; Gilleland, D.L.; Bonner, D.E.; Thomas, J.M.; Donley, D.A.; Lively, M.W.; et al. Resveratrol enhances exercise-induced cellular and functional adaptations of skeletal muscle in older men and women. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1595–1606. [Google Scholar] [CrossRef]
- Alway, S.E.; MacDougall, J.D.; Sale, D.G.; Sutton, J.R.; McComas, A.J. Functional and structural adaptations in skeletal muscle of trained athletes. J. Appl. Physiol. 1988, 64, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Alway, S.E. Is fiber mitochondrial volume density a good indicator of muscle fatigability to isometric exercise? J. Appl. Physiol. 1991, 70, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Delfinis, L.J.; Bellissimo, C.A.; Gandhi, S.; DiBenedetto, S.N.; Garibotti, M.C.; Thuhan, A.K.; Tsitkanou, S.; Rosa-Caldwell, M.E.; Rahman, F.A.; Cheng, A.J.; et al. Muscle weakness precedes atrophy during cancer cachexia and is linked to muscle-specific mitochondrial stress. JCI Insight 2022, 7, e155147. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Alway, S.E.; Mohamed, J.S.; Myers, M.J. Mitochondria Initiate and Regulate Sarcopenia. Exerc. Sport Sci. Rev. 2017, 45, 58–69. [Google Scholar] [CrossRef]
- Triolo, M.; Oliveira, A.N.; Kumari, R.; Hood, D.A. The influence of age, sex, and exercise on autophagy, mitophagy, and lysosome biogenesis in skeletal muscle. Skelet. Muscle 2022, 12, 13. [Google Scholar] [CrossRef]
- Triolo, M.; Slavin, M.; Moradi, N.; Hood, D.A. Time-dependent changes in autophagy, mitophagy and lysosomes in skeletal muscle during denervation-induced disuse. J. Physiol. 2022, 600, 1683–1701. [Google Scholar] [CrossRef]
- Slavin, M.B.; Memme, J.M.; Oliveira, A.N.; Moradi, N.; Hood, D.A. Regulatory networks coordinating mitochondrial quality control in skeletal muscle. Am. J. Physiol. Cell Physiol. 2022, 322, C913–C926. [Google Scholar] [CrossRef]
- Wescott, A.P.; Kao, J.P.Y.; Lederer, W.J.; Boyman, L. Voltage-energized Calcium-sensitive ATP Production by Mitochondria. Nat. Metab. 2019, 1, 975–984. [Google Scholar] [CrossRef]
- Voglhuber, J.; Holzer, M.; Radulovic, S.; Thai, P.N.; Djalinac, N.; Matzer, I.; Wallner, M.; Bugger, H.; Zirlik, A.; Leitinger, G.; et al. Functional remodelling of perinuclear mitochondria alters nucleoplasmic Ca(2+) signalling in heart failure. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2022, 377, 20210320. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef]
- Qiu, J.; Fang, Q.; Xu, T.; Wu, C.; Xu, L.; Wang, L.; Yang, X.; Yu, S.; Zhang, Q.; Ding, F.; et al. Mechanistic Role of Reactive Oxygen Species and Therapeutic Potential of Antioxidants in Denervation- or Fasting-Induced Skeletal Muscle Atrophy. Front. Physiol. 2018, 9, 215. [Google Scholar] [CrossRef]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef]
- Ryan, M.J.; Jackson, J.R.; Hao, Y.; Williamson, C.L.; Dabkowski, E.R.; Hollander, J.M.; Alway, S.E. Suppression of oxidative stress by resveratrol after isometric contractions in gastrocnemius muscles of aged mice. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 815–831. [Google Scholar] [CrossRef]
- Del Campo, A.; Contreras-Hernandez, I.; Castro-Sepulveda, M.; Campos, C.A.; Figueroa, R.; Tevy, M.F.; Eisner, V.; Casas, M.; Jaimovich, E. Muscle function decline and mitochondria changes in middle age precede sarcopenia in mice. Aging 2018, 10, 34–55. [Google Scholar] [CrossRef]
- Migliavacca, E.; Tay, S.K.H.; Patel, H.P.; Sonntag, T.; Civiletto, G.; McFarlane, C.; Forrester, T.; Barton, S.J.; Leow, M.K.; Antoun, E.; et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 2019, 10, 5808. [Google Scholar] [CrossRef]
- Capitanio, D.; Vasso, M.; De, P.S.; Fania, C.; Torretta, E.; Cammarata, F.P.; Magnaghi, V.; Procacci, P.; Gelfi, C. Specific protein changes contribute to the differential muscle mass loss during ageing. Proteomics 2016, 16, 645–656. [Google Scholar] [CrossRef]
- Myers, M.J.; Shepherd, D.L.; Durr, A.J.; Stanton, D.S.; Mohamed, J.S.; Hollander, J.M.; Alway, S.E. The role of SIRT1 in skeletal muscle function and repair of older mice. J. Cachexia Sarcopenia Muscle 2019, 10, 929–949. [Google Scholar] [CrossRef]
- Huang, Y.; Zhu, X.; Chen, K.; Lang, H.; Zhang, Y.; Hou, P.; Ran, L.; Zhou, M.; Zheng, J.; Yi, L.; et al. Resveratrol prevents sarcopenic obesity by reversing mitochondrial dysfunction and oxidative stress via the PKA/LKB1/AMPK pathway. Aging 2019, 11, 2217–2240. [Google Scholar] [CrossRef]
- de Castro, G.S.; Simoes, E.; Lima, J.; Ortiz-Silva, M.; Festuccia, W.T.; Tokeshi, F.; Alcantara, P.S.; Otoch, J.P.; Coletti, D.; Seelaender, M. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers 2019, 11, 1264. [Google Scholar] [CrossRef]
- Romanello, V.; Scalabrin, M.; Albiero, M.; Blaauw, B.; Scorrano, L.; Sandri, M. Inhibition of the Fission Machinery Mitigates OPA1 Impairment in Adult Skeletal Muscles. Cells 2019, 8, 597. [Google Scholar] [CrossRef] [PubMed]
- Abrigo, J.; Simon, F.; Cabrera, D.; Vilos, C.; Cabello-Verrugio, C. Mitochondrial Dysfunction in Skeletal Muscle Pathologies. Curr. Protein Pept. Sci. 2019, 20, 536–546. [Google Scholar] [CrossRef]
- Ji, L.L.; Yeo, D. Mitochondrial dysregulation and muscle disuse atrophy. F1000Researh 2019, 8, 1621. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, H.; Deminice, R.; Yoshihara, T.; Powers, S.K. Mitochondrial dysfunction induces muscle atrophy during prolonged inactivity: A review of the causes and effects. Arch. Biochem. Biophys. 2018, 662, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Adan, M.A.; Kapral, M.C.; Kressin, K.A.; Leathrum, C.M.; Seo, A.; Li, S.; Schaffrey, E.C. Neuromuscular adaptability of male and female rats to muscle unloading. J. Neurosci. Res. 2018, 96, 284–296. [Google Scholar] [CrossRef]
- Anderson, J.E. Key concepts in muscle regeneration: Muscle “cellular ecology” integrates a gestalt of cellular cross-talk, motility, and activity to remodel structure and restore function. Eur. J. Appl. Physiol. 2022, 122, 273–300. [Google Scholar] [CrossRef]
- Daneshvar, N.; Tatsumi, R.; Peeler, J.; Anderson, J.E. Premature satellite cell activation before injury accelerates myogenesis and disrupts neuromuscular junction maturation in regenerating muscle. Am. J. Physiol. Cell Physiol. 2020, 319, C116–C128. [Google Scholar] [CrossRef]
- Wei, X.; Luo, L.; Chen, J. Roles of mTOR Signaling in Tissue Regeneration. Cells 2019, 8, 1075. [Google Scholar] [CrossRef]
- Huard, J.; Lu, A.; Mu, X.; Guo, P.; Li, Y. Muscle Injuries and Repair: What’s New on the Horizon! Cells Tissues Organs. 2016, 202, 227–236. [Google Scholar] [CrossRef]
- Rahman, F.A.; Quadrilatero, J. Mitochondrial network remodeling: An important feature of myogenesis and skeletal muscle regeneration. Cell Mol. Life Sci. 2021, 78, 4653–4675. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, J.; Liu, Y. Skeletal Muscle Regeneration in Cardiotoxin-Induced Muscle Injury Models. Int. J. Mol. Sci. 2022, 23, 13380. [Google Scholar] [CrossRef]
- Brooks, M.J.; Hajira, A.; Mohamed, J.S.; Alway, S.E. Voluntary wheel running increases satellite cell abundance and improves recovery from disuse in gastrocnemius muscles from mice. J. Appl. Physiol. 2018, 124, 1616–1628. [Google Scholar] [CrossRef]
- Ye, Q.; Qiu, X.; Wang, J.; Xu, B.; Su, Y.; Zheng, C.; Gui, L.; Yu, L.; Kuang, H.; Liu, H.; et al. MSCs-derived apoptotic extracellular vesicles promote muscle regeneration by inducing Pannexin 1 channel-dependent creatine release by myoblasts. Int. J. Oral Sci. 2023, 15, 7. [Google Scholar] [CrossRef]
- Dubovskii, P.V.; Ignatova, A.A.; Alekseeva, A.S.; Starkov, V.G.; Boldyrev, I.A.; Feofanov, A.V.; Utkin, Y.N. Membrane-Disrupting Activity of Cobra Cytotoxins Is Determined by Configuration of the N-Terminal Loop. Toxins 2022, 15, 6. [Google Scholar] [CrossRef]
- Tarban, N.; Halasz, H.; Gogolak, P.; Garabuczi, E.; Moise, A.R.; Palczewski, K.; Sarang, Z.; Szondy, Z. Regenerating Skeletal Muscle Compensates for the Impaired Macrophage Functions Leading to Normal Muscle Repair in Retinol Saturase Null Mice. Cells 2022, 11, 1333. [Google Scholar] [CrossRef]
- Hardy, D.; Besnard, A.; Latil, M.; Jouvion, G.; Briand, D.; Thepenier, C.; Pascal, Q.; Guguin, A.; Gayraud-Morel, B.; Cavaillon, J.M.; et al. Comparative Study of Injury Models for Studying Muscle Regeneration in Mice. PLoS ONE 2016, 11, e0147198. [Google Scholar] [CrossRef]
- Morton, A.B.; Norton, C.E.; Jacobsen, N.L.; Fernando, C.A.; Cornelison, D.D.W.; Segal, S.S. Barium chloride injures myofibers through calcium-induced proteolysis with fragmentation of motor nerves and microvessels. Skelet. Muscle 2019, 9, 27. [Google Scholar] [CrossRef]
- George, R.M.; Biressi, S.; Beres, B.J.; Rogers, E.; Mulia, A.K.; Allen, R.E.; Rawls, A.; Rando, T.A.; Wilson-Rawls, J. Numb-deficient satellite cells have regeneration and proliferation defects. Proc. Natl. Acad. Sci. USA 2013, 110, 18549–18554. [Google Scholar] [CrossRef]
- Alway, S.E.; Paez, H.G.; Pitzer, C.R.; Ferrandi, P.J.; Khan, M.M.; Mohamed, J.S.; Carson, J.A.; Deschenes, M.R. Mitochondria transplant therapy improves regeneration and restoration of injured skeletal muscle. J. Cachexia Sarcopenia Muscle 2023, 14, 493–507. [Google Scholar] [CrossRef]
- Jung, H.W.; Choi, J.H.; Jo, T.; Shin, H.; Suh, J.M. Systemic and Local Phenotypes of Barium Chloride Induced Skeletal Muscle Injury in Mice. Ann. Geriatr. Med. Res. 2019, 23, 83–89. [Google Scholar] [CrossRef]
- Saito, Y.; Nonaka, I. Initiation of satellite cell replication in bupivacaine-induced myonecrosis. Acta Neuropathol. 1994, 88, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Corbiere, T.F.; Weinheimer-Haus, E.M.; Judex, S.; Koh, T.J. Low-Intensity Vibration Improves Muscle Healing in a Mouse Model of Laceration Injury. J. Funct. Morphol. Kinesiol. 2018, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.L.; Weinheimer-Haus, E.M.; Koh, T.J. Macrophage activation and skeletal muscle healing following traumatic injury. J. Pathol. 2014, 232, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Dongol, A.; Sapkota, A.; Devkota, R.; Pandey, A.; Bhattarai, T.R. Multiple Wasp Stings Leading to Rhabdomyolysis Induced Acute Kidney Injury with Incidental Ectopic Kidney: A Case Report. JNMA J. Nepal Med. Assoc. 2022, 60, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Gao, Z.; Chen, G.; Peng, C.; Sun, Y.; Jiang, B.; Zhou, H.; Cheng, Y.; Hu, F.; Zhang, Q. An integrative proteomics metabolomics based strategy reveals the mechanisms involved in wasp sting induced acute kidney injury. Toxicon 2022, 205, 1–10. [Google Scholar] [CrossRef]
- Ou, W.F.; Huang, W.H.; Chiu, H.F.; Mao, Y.C.; Wen, M.C.; Chen, C.H.; Hung, S.J.; Wu, M.J.; Wu, C.L.; Chao, W.C. Clinical manifestation of multiple wasp stings with details of whole transcriptome analysis: Two case reports. Medicine 2021, 100, e24492. [Google Scholar] [CrossRef]
- Nichenko, A.S.; Southern, W.M.; Tehrani, K.F.; Qualls, A.E.; Flemington, A.B.; Mercer, G.H.; Yin, A.; Mortensen, L.J.; Yin, H.; Call, J.A. Mitochondrial-specific autophagy linked to mitochondrial dysfunction following traumatic freeze injury in mice. Am. J. Physiol. Cell Physiol. 2020, 318, C242–C252. [Google Scholar] [CrossRef]
- Yoon, N.; Chu, V.; Gould, M.; Zhang, M. Spatial and temporal changes in myogenic protein expression by the microenvironment after freeze injury. J. Anat. 2019, 234, 359–367. [Google Scholar] [CrossRef]
- Le, G.; Lowe, D.A.; Kyba, M. Freeze Injury of the Tibialis Anterior Muscle. Methods Mol. Biol. 2016, 1460, 33–41. [Google Scholar] [CrossRef]
- Pollot, B.E.; Corona, B.T. Volumetric Muscle Loss. Methods Mol. Biol. 2016, 1460, 19–31. [Google Scholar] [CrossRef]
- Corona, B.T.; Rivera, J.C.; Owens, J.G.; Wenke, J.C.; Rathbone, C.R. Volumetric muscle loss leads to permanent disability following extremity trauma. J. Rehabil. Res. Dev. 2015, 52, 785–792. [Google Scholar] [CrossRef]
- Wang, H.D.; Lough, D.M.; Kurlander, D.E.; Lopez, J.; Quan, A.; Kumar, A.R. Muscle-Derived Stem Cell-Enriched Scaffolds Are Capable of Enhanced Healing of a Murine Volumetric Muscle Loss Defect. Plast. Reconstr. Surg. 2019, 143, 329e. [Google Scholar] [CrossRef]
- Raymond-Pope, C.J.; Basten, A.M.; Bruzina, A.S.; McFaline-Figueroa, J.; Lillquist, T.J.; Call, J.A.; Greising, S.M. Restricted physical activity after volumetric muscle loss alters whole-body and local muscle metabolism. J. Physiol. 2023, 601, 743–761. [Google Scholar] [CrossRef]
- Pavlath, G.K.; Dominov, J.A.; Kegley, K.M.; Miller, J.B. Regeneration of transgenic skeletal muscles with altered timing of expression of the basic helix-loop-helix muscle regulatory factor MRF4. Am. J. Pathol. 2003, 162, 1685–1691. [Google Scholar] [CrossRef]
- Nakasa, T.; Ishikawa, M.; Shi, M.; Shibuya, H.; Adachi, N.; Ochi, M. Acceleration of muscle regeneration by local injection of muscle-specific microRNAs in rat skeletal muscle injury model. J. Cell Mol. Med. 2010, 14, 2495–2505. [Google Scholar] [CrossRef]
- Tidball, J.G. Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol. 2011, 1, 2029–2062. [Google Scholar] [CrossRef]
- Thooyamani, A.S.; Mukhopadhyay, A. PDGFRalpha mediated survival of myofibroblasts inhibit satellite cell proliferation during aberrant regeneration of lacerated skeletal muscle. Sci. Rep. 2021, 11, 63. [Google Scholar] [CrossRef]
- Martin, K.S.; Kegelman, C.D.; Virgilio, K.M.; Passipieri, J.A.; Christ, G.J.; Blemker, S.S.; Peirce, S.M. In Silico and In Vivo Experiments Reveal M-CSF Injections Accelerate Regeneration Following Muscle Laceration. Ann. Biomed. Eng. 2017, 45, 747–760. [Google Scholar] [CrossRef]
- Park, J.K.; Ki, M.R.; Lee, E.M.; Kim, A.Y.; You, S.Y.; Han, S.Y.; Lee, E.J.; Hong, I.H.; Kwon, S.H.; Kim, S.J.; et al. Losartan improves adipose tissue-derived stem cell niche by inhibiting transforming growth factor-beta and fibrosis in skeletal muscle injury. Cell Transplant. 2012, 21, 2407–2424. [Google Scholar] [CrossRef]
- Quintana, H.T.; Baptista, V.I.A.; Lazzarin, M.C.; Antunes, H.K.M.; Le Sueur-Maluf, L.; de Oliveira, C.A.M.; de Oliveira, F. Insulin Modulates Myogenesis and Muscle Atrophy Resulting From Skin Scald Burn in Young Male Rats. J. Surg. Res. 2021, 257, 56–68. [Google Scholar] [CrossRef]
- Clark, A.T.; Song, J.; Yao, X.; Carlson, D.; Huebinger, R.M.; Mei Liu, M.; Madni, T.D.; Imran, J.B.; Taveras, L.R.; Weis, H.B.; et al. Muscle Homeostasis Is Disrupted in Burned Adults. J. Burn Care Res. 2020, 41, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Linard, C.; Brachet, M.; L’Homme, B.; Strup-Perrot, C.; Busson, E.; Bonneau, M.; Lataillade, J.J.; Bey, E.; Benderitter, M. Long-term effectiveness of local BM-MSCs for skeletal muscle regeneration: A proof of concept obtained on a pig model of severe radiation burn. Stem Cell Res. Ther. 2018, 9, 299. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Porter, C.; Sidossis, L.S.; Nieten, C.; Reidy, P.T.; Hundeshagen, G.; Mlcak, R.; Rasmussen, B.B.; Lee, J.O.; Suman, O.E.; et al. Satellite cell activation and apoptosis in skeletal muscle from severely burned children. J. Physiol. 2016, 594, 5223–5236. [Google Scholar] [CrossRef] [PubMed]
- Haugk, K.L.; Roeder, R.A.; Garber, M.J.; Schelling, G.T. Regulation of muscle cell proliferation by extracts from crushed muscle. J. Anim. Sci. 1995, 73, 1972–1981. [Google Scholar] [CrossRef] [PubMed]
- Schultz, E.; Jaryszak, D.L.; Valliere, C.R. Response of satellite cells to focal skeletal muscle injury. Muscle Nerve 1985, 8, 217–222. [Google Scholar] [CrossRef]
- Stauber, W.T.; Fritz, V.K.; Burkovskaya, T.E.; Ilyina-Kakueva, E.I. Effect of injury on mast cells of rat gastrocnemius muscle with respect to gravitational exposure. Exp. Mol. Pathol. 1993, 59, 87–94. [Google Scholar] [CrossRef]
- Bischoff, R. A satellite cell mitogen from crushed adult muscle. Dev. Biol. 1986, 115, 140–147. [Google Scholar] [CrossRef]
- Bischoff, R.; Heintz, C. Enhancement of skeletal muscle regeneration. Dev. Dyn. 1994, 201, 41–54. [Google Scholar] [CrossRef]
- Bischoff, R. Chemotaxis of skeletal muscle satellite cells. Dev. Dyn. 1997, 208, 505–515. [Google Scholar] [CrossRef]
- Gorecka, A.; Salemi, S.; Haralampieva, D.; Moalli, F.; Stroka, D.; Candinas, D.; Eberli, D.; Brugger, L. Autologous transplantation of adipose-derived stem cells improves functional recovery of skeletal muscle without direct participation in new myofiber formation. Stem Cell Res. Ther. 2018, 9, 195. [Google Scholar] [CrossRef]
- Hesselink, M.K.; Kuipers, H.; Geurten, P.; Van Straaten, H. Structural muscle damage and muscle strength after incremental number of isometric and forced lengthening contractions. J. Muscle Res. Cell Motil. 1996, 17, 335–341. [Google Scholar] [CrossRef]
- Clarkson, P.M.; Hubal, M.J. Exercise-induced muscle damage in humans. Am. J. Phys. Med. Rehabil. 2002, 81, S52–S69. [Google Scholar] [CrossRef]
- Kim, M.; Chun, J.; Jung, H.A.; Choi, J.S.; Kim, Y.S. Capillarisin attenuates exercise-induced muscle damage through MAPK and NF-kappaB signaling. Phytomedicine 2017, 32, 30–36. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, X.; Miao, R.; Cao, W.; Wei, H.; Jiang, W.; Gao, R.; Yang, Y.; Sun, H.; Qiu, J. Branched-chain amino acids promotes the repair of exercise-induced muscle damage via enhancing macrophage polarization. Front. Physiol. 2022, 13, 1037090. [Google Scholar] [CrossRef]
- Armstrong, R.B. Initial events in exercise-induced muscular injury. Med. Sci. Sports Exerc. 1990, 22, 429–435. [Google Scholar]
- Warren, G.L.; Ingalls, C.P.; Lowe, D.A.; Armstrong, R.B. What mechanisms contribute to the strength loss that occurs during and in the recovery from skeletal muscle injury? J. Orthop. Sports Phys. Ther. 2002, 32, 58–64. [Google Scholar] [CrossRef]
- Warren, G.L.; Lowe, D.A.; Hayes, D.A.; Karwoski, C.J.; Prior, B.M.; Armstrong, R.B. Excitation failure in eccentric contraction-induced injury of mouse soleus muscle. J. Physiol. 1993, 468, 487–499. [Google Scholar] [CrossRef]
- Krajnak, K.; Waugh, S.; Miller, R.; Baker, B.; Geronilla, K.; Alway, S.E.; Cutlip, R.G. Proapoptotic factor Bax is increased in satellite cells in the tibialis anterior muscles of old rats. Muscle Nerve 2006, 34, 720–730. [Google Scholar] [CrossRef]
- Bryner, R.W.; Donley, D.A.; Cutlip, R.G.; Wirth, O.; Alway, S.E. Effects of downhill treadmill running on uncoupling protein 3 mRNA expression. Int. J. Sports Med. 2004, 25, 433–437. [Google Scholar] [CrossRef]
- Stauber, W.T.; Clarkson, P.M.; Fritz, V.K.; Evans, W.J. Extracellular matrix disruption and pain after eccentric muscle action. J. Appl. Physiol. 1990, 69, 868–874. [Google Scholar] [CrossRef]
- Smith, C.A.; Stauber, F.; Waters, C.; Alway, S.E.; Stauber, W.T. Transforming growth factor-beta following skeletal muscle strain injury in rats. J. Appl. Physiol. 2007, 102, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Murlasits, Z.; Cutlip, R.G.; Geronilla, K.B.; Rao, K.M.; Wonderlin, W.F.; Alway, S.E. Resistance training increases heat shock protein levels in skeletal muscle of young and old rats. Exp. Gerontol 2006, 41, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Rader, E.P.; Baker, B.A. Inflammaging and the Age-Specific Responsiveness to Stretch-Shortening Contractions. Exerc. Sport Sci. Rev. 2017, 45, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.A.; Cutlip, R.G. Skeletal muscle injury versus adaptation with aging: Novel insights on perplexing paradigms. Exerc. Sport Sci. Rev. 2010, 38, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.A.; Mercer, R.R.; Geronilla, K.B.; Kashon, M.L.; Miller, G.R.; Cutlip, R.G. Impact of repetition number on muscle performance and histological response. Med. Sci. Sports Exerc. 2007, 39, 1275–1281. [Google Scholar] [CrossRef]
- Cutlip, R.G.; Hollander, M.S.; Johnson, G.A.; Johnson, B.W.; Friend, S.A.; Baker, B.A. Magnetic resonance imaging of graded skeletal muscle injury in live rats. Environ. Health Insights 2014, 8, 31–39. [Google Scholar] [CrossRef]
- Waldemer-Streyer, R.J.; Kim, D.; Chen, J. Muscle cell-derived cytokines in skeletal muscle regeneration. FEBS J. 2022, 289, 6463–6483. [Google Scholar] [CrossRef]
- Mosele, F.C.; Bissi Ricci, R.; Abreu, P.; Rosa Neto, J.C. Muscle regeneration in adiponectin knockout mice showed early activation of anti-inflammatory response with perturbations in myogenesis. J. Cell Physiol. 2020, 235, 6183–6193. [Google Scholar] [CrossRef]
- Caballero-Sanchez, N.; Alonso-Alonso, S.; Nagy, L. Regenerative inflammation: When immune cells help to re-build tissues. FEBS J. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Tidball, J.G.; Flores, I.; Welc, S.S.; Wehling-Henricks, M.; Ochi, E. Aging of the immune system and impaired muscle regeneration: A failure of immunomodulation of adult myogenesis. Exp. Gerontol. 2021, 145, 111200. [Google Scholar] [CrossRef]
- Welc, S.S.; Wehling-Henricks, M.; Kuro, O.M.; Thomas, K.A.; Tidball, J.G. Modulation of Klotho expression in injured muscle perturbs Wnt signalling and influences the rate of muscle growth. Exp. Physiol. 2020, 105, 132–147. [Google Scholar] [CrossRef]
- Welc, S.S.; Wehling-Henricks, M.; Antoun, J.; Ha, T.T.; Tous, I.; Tidball, J.G. Differential Effects of Myeloid Cell PPARdelta and IL-10 in Regulating Macrophage Recruitment, Phenotype, and Regeneration following Acute Muscle Injury. J. Immunol. 2020, 205, 1664–1677. [Google Scholar] [CrossRef]
- Tidball, J.G.; Villalta, S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef]
- Le Moal, E.; Pialoux, V.; Juban, G.; Groussard, C.; Zouhal, H.; Chazaud, B.; Mounier, R. Redox Control of Skeletal Muscle Regeneration. Antioxid. Redox Signal. 2017, 27, 276–310. [Google Scholar] [CrossRef]
- Minari, A.L.A.; Thomatieli-Santos, R.V. From skeletal muscle damage and regeneration to the hypertrophy induced by exercise: What is the role of different macrophage subsets? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2022, 322, R41–R54. [Google Scholar] [CrossRef]
- Miyazaki, A.; Kawashima, M.; Nagata, I.; Miyoshi, M.; Miyakawa, M.; Sugiyama, M.; Sakuraya, T.; Sonomura, T.; Arakawa, T. Icing after skeletal muscle injury decreases M1 macrophage accumulation and TNF-alpha expression during the early phase of muscle regeneration in rats. Histochem. Cell Biol. 2022, 159, 77–89. [Google Scholar] [CrossRef]
- Walton, R.G.; Kosmac, K.; Mula, J.; Fry, C.S.; Peck, B.D.; Groshong, J.S.; Finlin, B.S.; Zhu, B.; Kern, P.A.; Peterson, C.A. Human skeletal muscle macrophages increase following cycle training and are associated with adaptations that may facilitate growth. Sci. Rep. 2019, 9, 969. [Google Scholar] [CrossRef]
- Szondy, Z.; Al-Zaeed, N.; Tarban, N.; Fige, E.; Garabuczi, E.; Sarang, Z. Involvement of phosphatidylserine receptors in the skeletal muscle regeneration: Therapeutic implications. J. Cachexia Sarcopenia Muscle 2022, 13, 1961–1973. [Google Scholar] [CrossRef]
- Hwang, A.B.; Brack, A.S. Muscle Stem Cells and Aging. Curr. Top. Dev. Biol. 2018, 126, 299–322. [Google Scholar] [CrossRef]
- Chang, N.C.; Rudnicki, M.A. Satellite cells: The architects of skeletal muscle. Curr. Top. Dev. Biol. 2014, 107, 161–181. [Google Scholar]
- von Maltzahn, J.; Jones, A.E.; Parks, R.J.; Rudnicki, M.A. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. USA 2013, 110, 16474–16479. [Google Scholar] [CrossRef] [PubMed]
- Winchester, P.K.; Davis, M.E.; Alway, S.E.; Gonyea, W.J. Satellite cell activation in the stretch-enlarged anterior latissimus dorsi muscle of the adult quail. Am. J. Physiol. Cell Physiol. 1991, 260, C206–C212. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Y.; Wang, Y.; Chen, X.; Wei, L.S.; Wang, H.; Tao, T.; Zhou, Y.W.; Jiang, Z.H.; Qiu, T.T.; Sun, Z.Y.; et al. The thymus regulates skeletal muscle regeneration by directly promoting satellite cell expansion. J. Biol. Chem. 2022, 298, 101516. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.A.; Alway, S.E. Stretch overload-induced satellite cell activation in slow tonic muscle from adult and aged Japanese quail. Am. J. Physiol. Cell Physiol. 1996, 270, C578–C584. [Google Scholar] [CrossRef]
- Hawke, T.J.; Garry, D.J. Myogenic satellite cells: Physiology to molecular biology. J. Appl. Physiol. 2001, 91, 534–551. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef]
- Alway, S.E.; Martyn, J.K.; Ouyang, J.; Chaudhrai, A.; Murlasits, Z.S. Id2 expression during apoptosis and satellite cell activation in unloaded and loaded quail skeletal muscles. Am. J. Physiol. Regul. Integr Comp. Physiol. 2003, 284, R540–R549. [Google Scholar] [CrossRef]
- Collins-Hooper, H.; Woolley, T.E.; Dyson, L.; Patel, A.; Potter, P.; Baker, R.E.; Gaffney, E.A.; Maini, P.K.; Dash, P.R.; Patel, K. Age-related changes in speed and mechanism of adult skeletal muscle stem cell migration. Stem Cells 2012, 30, 1182–1195. [Google Scholar] [CrossRef]
- Collins, C.A.; Olsen, I.; Zammit, P.S.; Heslop, L.; Petrie, A.; Partridge, T.A.; Morgan, J.E. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 2005, 122, 289–301. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Neves, J.; Munoz-Canoves, P. Muscle stem cell aging: Identifying ways to induce tissue rejuvenation. Mech. Ageing Dev. 2020, 188, 111246. [Google Scholar] [CrossRef]
- Peterson, J.M.; Bryner, R.; Alway, S.E. Satellite cell proliferation is reduced in muscles of obese Zucker rats, but restored with loading. Am. J. Physiol. Cell Physiol. 2008, 295, C521–C528. [Google Scholar] [CrossRef]
- Ji, S.; Ma, P.; Cao, X.; Wang, J.; Yu, X.; Luo, X.; Lu, J.; Hou, W.; Zhang, Z.; Yan, Y.; et al. Myoblast-derived exosomes promote the repair and regeneration of injured skeletal muscle in mice. FEBS Open Bio 2022, 12, 2213–2226. [Google Scholar] [CrossRef]
- Alway, S.E.; Winchester, P.K.; Davis, M.E.; Gonyea, W.J. Regionalized adaptations and muscle fiber proliferation in stretch- induced enlargement. J. Appl. Physiol. 1989, 66, 771–781. [Google Scholar] [CrossRef]
- Alway, S.E. Perpetuation of muscle fibers after removal of stretch in the Japanese quail. Am. J. Physiol. Cell Physiol. 1991, 260, C400–C408. [Google Scholar] [CrossRef]
- Alway, S.E.; Starkweather, L.A. Effect of anabolic steroids on new fiber formation and fiber area during stretch-overload. J. Appl Physiol. 1993, 74, 832–837. [Google Scholar] [CrossRef]
- Carson, J.A.; Alway, S.E.; Yamaguchi, M. Time course of hypertrophic adaptations of the anterior latissimus dorsi muscle to stretch overload in aged Japanese quail. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, B391–B398. [Google Scholar] [CrossRef]
- Cai, A.; Hardt, M.; Schneider, P.; Schmid, R.; Lange, C.; Dippold, D.; Schubert, D.W.; Boos, A.M.; Weigand, A.; Arkudas, A.; et al. Myogenic differentiation of primary myoblasts and mesenchymal stromal cells under serum-free conditions on PCL-collagen I-nanoscaffolds. BMC Biotechnol. 2018, 18, 75. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; McNally, E.M. Muscle cell communication in development and repair. Curr. Opin. Pharmacol. 2017, 34, 7–14. [Google Scholar] [CrossRef]
- Fry, C.S.; Kirby, T.J.; Kosmac, K.; McCarthy, J.J.; Peterson, C.A. Myogenic Progenitor Cells Control Extracellular Matrix Production by Fibroblasts during Skeletal Muscle Hypertrophy. Cell Stem Cell 2017, 20, 56–69. [Google Scholar] [CrossRef]
- Alway, S.E. Contractile properties of aged avian muscle after stretch-overload. Mech. Ageing Dev. 1994, 73, 97–112. [Google Scholar] [CrossRef]
- Alway, S.E. Force and contractile characteristics after stretch overload in quail anterior latissimus dorsi muscle. J. Appl. Physiol. 1994, 77, 135–141. [Google Scholar] [CrossRef]
- Alway, S.E.; Coggan, A.R.; Sproul, M.S.; Abduljalil, A.M.; Robitaille, P.M. Muscle torque in young and older untrained and endurance-trained men. J. Gerontol. A Biol. Sci. Med. Sci. 1996, 51, B195–B201. [Google Scholar] [CrossRef] [PubMed]
- Alway, S.E. Attenuation of Ca(2+)-activated ATPase and shortening velocity in hypertrophied fast twitch skeletal muscle from aged Japanese quail. Exp. Gerontol. 2002, 37, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Tomaz da Silva, M.; Santos, A.R.; Koike, T.E.; Nascimento, T.L.; Rozanski, A.; Bosnakovski, D.; Pereira, L.V.; Kumar, A.; Kyba, M.; Miyabara, E.H. The fibrotic niche impairs satellite cell function and muscle regeneration in mouse models of Marfan syndrome. Acta Physiol. 2022, 237, e13889. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Victor, P.; Munoz-Canoves, P. Regenerative decline of stem cells in sarcopenia. Mol. Asp. Med. 2016, 50, 109–117. [Google Scholar] [CrossRef]
- Kim, B.; Kim, J.S.; Yoon, Y.; Santiago, M.C.; Brown, M.D.; Park, J.Y. Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R927–R938. [Google Scholar] [CrossRef]
- Rahman, F.A.; Quadrilatero, J. Emerging role of mitophagy in myoblast differentiation and skeletal muscle remodeling. Semin. Cell Dev. Biol. 2021, 143, 54–65. [Google Scholar] [CrossRef]
- Baechler, B.L.; Bloemberg, D.; Quadrilatero, J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy 2019, 15, 1606–1619. [Google Scholar] [CrossRef]
- Bloemberg, D.; Quadrilatero, J. Autophagy displays divergent roles during intermittent amino acid starvation and toxic stress-induced senescence in cultured skeletal muscle cells. J. Cell Physiol. 2021, 236, 3099–3113. [Google Scholar] [CrossRef]
- Hong, X.; Isern, J.; Campanario, S.; Perdiguero, E.; Ramirez-Pardo, I.; Segales, J.; Hernansanz-Agustin, P.; Curtabbi, A.; Deryagin, O.; Pollan, A.; et al. Mitochondrial dynamics maintain muscle stem cell regenerative competence throughout adult life by regulating metabolism and mitophagy. Cell Stem Cell 2022, 29, 1298–1314.e10. [Google Scholar] [CrossRef]
- Banerji, C.R.S.; Panamarova, M.; Pruller, J.; Figeac, N.; Hebaishi, H.; Fidanis, E.; Saxena, A.; Contet, J.; Sacconi, S.; Severini, S.; et al. Dynamic transcriptomic analysis reveals suppression of PGC1alpha/ERRalpha drives perturbed myogenesis in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2019, 28, 1244–1259. [Google Scholar] [CrossRef]
- Beltra, M.; Pin, F.; Ballaro, R.; Costelli, P.; Penna, F. Mitochondrial Dysfunction in Cancer Cachexia: Impact on Muscle Health and Regeneration. Cells 2021, 10, 3150. [Google Scholar] [CrossRef]
- Silvennoinen, M.; Ahtiainen, J.P.; Hulmi, J.J.; Pekkala, S.; Taipale, R.S.; Nindl, B.C.; Laine, T.; Hakkinen, K.; Selanne, H.; Kyrolainen, H.; et al. PGC-1 isoforms and their target genes are expressed differently in human skeletal muscle following resistance and endurance exercise. Physiol. Rep. 2015, 3, e12563. [Google Scholar] [CrossRef]
- Irazoki, A.; Gordaliza-Alaguero, I.; Frank, E.; Giakoumakis, N.N.; Seco, J.; Palacin, M.; Guma, A.; Sylow, L.; Sebastian, D.; Zorzano, A. Disruption of mitochondrial dynamics triggers muscle inflammation through interorganellar contacts and mitochondrial DNA mislocation. Nat. Commun. 2023, 14, 108. [Google Scholar] [CrossRef]
- Rodriguez-Nuevo, A.; Diaz-Ramos, A.; Noguera, E.; Diaz-Saez, F.; Duran, X.; Munoz, J.P.; Romero, M.; Plana, N.; Sebastian, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37, e96553. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Huang, D.D.; Fan, S.D.; Chen, X.Y.; Yan, X.L.; Zhang, X.Z.; Ma, B.W.; Yu, D.Y.; Xiao, W.Y.; Zhuang, C.L.; Yu, Z. Nrf2 deficiency exacerbates frailty and sarcopenia by impairing skeletal muscle mitochondrial biogenesis and dynamics in an age-dependent manner. Exp. Gerontol. 2019, 119, 61–73. [Google Scholar] [CrossRef]
- Irazoki, A.; Martinez-Vicente, M.; Aparicio, P.; Aris, C.; Alibakhshi, E.; Rubio-Valera, M.; Castellanos, J.; Lores, L.; Palacin, M.; Guma, A.; et al. Coordination of mitochondrial and lysosomal homeostasis mitigates inflammation and muscle atrophy during aging. Aging Cell 2022, 21, e13583. [Google Scholar] [CrossRef]
- Sebastian, D.; Sorianello, E.; Segales, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Munoz, J.P.; Sanchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef]
- Arribat, Y.; Broskey, N.T.; Greggio, C.; Boutant, M.; Conde Alonso, S.; Kulkarni, S.S.; Lagarrigue, S.; Carnero, E.A.; Besson, C.; Canto, C.; et al. Distinct patterns of skeletal muscle mitochondria fusion, fission and mitophagy upon duration of exercise training. Acta Physiol. 2019, 225, e13179. [Google Scholar] [CrossRef] [PubMed]
- Hamel, Y.; Mauvais, F.X.; Madrange, M.; Renard, P.; Lebreton, C.; Nemazanyy, I.; Pelle, O.; Goudin, N.; Tang, X.; Rodero, M.P.; et al. Compromised mitochondrial quality control triggers lipin1-related rhabdomyolysis. Cell Rep. Med. 2021, 2, 100370. [Google Scholar] [CrossRef] [PubMed]
- Monir, N.; Saber, M.M.; Awad, A.S.; Elsherbiny, M.E.; Zaki, H.F. Repression of inflammatory pathways with Boswellia for alleviation of liver injury after renal ischemia reperfusion. Life Sci. 2022, 306, 120799. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Qu, H.; Zhang, H.; Zhong, X. Prunella vulgaris L. Attenuates Experimental Autoimmune Thyroiditis by Inhibiting HMGB1/TLR9 Signaling. Drug Des. Devel Ther. 2021, 15, 4559–4574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sliter, D.A.; Bleck, C.K.E.; Ding, S. Fis1 deficiencies differentially affect mitochondrial quality in skeletal muscle. Mitochondrion 2019, 49, 217–226. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Marzetti, E. Cell Death and Inflammation: The Role of Mitochondria in Health and Disease. Cells 2021, 10, 537. [Google Scholar] [CrossRef]
- Tumburu, L.; Ghosh-Choudhary, S.; Seifuddin, F.T.; Barbu, E.A.; Yang, S.; Ahmad, M.M.; Wilkins, L.H.W.; Tunc, I.; Sivakumar, I.; Nichols, J.S.; et al. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood 2021, 137, 3116–3126. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Bossola, M.; Manes-Gravina, E.; Landi, F.; Bernabei, R.; Marzetti, E. Circulating Mitochondrial DNA at the Crossroads of Mitochondrial Dysfunction and Inflammation During Aging and Muscle Wasting Disorders. Rejuvenation Res. 2018, 21, 350–359. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, C.; Fang, X.; Zhang, N.; Zhang, X.; Zhu, Z. Activation of autophagy inhibits the activation of NLRP3 inflammasome and alleviates sevoflurane-induced cognitive dysfunction in elderly rats. BMC Neurosci. 2023, 24, 9. [Google Scholar] [CrossRef]
- Xia, P.; Shao, Y.Q.; Yu, C.C.; Xie, Y.; Zhou, Z.J. NLRP3 inflammasome up-regulates major histocompatibility complex class I expression and promotes inflammatory infiltration in polymyositis. BMC Immunol. 2022, 23, 39. [Google Scholar] [CrossRef]
- Shi, X.; Qiu, S.; Zhuang, W.; Yuan, N.; Wang, C.; Zhang, S.; Sun, T.; Guo, W.; Gao, F.; Yang, S.; et al. NLRP3-inflammasomes are triggered by age-related hearing loss in the inner ear of mice. Am. J. Transl. Res. 2017, 9, 5611–5618. [Google Scholar]
- Fallaize, D.; Chin, L.-S.; Li, L. Differential submitochondrial localization of PINK1 as a molecular switch for mediating distinct mitochondrial signaling pathways. Cell. Signal. 2015, 27, 2543–2554. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-s.; Saiki, S.; Kawajiri, S.; Sato, F. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Sun, L.; Shen, R.; Agnihotri, S.K.; Chen, Y.; Huang, Z.; Bueler, H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci. Rep. 2018, 8, 383. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Rosazza, T.; Sepulveda-Diaz, J.; Ieang, A.; Hassoun, S.M.; Claire, E.; Mangone, G.; Brice, A.; Michel, P.P.; Corvol, J.C.; et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 2018, 66, 1736–1751. [Google Scholar] [CrossRef]
- Haramizu, S.; Asano, S.; Butler, D.C.; Stanton, D.A.; Hajira, A.; Mohamed, J.S.; Alway, S.E. Dietary resveratrol confers apoptotic resistance to oxidative stress in myoblasts. J. Nutr. Biochem. 2017, 50, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J.; Jackson, J.R.; Hao, Y.; Leonard, S.S.; Alway, S.E. Inhibition of xanthine oxidase reduces oxidative stress and improves skeletal muscle function in response to electrically stimulated isometric contractions in aged mice. Free Radic. Biol. Med. 2011, 51, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Quadrilatero, J.; Alway, S.E.; Dupont-Versteegden, E.E. Skeletal muscle apoptotic response to physical activity: Potential mechanisms for protection. Appl. Physiol. Nutr. Metab. 2011, 36, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Jackson, J.R.; Wang, Y.; Edens, N.; Pereira, S.L.; Alway, S.E. β-Hydroxy-β-methylbutyrate reduces myonuclear apoptosis during recovery from hind limb suspension-induced muscle fiber atrophy in aged rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R701–R715. [Google Scholar] [CrossRef]
- Siu, P.M.; Alway, S.E. Aging alters the reduction of pro-apoptotic signaling in response to loading-induced hypertrophy. Exp. Gerontol. 2006, 41, 175–188. [Google Scholar] [CrossRef]
- Siu, P.M.; Pistilli, E.E.; Alway, S.E. Apoptotic responses to hindlimb suspension in gastrocnemius muscles from young adult and aged rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1015–R1026. [Google Scholar] [CrossRef]
- Siu, P.M.; Alway, S.E. Age-related apoptotic responses to stretch-induced hypertrophy in quail slow-tonic skeletal muscle. Am. J. Physiol. Cell Physiol. 2005, 289, C1105–C1113. [Google Scholar] [CrossRef]
- Oliveira, J.R.S.; Mohamed, J.S.; Myers, M.J.; Brooks, M.J.; Alway, S.E. Effects of hindlimb suspension and reloading on gastrocnemius and soleus muscle mass and function in geriatric mice. Exp. Gerontol. 2019, 115, 19–31. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Joshi, B.; Joshi, J.C.; Mehta, D. Regulation of cGAS Activity and Downstream Signaling. Cells 2022, 11, 2812. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Rongvaux, A. Innate immunity and tolerance toward mitochondria. Mitochondrion 2018, 41, 14–20. [Google Scholar] [CrossRef]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef]
- Song, J.X.; Villagomes, D.; Zhao, H.; Zhu, M. cGAS in nucleus: The link between immune response and DNA damage repair. Front. Immunol. 2022, 13, 1076784. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Fu, Y.; Fang, Y.; Lin, Z.; Yang, L.; Zheng, L.; Hu, H.; Yu, T.; Huang, B.; Chen, S.; Wang, H.; et al. Inhibition of cGAS-Mediated Interferon Response Facilitates Transgene Expression. iScience 2020, 23, 101026. [Google Scholar] [CrossRef]
- Ding, L.; Wang, Q.; Martincuks, A.; Kearns, M.J.; Jiang, T.; Lin, Z.; Cheng, X.; Qian, C.; Xie, S.; Kim, H.J.; et al. STING agonism overcomes STAT3-mediated immunosuppression and adaptive resistance to PARP inhibition in ovarian cancer. J. Immunother. Cancer 2023, 11, e005627. [Google Scholar] [CrossRef]
- Badu-Mensah, A.; Valinski, P.; Parsaud, H.; Hickman, J.J.; Guo, X. Hyperglycemia Negatively Affects IPSC-Derived Myoblast Proliferation and Skeletal Muscle Regeneration and Function. Cells 2022, 11, 3674. [Google Scholar] [CrossRef]
- Yucel, N.; Wang, Y.X.; Mai, T.; Porpiglia, E.; Lund, P.J.; Markov, G.; Garcia, B.A.; Bendall, S.C.; Angelo, M.; Blau, H.M. Glucose Metabolism Drives Histone Acetylation Landscape Transitions that Dictate Muscle Stem Cell Function. Cell Rep. 2019, 27, 3939–3955.e6. [Google Scholar] [CrossRef]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef]
- Saini, A.; Al-Shanti, N.; Sharples, A.P.; Stewart, C.E. Sirtuin 1 regulates skeletal myoblast survival and enhances differentiation in the presence of resveratrol. Exp. Physiol. 2012, 97, 400–418. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef]
- Mathis, D.; Shoelson, S.E. Immunometabolism: An emerging frontier. Nat. Rev. Immunol. 2011, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Mathis, D. Organismal immunometabolism: Advances in both directions. Nat. Rev. Immunol. 2019, 19, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases? Endocr. Rev. 2020, 41, bnaa005. [Google Scholar] [CrossRef] [PubMed]
- Chella Krishnan, K.; Vergnes, L.; Acin-Perez, R.; Stiles, L.; Shum, M.; Ma, L.; Mouisel, E.; Pan, C.; Moore, T.M.; Peterfy, M.; et al. Sex-specific genetic regulation of adipose mitochondria and metabolic syndrome by Ndufv2. Nat. Metab. 2021, 3, 1552–1568. [Google Scholar] [CrossRef] [PubMed]
- Benmoussa, K.; Garaude, J.; Acin-Perez, R. How Mitochondrial Metabolism Contributes to Macrophage Phenotype and Functions. J. Mol. Biol 2018, 430, 3906–3921. [Google Scholar] [CrossRef]
- Curat, C.A.; Miranville, A.; Sengenes, C.; Diehl, M.; Tonus, C.; Busse, R.; Bouloumie, A. From blood monocytes to adipose tissue-resident macrophages: Induction of diapedesis by human mature adipocytes. Diabetes 2004, 53, 1285–1292. [Google Scholar] [CrossRef]
- Garaude, J.; Acin-Perez, R.; Martinez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villan, E.; Hervas-Stubbs, S.; Pelegrin, P.; Sander, L.E.; Enriquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Iborra, S.; Marti-Mateos, Y.; Cook, E.C.L.; Conde-Garrosa, R.; Petcherski, A.; Munoz, M.D.M.; Martinez de Mena, R.; Krishnan, K.C.; Jimenez, C.; et al. Fgr kinase is required for proinflammatory macrophage activation during diet-induced obesity. Nat. Metab. 2020, 2, 974–988. [Google Scholar] [CrossRef]
- Casuso, R.A.; Huertas, J.R. Mitochondrial Functionality in Inflammatory Pathology-Modulatory Role of Physical Activity. Life 2021, 11, 61. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martinez, L.; Sanchez-Diaz, M.; Diaz-Garcia, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e123. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Hidalgo, A. TREM2(+) macrophages are guardians of the heart. Nat. Metab. 2023, 5, 13–15. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Pena-Couso, L.; Munoz-Canoves, P.; Hidalgo, A. Macrophages, Metabolism and Heterophagy in the Heart. Circ. Res. 2022, 130, 418–431. [Google Scholar] [CrossRef]
- Runyan, C.E.; Welch, L.C.; Lecuona, E.; Shigemura, M.; Amarelle, L.; Abdala-Valencia, H.; Joshi, N.; Lu, Z.; Nam, K.; Markov, N.S.; et al. Impaired phagocytic function in CX3CR1(+) tissue-resident skeletal muscle macrophages prevents muscle recovery after influenza A virus-induced pneumonia in old mice. Aging Cell 2020, 19, e13180. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef]
- Mendivil-Alvarado, H.; Sosa-Leon, L.A.; Carvajal-Millan, E.; Astiazaran-Garcia, H. Malnutrition and Biomarkers: A Journey through Extracellular Vesicles. Nutrients 2022, 14, 1002. [Google Scholar] [CrossRef]
- Choi, D.; Kim, S.; Woo, J.; Lee, H.; Kim, H.; Jeon, J.S.; Noh, H.; Han, D.C.; Kim, S.H.; Cho, H.C.; et al. Weight Change Alters the Small RNA Profile of Urinary Extracellular Vesicles in Obesity. Obes. Facts 2022, 15, 292–301. [Google Scholar] [CrossRef]
- Kahn, D.; Perreault, L.; Macias, E.; Zarini, S.; Newsom, S.A.; Strauss, A.; Kerege, A.; Harrison, K.; Snell-Bergeon, J.; Bergman, B.C. Subcellular localisation and composition of intramuscular triacylglycerol influence insulin sensitivity in humans. Diabetologia 2021, 64, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Pathan, M.; Fonseka, P.; Chitti, S.V.; Kang, T.; Sanwlani, R.; Van Deun, J.; Hendrix, A.; Mathivanan, S. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019, 47, D516–D519. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Patel, T.; Freedman, J.E. Circulating Extracellular Vesicles in Human Disease. N. Engl. J. Med. 2018, 379, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, V.A.; Mancini, E.; Brunet, A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J. Cell Sci. 2012, 125, 5597–5608. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.C.; Crescitelli, R.; Cvjetkovic, A.; Belgrano, V.; Olofsson Bagge, R.; Sundfeldt, K.; Ochiya, T.; Kalluri, R.; Lotvall, J. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J. Extracell Vesicles 2019, 8, 1635420. [Google Scholar] [CrossRef]
- Hegyesi, H.; Pallinger, E.; Mecsei, S.; Hornyak, B.; Kovacshazi, C.; Brenner, G.B.; Giricz, Z.; Paloczi, K.; Kittel, A.; Tovari, J.; et al. Circulating cardiomyocyte-derived extracellular vesicles reflect cardiac injury during systemic inflammatory response syndrome in mice. Cell Mol. Life Sci. 2022, 79, 84. [Google Scholar] [CrossRef]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef]
- Lovett, J.A.C.; Durcan, P.J.; Myburgh, K.H. Investigation of Circulating Extracellular Vesicle MicroRNA Following Two Consecutive Bouts of Muscle-Damaging Exercise. Front. Physiol. 2018, 9, 1149. [Google Scholar] [CrossRef]
- Maligianni, I.; Yapijakis, C.; Bacopoulou, F.; Chrousos, G. The Potential Role of Exosomes in Child and Adolescent Obesity. Children 2021, 8, 196. [Google Scholar] [CrossRef]
- Benkafadar, N.; Francois, F.; Affortit, C.; Casas, F.; Ceccato, J.C.; Menardo, J.; Venail, F.; Malfroy-Camine, B.; Puel, J.L.; Wang, J. ROS-Induced Activation of DNA Damage Responses Drives Senescence-Like State in Postmitotic Cochlear Cells: Implication for Hearing Preservation. Mol. Neurobiol. 2019, 56, 5950–5969. [Google Scholar] [CrossRef]
- Harman, D.A. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Vezzoli, M.; Castellani, P.; Corna, G.; Castiglioni, A.; Bosurgi, L.; Monno, A.; Brunelli, S.; Manfredi, A.A.; Rubartelli, A.; Rovere-Querini, P. High-Mobility Group Box 1 Release and Redox Regulation Accompany Regeneration and Remodeling of Skeletal Muscle. Antioxid. Redox Signal. 2011, 15, 2161–2174. [Google Scholar] [CrossRef]
- Le Moal, E.; Juban, G.; Bernard, A.S.; Varga, T.; Policar, C.; Chazaud, B.; Mounier, R. Macrophage-derived superoxide production and antioxidant response following skeletal muscle injury. Free Radic. Biol. Med. 2018, 120, 33–40. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation Mechanisms involved in H2O2 signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Bellot, G.L.; Dong, X.; Lahiri, A.; Sebastin, S.J.; Batinic-Haberle, I.; Pervaiz, S.; Puhaindran, M.E. MnSOD is implicated in accelerated wound healing upon Negative Pressure Wound Therapy (NPWT): A case in point for MnSOD mimetics as adjuvants for wound management. Redox Biol. 2019, 20, 307–320. [Google Scholar] [CrossRef]
- Moreira, O.C.; Estebanez, B.; Martinez-Florez, S.; de Paz, J.A.; Cuevas, M.J.; Gonzalez-Gallego, J. Mitochondrial Function and Mitophagy in the Elderly: Effects of Exercise. Oxid. Med. Cell Longev. 2017, 2017, 2012798. [Google Scholar] [CrossRef]
- Porter, C.; Hurren, N.M.; Cotter, M.V.; Bhattarai, N.; Reidy, P.T.; Dillon, E.L.; Durham, W.J.; Tuvdendorj, D.; Sheffield-Moore, M.; Volpi, E.; et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E224–E232. [Google Scholar] [CrossRef]
- Yan, Z.; Lira, V.A.; Greene, N.P. Exercise training-induced regulation of mitochondrial quality. Exerc. Sport Sci. Rev. 2012, 40, 159–164. [Google Scholar] [CrossRef]
- Ryan, M.J.; Dudash, H.J.; Docherty, M.; Geronilla, K.B.; Baker, B.A.; Haff, G.G.; Cutlip, R.G.; Alway, S.E. Vitamin E and C supplementation reduces oxidative stress, improves antioxidant enzymes, and postive muscle work in chronically loaded muscles of aged rats. Exp. Gerontol. 2010, 45, 882–895. [Google Scholar] [CrossRef]
- Jackson, J.R.; Ryan, M.J.; Alway, S.E. Long-term supplementation with resveratrol alleviates oxidative stress but does not attenuate sarcopenia in aged mice. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, S.D.; Rando, T.A. Stem cell review series: Aging of the skeletal muscle stem cell niche. Aging Cell 2008, 7, 590–598. [Google Scholar] [CrossRef]
- Day, K.; Shefer, G.; Shearer, A.; Yablonka-Reuveni, Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev. Biol. 2010, 340, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Ciciliot, S.; Schiaffino, S. Regeneration of mammalian skeletal muscle. Basic mechanisms and clinical implications. Curr. Pharm. Des. 2010, 16, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Yeo, D.; Kang, C.; Zhang, T. The role of mitochondria in redox signaling of muscle homeostasis. J. Sport Health Sci. 2020, 9, 386–393. [Google Scholar] [CrossRef]
- Siu, P.M.; Wang, Y.; Alway, S.E. Apoptotic signaling induced by H2O2-mediated oxidative stress in differentiated C2C12 myotubes. Life Sci. 2009, 84, 468–481. [Google Scholar] [CrossRef]
- Siu, P.M.; Alway, S.E. Deficiency of the Bax gene attenuates denervation-induced apoptosis. Apoptosis 2006, 11, 967–981. [Google Scholar] [CrossRef]
- Degens, H.; Moore, J.A.; Alway, S.E. Vascular endothelial growth factor, capillarization, and function of the rat plantaris muscle at the onset of hypertrophy. Jpn. J. Physiol. 2003, 53, 181–191. [Google Scholar] [CrossRef]
- Ji, L.L.; Kang, C.; Zhang, Y. Exercise-induced hormesis and skeletal muscle health. Free Radic Biol Med. 2016, 98, 113–122. [Google Scholar] [CrossRef]
- Pearson, T.; McArdle, A.; Jackson, M.J. Nitric oxide availability is increased in contracting skeletal muscle from aged mice, but does not differentially decrease muscle superoxide. Free Radic. Biol. Med. 2015, 78, 82–88. [Google Scholar] [CrossRef]
- Siu, P.M.; Alway, S.E. Response and adaptation of skeletal muscle to denervation stress: The role of apoptosis in muscle loss. Front. Biosci. 2009, 14, 432–452. [Google Scholar] [CrossRef]
- Alway, S.E.; Degens, H.; Krishnamurthy, G.; Chaudhrai, A. Denervation stimulates apoptosis but not Id2 expression in hindlimb muscles of aged rats. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, B687–B697. [Google Scholar] [CrossRef]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef]
- Pollock, N.; Staunton, C.A.; Vasilaki, A.; McArdle, A.; Jackson, M.J. Denervated muscle fibers induce mitochondrial peroxide generation in neighboring innervated fibers: Role in muscle aging. Free Radic. Biol. Med. 2017, 112, 84–92. [Google Scholar] [CrossRef]
- Liu, J.; Peng, Y.; Feng, Z.; Shi, W.; Qu, L.; Li, Y.; Liu, J.; Long, J. Reloading functionally ameliorates disuse-induced muscle atrophy by reversing mitochondrial dysfunction, and similar benefits are gained by administering a combination of mitochondrial nutrients. Free Radic. Biol. Med. 2014, 69, 116–128. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef]
- Jackson, M.J.; Stretton, C.; McArdle, A. Hydrogen peroxide as a signal for skeletal muscle adaptations to exercise: What do concentrations tell us about potential mechanisms? Redox Biol. 2020, 35, 101484. [Google Scholar] [CrossRef]
- Spendiff, S.; Vuda, M.; Gouspillou, G.; Aare, S.; Perez, A.; Morais, J.A.; Jagoe, R.T.; Filion, M.E.; Glicksman, R.; Kapchinsky, S.; et al. Denervation drives mitochondrial dysfunction in skeletal muscle of octogenarians. J. Physiol. 2016, 594, 7361–7379. [Google Scholar] [CrossRef] [PubMed]
- Hepple, R.T.; Rice, C.L. Innervation and neuromuscular control in ageing skeletal muscle. J. Physiol. 2016, 594, 1965–1978. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Quijano, C.; Chen, E.; Liu, H.; Cao, L.; Fergusson, M.M.; Rovira, I.I.; Gutkind, S.; Daniels, M.P.; Komatsu, M.; et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging 2009, 1, 425–437. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by mitochondrial oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef]
- Jackson, M.J. Reactive oxygen species in sarcopenia: Should we focus on excess oxidative damage or defective redox signalling? Mol. Aspects Med. 2016, 50, 33–40. [Google Scholar] [CrossRef]
- Adhihetty, P.J.; O’Leary, M.F.; Chabi, B.; Wicks, K.L.; Hood, D.A. Effect of denervation on mitochondrially mediated apoptosis in skeletal muscle. J. Appl. Physiol. 2007, 102, 1143–1151. [Google Scholar] [CrossRef]
- Scalabrin, M.; Pollock, N.; Staunton, C.A.; Brooks, S.V.; McArdle, A.; Jackson, M.J.; Vasilaki, A. Redox responses in skeletal muscle following denervation. Redox Biol. 2019, 26, 101294. [Google Scholar] [CrossRef]
- Staunton, C.A.; Owen, E.D.; Pollock, N.; Vasilaki, A.; Barrett-Jolley, R.; McArdle, A.; Jackson, M.J. HyPer2 imaging reveals temporal and heterogeneous hydrogen peroxide changes in denervated and aged skeletal muscle fibers in vivo. Sci. Rep. 2019, 9, 14461. [Google Scholar] [CrossRef]
- Cutlip, R.G.; Baker, B.A.; Geronilla, K.B.; Mercer, R.R.; Kashon, M.L.; Miller, G.R.; Murlasits, Z.; Alway, S.E. Chronic exposure to stretch-shortening contractions results in skeletal muscle adaptation in young rats and maladaptation in old rats. Appl. Physiol. Nutr. Metab. 2006, 31, 573–587. [Google Scholar] [CrossRef]
- Escobedo, J.; Pucci, A.M.; Koh, T.J. HSP25 protects skeletal muscle cells against oxidative stress. Free Radic. Biol. Med. 2004, 37, 1455–1462. [Google Scholar] [CrossRef]
- Yang, Y.X.; Fei, W.Y.; Liu, M.S.; Zhang, Y.C.; Gao, R.S.; Hu, Y.Y.; Pang, E.K.; Hou, L. Molecular Hydrogen Promotes Adipose-derived Stem Cell Myogenic Differentiation via Regulation of Mitochondria. Curr. Stem Cell Res. Ther. 2022, 17, 207–213. [Google Scholar] [CrossRef]
- Somwar, R.; Koterski, S.; Sweeney, G.; Sciotti, R.; Djuric, S.; Berg, C.; Trevillyan, J.; Scherer, P.E.; Rondinone, C.M.; Klip, A. A dominant-negative p38 MAPK mutant and novel selective inhibitors of p38 MAPK reduce insulin-stimulated glucose uptake in 3T3-L1 adipocytes without affecting GLUT4 translocation. J. Biol. Chem. 2002, 277, 50386–50395. [Google Scholar] [CrossRef]
- Somwar, R.; Perreault, M.; Kapur, S.; Taha, C.; Sweeney, G.; Ramlal, T.; Kim, D.Y.; Keen, J.; Cote, C.H.; Klip, A.; et al. Activation of p38 mitogen-activated protein kinase alpha and beta by insulin and contraction in rat skeletal muscle: Potential role in the stimulation of glucose transport. Diabetes 2000, 49, 1794–1800. [Google Scholar] [CrossRef]
- Sweeney, G.; Somwar, R.; Ramlal, T.; Volchuk, A.; Ueyama, A.; Klip, A. An inhibitor of p38 mitogen-activated protein kinase prevents insulin-stimulated glucose transport but not glucose transporter translocation in 3T3-L1 adipocytes and L6 myotubes. J. Biol. Chem. 1999, 274, 10071–10078. [Google Scholar] [CrossRef]
- Olson, L.C.; Redden, J.T.; Gilliam, L.; Nguyen, T.M.; Vossen, J.A.; Cohen, D.J.; Schwartz, Z.; McClure, M.J. Human Adipose-Derived Stromal Cells Delivered on Decellularized Muscle Improve Muscle Regeneration and Regulate RAGE and P38 MAPK. Bioengineering 2022, 9, 426. [Google Scholar] [CrossRef]
- Motawi, T.K.; El-Maraghy, S.A.; Kamel, A.S.; Said, S.E.; Kortam, M.A. Modulation of p38 MAPK and Nrf2/HO-1/NLRP3 inflammasome signaling and pyroptosis outline the anti-neuroinflammatory and remyelinating characters of Clemastine in EAE rat model. Biochem. Pharmacol. 2023, 209, 115435. [Google Scholar] [CrossRef]
- Liu, X.; Gao, J.; Yan, Y.; Georgiou, E.A.; Lou, J.; Feng, M.; Zhang, X.; Gao, F.; Liu, J.; Kostakis, I.K.; et al. Mitochondria-Targeted Triphenylphosphonium-Hydroxytyrosol Prevents Lipotoxicity-Induced Endothelial Injury by Enhancing Mitochondrial Function and Redox Balance via Promoting FoxO1 and Nrf2 Nuclear Translocation and Suppressing Inflammation via Inhibiting p38/NF-small ka, CyrillicB Pathway. Antioxidants 2023, 12, 175. [Google Scholar] [CrossRef]
- Pryce, B.R.; Labreche, C.; Hamoudi, D.; Abou-Hamad, J.; Al-Zahrani, K.N.; Hodgins, J.J.; Boulanger-Piette, A.; Bosse, S.; Balog-Alvarez, C.; Frenette, J.; et al. Muscle-specific deletion of SLK/Stk2 enhances p38 activity and myogenesis in mdx mice. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118917. [Google Scholar] [CrossRef]
- Ryu, M.; Kim, M.; Jung, H.Y.; Kim, C.H.; Jo, C. Effect of p38 inhibitor on the proliferation of chicken muscle stem cells and differentiation into muscle and fat. Anim Biosci. 2023, 36, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.M.; Emerson, C.P., Jr.; Owens, J.; Christoforou, N. p38 MAPKs—Roles in skeletal muscle physiology, disease mechanisms, and as potential therapeutic targets. JCI Insight 2021, 6, e149915. [Google Scholar] [CrossRef] [PubMed]
- Nakai, N.; Kawano, F.; Oke, Y.; Nomura, S.; Ohira, T.; Fujita, R.; Ohira, Y. Mechanical stretch activates signaling events for protein translation initiation and elongation in C2C12 myoblasts. Mol. Cells 2010, 30, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Liu, D.; Liu, J.; Gao, H.; Yuan, X.; Shen, G. p38 mitogen-activated protein kinase up-regulates NF-kappaB transcriptional activation through RelA phosphorylation during stretch-induced myogenesis. Biochem. Biophys. Res. Commun. 2010, 391, 547–551. [Google Scholar] [CrossRef]
- Troy, A.; Cadwallader, A.B.; Fedorov, Y.; Tyner, K.; Tanaka, K.K.; Olwin, B.B. Coordination of Satellite Cell Activation and Self-Renewal by Par-Complex-Dependent Asymmetric Activation of p38alpha/beta MAPK. Cell Stem Cell 2012, 11, 541–553. [Google Scholar] [CrossRef]
- Shirvani, H.; Mirnejad, R.; Soleimani, M.; Arabzadeh, E. Swimming exercise improves gene expression of PPAR-gamma and downregulates the overexpression of TLR4, MyD88, IL-6, and TNF-alpha after high-fat diet in rat skeletal muscle cells. Gene 2021, 775, 145441. [Google Scholar] [CrossRef]
- Thirupathi, A.; de Souza, C.T. Multi-regulatory network of ROS: The interconnection of ROS, PGC-1 alpha, and AMPK-SIRT1 during exercise. J. Physiol. Biochem. 2017, 73, 487–494. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef]
- Nalbandian, M.; Takeda, M. Lactate as a Signaling Molecule That Regulates Exercise-Induced Adaptations. Biology 2016, 5, 38. [Google Scholar] [CrossRef]
- Broome, S.C.; Pham, T.; Braakhuis, A.J.; Narang, R.; Wang, H.W.; Hickey, A.J.R.; Mitchell, C.J.; Merry, T.L. MitoQ supplementation augments acute exercise-induced increases in muscle PGC1alpha mRNA and improves training-induced increases in peak power independent of mitochondrial content and function in untrained middle-aged men. Redox Biol. 2022, 53, 102341. [Google Scholar] [CrossRef]
- Kang, C.; O’Moore, K.M.; Dickman, J.R.; Ji, L.L. Exercise activation of muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling is redox sensitive. Free Radic. Biol. Med. 2009, 47, 1394–1400. [Google Scholar] [CrossRef]
- Yuan, Z.; Yang, M.; Liang, Z.; Yang, C.; Kong, X.; Wu, Y.; Wang, S.; Fan, H.; Ning, C.; Xiao, W.; et al. PI3K/AKT/mTOR, NF-kappaB and ERS pathway participated in the attenuation of H(2)O(2)-induced IPEC-J2 cell injury by koumine. J. Ethnopharmacol. 2022, 304, 116028. [Google Scholar] [CrossRef]
- Wang, Q.; Shi, Q.; Liu, L.; Qian, Y.; Dong, N. FGF10 mediates protective anti-oxidative effects in particulate matter-induced lung injury through Nrf2 and NF-kappaB signaling. Ann. Transl. Med. 2022, 10, 1203. [Google Scholar] [CrossRef]
- Liu, J.; Jia, S.; Yang, Y.; Piao, L.; Wang, Z.; Jin, Z.; Bai, L. Exercise induced meteorin-like protects chondrocytes against inflammation and pyroptosis in osteoarthritis by inhibiting PI3K/Akt/NF-kappaB and NLRP3/caspase-1/GSDMD signaling. Biomed. Pharmacother. 2023, 158, 114118. [Google Scholar] [CrossRef]
- Li, W.; Jin, K.; Luo, J.; Xu, W.; Wu, Y.; Zhou, J.; Wang, Y.; Xu, R.; Jiao, L.; Wang, T.; et al. NF-kappaB and its crosstalk with endoplasmic reticulum stress in atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 988266. [Google Scholar] [CrossRef]
- Gupta, A.; Brooks, C.; Storey, K.B. Regulation of NF-kappaB, FHC and SOD2 in response to oxidative stress in the freeze tolerant wood frog, Rana sylvatica. Cryobiology 2020, 97, 28–36. [Google Scholar] [CrossRef]
- Valduga, A.H.; Mizobuti, D.S.; Moraes, F.; Mancio, R.D.; Moraes, L.H.R.; Hermes, T.A.; Macedo, A.B.; Minatel, E. Protection of dystrophic muscle cells using Idebenone correlates with the interplay between calcium, oxidative stress and inflammation. Int. J. Exp. Pathol. 2023, 104, 4–12. [Google Scholar] [CrossRef]
- Indo, H.P.; Hawkins, C.L.; Nakanishi, I.; Matsumoto, K.I.; Matsui, H.; Suenaga, S.; Davies, M.J.; St Clair, D.K.; Ozawa, T.; Majima, H.J. Role of Mitochondrial Reactive Oxygen Species in the Activation of Cellular Signals, Molecules, and Function. Handb. Exp. Pharmacol. 2017, 240, 439–456. [Google Scholar] [CrossRef]
- Muri, J.; Kopf, M. The thioredoxin system: Balancing redox responses in immune cells and tumors. Eur. J. Immunol. 2023, 53, e2249948. [Google Scholar] [CrossRef]
- Hollander, J.M.; Lin, K.M.; Scott, B.T.; Dillmann, W.H. Overexpression of PHGPx and HSP60/10 protects against ischemia/reoxygenation injury. Free Radic. Biol. Med. 2003, 35, 742–751. [Google Scholar] [CrossRef]
- Gloire, G.; Piette, J. Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid. Redox Signal. 2009, 11, 2209–2222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Johnston, G.; Stebler, B.; Keller, E.T. Hydrogen peroxide activates NFkappaB and the interleukin-6 promoter through NFkappaB-inducing kinase. Antioxid. Redox Signal. 2001, 3, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Liu, C.; Tu, Z.; Xu, Q.; Chen, S.; Zhang, Y.; Wang, X.; Zhang, J.; Hu, C.A.; Liu, Y. Activation of the NF-kappaB and MAPK Signaling Pathways Contributes to the Inflammatory Responses, but Not Cell Injury, in IPEC-1 Cells Challenged with Hydrogen Peroxide. Oxid. Med. Cell Longev. 2020, 2020, 5803639. [Google Scholar] [CrossRef] [PubMed]
- Antinozzi, C.; Duranti, G.; Ceci, R.; Lista, M.; Sabatini, S.; Caporossi, D.; Di Luigi, L.; Sgro, P.; Dimauro, I. Hydrogen Peroxide Stimulates Dihydrotestosterone Release in C2C12 Myotubes: A New Perspective for Exercise-Related Muscle Steroidogenesis? Int. J. Mol. Sci. 2022, 23, 6566. [Google Scholar] [CrossRef]
- Asanuma, K.; Yokota, S.; Chosa, N.; Kamo, M.; Ibi, M.; Mayama, H.; Irie, T.; Satoh, K.; Ishisaki, A. Hydrogen peroxide-induced oxidative stress promotes expression of CXCL15/Lungkine mRNA in a MEK/ERK-dependent manner in fibroblast-like synoviocytes derived from mouse temporomandibular joint. J. Oral Biosci. 2022, 65, 97–103. [Google Scholar] [CrossRef]
- Koufos, O.; Mailloux, R.J. Protein S-glutathionylation and sex dimorphic effects on hydrogen peroxide production by dihydroorotate dehydrogenase in liver mitochondria. Free Radic. Biol. Med. 2023, 194, 123–130. [Google Scholar] [CrossRef]
- Chen, Z.; Xing, T.; Li, J.; Zhang, L.; Jiang, Y.; Gao, F. Hydrogen peroxide-induced oxidative stress impairs redox status and damages aerobic metabolism of breast muscle in broilers. Poult. Sci. 2021, 100, 918–925. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Grayson, C.; Koufos, O. Regulation of Mitochondrial Hydrogen Peroxide Availability by Protein S-glutathionylation. Cells 2022, 12, 107. [Google Scholar] [CrossRef]
- Loboda, A.; Dulak, J. NRF2 and its targets in skeletal muscle repair and regeneration. Antioxid. Redox Signal. 2023, 38, 619–642. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Nyul-Toth, A.; Kiss, T.; Yabluchanskiy, A.; Csipo, T.; Balasubramanian, P.; Lipecz, A.; Benyo, Z.; Csiszar, A. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: From increased cellular senescence to the pathogenesis of age-related vascular diseases. Geroscience 2019, 41, 727–738. [Google Scholar] [CrossRef]
- Abreu, C.C.; Cardozo, L.; Stockler-Pinto, M.B.; Esgalhado, M.; Barboza, J.E.; Frauches, R.; Mafra, D. Does resistance exercise performed during dialysis modulate Nrf2 and NF-kappaB in patients with chronic kidney disease? Life Sci. 2017, 188, 192–197. [Google Scholar] [CrossRef]
- Wang, L.X.; Zhu, X.M.; Yao, Y.M. Sestrin2: Its Potential Role and Regulatory Mechanism in Host Immune Response in Diseases. Front. Immunol. 2019, 10, 2797. [Google Scholar] [CrossRef]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef]
- Rhee, S.G.; Bae, S.H. The antioxidant function of sestrins is mediated by promotion of autophagic degradation of Keap1 and Nrf2 activation and by inhibition of mTORC1. Free Radic. Biol. Med. 2015, 88, 205–211. [Google Scholar] [CrossRef]
- Liu, W.; Xu, C.; Zou, Z.; Weng, Q.; Xiao, Y. Sestrin2 suppresses ferroptosis to alleviate septic intestinal inflammation and barrier dysfunction. Immunopharmacol. Immunotoxicol. 2022, 2022, 1–10. [Google Scholar] [CrossRef]
- Park, H.J.; Yang, S.G.; Koo, D.B. SESN2/NRF2 signaling activates as a direct downstream regulator of the PERK pathway against endoplasmic reticulum stress to improve the in vitro maturation of porcine oocytes. Free Radic. Biol. Med. 2022, 178, 413–427. [Google Scholar] [CrossRef]
- Piochi, L.F.; Machado, I.F.; Palmeira, C.M.; Rolo, A.P. Sestrin2 and mitochondrial quality control: Potential impact in myogenic differentiation. Ageing Res. Rev. 2021, 67, 101309. [Google Scholar] [CrossRef]
- Rongjin, H.; Feng, C.; Jun, K.; Shirong, L. Oxidative Stress-Induced Protein of SESTRIN2 in Cardioprotection Effect. Dis. Markers 2022, 2022, 7439878. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, X.; Ye, H.; Chen, Z.; Li, W. Inhibition of Sestrin2 overexpression in diabetic cardiomyopathy ameliorates cardiac injury via restoration of mitochondrial function. Exp. Ther. Med. 2022, 23, 265. [Google Scholar] [CrossRef]
- Brearley, M.C.; Li, C.; Daniel, Z.; Loughna, P.T.; Parr, T.; Brameld, J.M. Changes in expression of serine biosynthesis and integrated stress response genes during myogenic differentiation of C2C12 cells. Biochem. Biophys. Rep. 2019, 20, 100694. [Google Scholar] [CrossRef]
- Touron, J.; Perrault, H.; Maisonnave, L.; Patrac, V.; Walrand, S.; Malpuech-Brugere, C.; Pereira, B.; Burelle, Y.; Costes, F.; Richard, R. Effects of exercise-induced metabolic and mechanical loading on skeletal muscle mitochondrial function in male rats. J. Appl. Physiol. 2022, 133, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yu, W.; Xu, H.; Li, C.; Zou, R.; Wu, N.N.; Wang, L.; Ge, J.; Ren, J.; Zhang, Y. TBC1D15-Drp1 interaction-mediated mitochondrial homeostasis confers cardioprotection against myocardial ischemia/reperfusion injury. Metabolism 2022, 134, 155239. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, T.; Ancel, S.; Karaz, S.; Cichosz, P.; Jacot, G.; Giner, M.P.; Sanchez-Garcia, J.L.; Pannerec, A.; Moco, S.; Sorrentino, V.; et al. Nicotinamide riboside kinases regulate skeletal muscle fiber-type specification and are rate-limiting for metabolic adaptations during regeneration. Front. Cell Dev. Biol. 2022, 10, 1049653. [Google Scholar] [CrossRef] [PubMed]
- Shimura, D.; Nuebel, E.; Baum, R.; Valdez, S.E.; Xiao, S.; Warren, J.S.; Palatinus, J.A.; Hong, T.; Rutter, J.; Shaw, R.M. Protective mitochondrial fission induced by stress-responsive protein GJA1-20k. Elife 2021, 10, e69207. [Google Scholar] [CrossRef]
- Cho, Y.; Tachibana, S.; Lam, K.; Arita, Y.; Khosrowjerdi, S.; Zhang, O.; Liang, A.; Li, R.; Andreyev, A.; Murphy, A.N.; et al. Perm1 promotes cardiomyocyte mitochondrial biogenesis and protects against hypoxia/reoxygenation-induced damage in mice. J. Biol. Chem. 2021, 297, 100825. [Google Scholar] [CrossRef]
- Cho, Y.; Tachibana, S.; Hazen, B.C.; Moresco, J.J.; Yates, J.R., 3rd; Kok, B.; Saez, E.; Ross, R.S.; Russell, A.P.; Kralli, A. Perm1 regulates CaMKII activation and shapes skeletal muscle responses to endurance exercise training. Mol. Metab. 2019, 23, 88–97. [Google Scholar] [CrossRef]
- Cho, Y.; Hazen, B.C.; Gandra, P.G.; Ward, S.R.; Schenk, S.; Russell, A.P.; Kralli, A. Perm1 enhances mitochondrial biogenesis, oxidative capacity, and fatigue resistance in adult skeletal muscle. FASEB J. 2016, 30, 674–687. [Google Scholar] [CrossRef]
- Qin, C.; Wu, X.L.; Gu, J.; Du, D.; Guo, Y. Mitochondrial Dysfunction Secondary to Endoplasmic Reticulum Stress in Acute Myocardial Ischemic Injury in Rats. Med. Sci. Monit. 2020, 26, e923124. [Google Scholar] [CrossRef]
- Yang, Y.X.; Liu, M.S.; Liu, X.J.; Zhang, Y.C.; Hu, Y.Y.; Gao, R.S.; Pang, E.K.; Hou, L.; Wang, J.C.; Fei, W.Y. Porous Se@SiO(2) nanoparticles improve oxidative injury to promote muscle regeneration via modulating mitochondria. Nanomedicine 2022, 17, 1547–1565. [Google Scholar] [CrossRef]
- Wagatsuma, A.; Kotake, N.; Yamada, S. Muscle regeneration occurs to coincide with mitochondrial biogenesis. Mol. Cell Biochem. 2011, 349, 139–147. [Google Scholar] [CrossRef]
- Wagatsuma, A.; Sakuma, K. Mitochondria as a potential regulator of myogenesis. Sci. World J. 2013, 2013, 593267. [Google Scholar] [CrossRef]
- Moyes, C.D.; Mathieu-Costello, O.A.; Tsuchiya, N.; Filburn, C.; Hansford, R.G. Mitochondrial biogenesis during cellular differentiation. Am. J. Physiol. 1997, 272, C1345–C1351. [Google Scholar] [CrossRef]
- Kraft, C.S.; LeMoine, C.M.; Lyons, C.N.; Michaud, D.; Mueller, C.R.; Moyes, C.D. Control of mitochondrial biogenesis during myogenesis. Am. J. Physiol. Cell Physiol. 2006, 290, C1119–C1127. [Google Scholar] [CrossRef]
- Nichenko, A.S.; Southern, W.M.; Atuan, M.; Luan, J.; Peissig, K.B.; Foltz, S.J.; Beedle, A.M.; Warren, G.L.; Call, J.A. Mitochondrial maintenance via autophagy contributes to functional skeletal muscle regeneration and remodeling. Am. J. Physiol. Cell Physiol. 2016, 311, C190–C200. [Google Scholar] [CrossRef]
- Alway, S.E.; Degens, H.; Krishnamurthy, G.; Smith, C.A. Potential role for Id myogenic repressors in apoptosis and attenuation of hypertrophy in muscles of aged rats. Am. J. Physiol. Cell Physiol. 2002, 283, C66–C76. [Google Scholar] [CrossRef]
- Siu, P.M.; Alway, S.E. Id2 and p53 participate in apoptosis during unloading-induced muscle atrophy. Am. J. Physiol. Cell Physiol. 2005, 288, C1058–C1073. [Google Scholar] [CrossRef]
- Cho, Y.; Hazen, B.C.; Russell, A.P.; Kralli, A. Peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1)- and estrogen-related receptor (ERR)-induced regulator in muscle 1 (Perm1) is a tissue-specific regulator of oxidative capacity in skeletal muscle cells. J. Biol. Chem. 2013, 288, 25207–25218. [Google Scholar] [CrossRef]
- Sasaki, Y.; Vohra, B.P.; Lund, F.E.; Milbrandt, J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J. Neurosci. 2009, 29, 5525–5535. [Google Scholar] [CrossRef]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef]
- Sasaki, Y.; Araki, T.; Milbrandt, J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J. Neurosci. 2006, 26, 8484–8491. [Google Scholar] [CrossRef]
- Sasaki, Y.; Milbrandt, J. Axonal degeneration is blocked by nicotinamide mononucleotide adenylyltransferase (Nmnat) protein transduction into transected axons. J. Biol. Chem. 2010, 285, 41211–41215. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y. Metabolic aspects of neuronal degeneration: From a NAD(+) point of view. Neurosci. Res. 2019, 139, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Wallace, G.Q.; McNally, E.M. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 2009, 71, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Monceau, A.; Moutachi, D.; Lemaitre, M.; Garcia, L.; Trollet, C.; Furling, D.; Klein, A.; Ferry, A. Dystrophin Restoration after Adeno-Associated Virus U7-Mediated Dmd Exon Skipping Is Modulated by Muscular Exercise in the Severe D2-Mdx Duchenne Muscular Dystrophy Murine Model. Am. J. Pathol. 2022, 192, 1604–1618. [Google Scholar] [CrossRef] [PubMed]
- Mollard, A.; Peccate, C.; Forand, A.; Chassagne, J.; Julien, L.; Meunier, P.; Guesmia, Z.; Marais, T.; Bitoun, M.; Pietri-Rouxel, F.; et al. Muscle regeneration affects Adeno Associated Virus 1 mediated transgene transcription. Sci. Rep. 2022, 12, 9674. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Tasfaout, H.; Hauschka, S.D.; Chamberlain, J.S. Dystrophin Gene-Editing Stability Is Dependent on Dystrophin Levels in Skeletal but Not Cardiac Muscles. Mol. Ther. 2021, 29, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Zhao, P.; Borup, R.; Hoffman, E.P. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J. Cell Biol. 2000, 151, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.R.; von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef]
- Samaha, F.J.; Davis, B.; Nagy, B. Duchenne muscular dystrophy: Adenosine triphosphate and creatine phosphate content in muscle. Neurology 1981, 31, 916–919. [Google Scholar] [CrossRef]
- Samaha, F.J.; Schroeder, J.M.; Rebeiz, J.; Adams, R.D. Studies on myotonia. Biochemical and electron microscopic studies on myotonia congenita and myotonia dystrophica. Arch. Neurol. 1967, 17, 22–33. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef]
- Giovarelli, M.; Zecchini, S.; Catarinella, G.; Moscheni, C.; Sartori, P.; Barbieri, C.; Roux-Biejat, P.; Napoli, A.; Vantaggiato, C.; Cervia, D.; et al. Givinostat as metabolic enhancer reverting mitochondrial biogenesis deficit in Duchenne Muscular Dystrophy. Pharmacol. Res. 2021, 170, 105751. [Google Scholar] [CrossRef]
- Timpani, C.A.; Goodman, C.A.; Stathis, C.G.; White, J.D.; Mamchaoui, K.; Butler-Browne, G.; Gueven, N.; Hayes, A.; Rybalka, E. Adenylosuccinic acid therapy ameliorates murine Duchenne Muscular Dystrophy. Sci. Rep. 2020, 10, 1125. [Google Scholar] [CrossRef]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef]
- Ljubicic, V.; Burt, M.; Jasmin, B.J. The therapeutic potential of skeletal muscle plasticity in Duchenne muscular dystrophy: Phenotypic modifiers as pharmacologic targets. FASEB J. 2014, 28, 548–568. [Google Scholar] [CrossRef]
- Ljubicic, V.; Burt, M.; Lunde, J.A.; Jasmin, B.J. Resveratrol induces expression of the slow, oxidative phenotype in mdx mouse muscle together with enhanced activity of the SIRT1-PGC-1alpha axis. Am. J. Physiol. Cell Physiol. 2014, 307, C66–C82. [Google Scholar] [CrossRef]
- Marchioretti, C.; Zanetti, G.; Pirazzini, M.; Gherardi, G.; Nogara, L.; Andreotti, R.; Martini, P.; Marcucci, L.; Canato, M.; Nath, S.R.; et al. Defective excitation-contraction coupling and mitochondrial respiration precede mitochondrial Ca(2+) accumulation in spinobulbar muscular atrophy skeletal muscle. Nat. Commun. 2023, 14, 602. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Belosludtseva, N.V.; Mikheeva, I.B.; Chelyadnikova, Y.A.; Igoshkina, A.D.; Vafina, A.B.; Vedernikov, A.A.; Belosludtsev, K.N. BK(Ca) Activator NS1619 Improves the Structure and Function of Skeletal Muscle Mitochondria in Duchenne Dystrophy. Pharmaceutics 2022, 14, 2336. [Google Scholar] [CrossRef]
- Shkryl, V.M.; Martins, A.S.; Ullrich, N.D.; Nowycky, M.C.; Niggli, E.; Shirokova, N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflug. Arch. 2009, 458, 915–928. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Belosludtseva, N.V.; Mikheeva, I.B.; Chelyadnikova, Y.A.; Penkina, D.K.; Vedernikov, A.A.; Belosludtsev, K.N. The Effect of Uridine on the State of Skeletal Muscles and the Functioning of Mitochondria in Duchenne Dystrophy. Int. J. Mol. Sci. 2022, 23, 10660. [Google Scholar] [CrossRef]
- Rocha, G.L.D.; Rupcic, I.F.; Mizobuti, D.S.; Hermes, T.A.; Covatti, C.; Silva, H.; Araujo, H.N.; Lourenco, C.C.; Silveira, L.D.R.; Pereira, E.C.L.; et al. Cross-talk between TRPC-1, mTOR, PGC-1alpha and PPARdelta in the dystrophic muscle cells treated with tempol. Free Radic Res. 2022, 56, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Nogami, K.; Maruyama, Y.; Sakai-Takemura, F.; Motohashi, N.; Elhussieny, A.; Imamura, M.; Miyashita, S.; Ogawa, M.; Noguchi, S.; Tamura, Y.; et al. Pharmacological activation of SERCA ameliorates dystrophic phenotypes in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2021, 30, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, N.; Mareedu, S.; Tian, Y.; Kodippili, K.; Fefelova, N.; Voit, A.; Xie, L.H.; Duan, D.; Babu, G.J. Sarcolipin overexpression impairs myogenic differentiation in Duchenne muscular dystrophy. Am. J. Physiol. Cell Physiol. 2019, 317, C813–C824. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef]
- Duelen, R.; Costamagna, D.; Gilbert, G.; De Waele, L.; Goemans, N.; Desloovere, K.; Verfaillie, C.M.; Sipido, K.R.; Buyse, G.M.; Sampaolesi, M. Human iPSC model reveals a central role for NOX4 and oxidative stress in Duchenne cardiomyopathy. Stem Cell Rep. 2022, 17, 352–368. [Google Scholar] [CrossRef]
- Alves, F.M.; Kysenius, K.; Caldow, M.K.; Hardee, J.P.; Chung, J.D.; Trieu, J.; Hare, D.J.; Crouch, P.J.; Ayton, S.; Bush, A.I.; et al. Iron overload and impaired iron handling contribute to the dystrophic pathology in models of Duchenne muscular dystrophy. J. Cachexia Sarcopenia Muscle 2022, 13, 1541–1553. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca(2+) and Ca(2+)-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef]
- Alway, S.E.; Myers, M.J.; Mohamed, J.S. Regulation of satellite cell function in sarcopenia. Front. Aging Neurosci. 2014, 6, 246. [Google Scholar] [CrossRef]
- Alway, S.E.; Siu, P.M. Nuclear Apoptosis Contributes to Sarcopenia. Exerc. Sport Sci. Rev. 2008, 36, 51–57. [Google Scholar] [CrossRef]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Sileikyte, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapje zebrafish lacking dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef]
- Morotti, M.; Garofalo, S.; Cocozza, G.; Antonangeli, F.; Bianconi, V.; Mozzetta, C.; De Stefano, M.E.; Capitani, R.; Wulff, H.; Limatola, C.; et al. Muscle Damage in Dystrophic mdx Mice Is Influenced by the Activity of Ca(2+)-Activated K(Ca)3.1 Channels. Life 2022, 12, 538. [Google Scholar] [CrossRef]
- Sincennes, M.C.; Brun, C.E.; Lin, A.Y.T.; Rosembert, T.; Datzkiw, D.; Saber, J.; Ming, H.; Kawabe, Y.I.; Rudnicki, M.A. Acetylation of PAX7 controls muscle stem cell self-renewal and differentiation potential in mice. Nat. Commun. 2021, 12, 3253. [Google Scholar] [CrossRef]
- Seale, P.; Ishibashi, J.; Scime, A.; Rudnicki, M.A. Pax7 is necessary and sufficient for the myogenic specification of CD45+:Sca1+ stem cells from injured muscle. PLoS Biol. 2004, 2, E130. [Google Scholar] [CrossRef]
- Butler, D.C.; Haramizu, S.; Williamson, D.L.; Alway, S.E. Phospho-ablated Id2 is growth suppressive and pro-apoptotic in proliferating myoblasts. PLoS ONE 2009, 4, e6302. [Google Scholar] [CrossRef]
- Siu, P.M.; Alway, S.E. Subcellular responses of p53 and Id2 in fast and slow skeletal muscle in response to stretch-induced overload. J. Appl. Physiol. 2005, 99, 1897–1904. [Google Scholar] [CrossRef]
- Murach, K.A.; Liu, Z.; Jude, B.; Figueiredo, V.C.; Wen, Y.; Khadgi, S.; Lim, S.; Morena da Silva, F.; Greene, N.P.; Lanner, J.T.; et al. Multi-transcriptome analysis following an acute skeletal muscle growth stimulus yields tools for discerning global and MYC regulatory networks. J. Biol. Chem. 2022, 298, 102515. [Google Scholar] [CrossRef]
- Figueiredo, V.C.; Wen, Y.; Alkner, B.; Fernandez-Gonzalo, R.; Norrbom, J.; Vechetti, I.J., Jr.; Valentino, T.; Mobley, C.B.; Zentner, G.E.; Peterson, C.A.; et al. Genetic and epigenetic regulation of skeletal muscle ribosome biogenesis with exercise. J. Physiol. 2021, 599, 3363–3384. [Google Scholar] [CrossRef]
- Alway, S.E. Overload-induced C-Myc oncoprotein is reduced in aged skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52, B203–B211. [Google Scholar] [CrossRef]
- Dam, A.D.; Mitchell, A.S.; Quadrilatero, J. Induction of mitochondrial biogenesis protects against caspase-dependent and caspase-independent apoptosis in L6 myoblasts. Biochim. Biophys. Acta 2013, 1833, 3426–3435. [Google Scholar] [CrossRef]
- Niu, W.; Wang, H.; Wang, B.; Mao, X.; Du, M. Resveratrol improves muscle regeneration in obese mice through enhancing mitochondrial biogenesis. J. Nutr. Biochem. 2021, 98, 108804. [Google Scholar] [CrossRef] [PubMed]
- Rathbone, C.R.; Booth, F.W.; Lees, S.J. Sirt1 increases skeletal muscle precursor cell proliferation. Eur. J. Cell. Biol. 2009, 88, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Call, J.A.; Nichenko, A.S. Autophagy: An essential but limited cellular process for timely skeletal muscle recovery from injury. Autophagy 2020, 16, 1344–1347. [Google Scholar] [CrossRef] [PubMed]
- Nakagawara, K.; Takeuchi, C.; Ishige, K. 5’-CMP and 5’-UMP promote myogenic differentiation and mitochondrial biogenesis by activating myogenin and PGC-1alpha in a mouse myoblast C2C12 cell line. Biochem. Biophys. Rep. 2022, 31, 101309. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; Kim, W.; Davie, J. Myogenin is required for assembly of the transcription machinery on muscle genes during skeletal muscle differentiation. PLoS ONE 2021, 16, e0245618. [Google Scholar] [CrossRef]
- Siu, P.M.; Donley, D.A.; Bryner, R.W.; Alway, S.E. Myogenin and oxidative enzyme gene expression levels are elevated in rat soleus muscles after endurance training. J. Appl. Physiol. 2004, 97, 277–285. [Google Scholar] [CrossRef]
- de la Vega, E.; Gonzalez, N.; Cabezas, F.; Montecino, F.; Blanco, N.; Olguin, H. USP7-dependent control of myogenin stability is required for terminal differentiation in skeletal muscle progenitors. FEBS J. 2020, 287, 4659–4677. [Google Scholar] [CrossRef]
- Jones, F.K.; Phillips, A.M.; Jones, A.R.; Pisconti, A. The INSR/AKT/mTOR pathway regulates the pace of myogenesis in a syndecan-3-dependent manner. Matrix Biol. 2022, 113, 61–82. [Google Scholar] [CrossRef]
- Wang, Y.; Mohamed, J.S.; Alway, S.E. M-cadherin-inhibited phosphorylation of B-catenin augments differentiation of mouse myoblasts. Cell Tissue Res. 2013, 351, 183–200. [Google Scholar] [CrossRef]
- Jones, A.E.; Price, F.D.; Le, G.F.; Soleimani, V.D.; Dick, S.A.; Megeney, L.A.; Rudnicki, M.A. Wnt/beta-catenin controls follistatin signalling to regulate satellite cell myogenic potential. Skelet. Muscle 2015, 5, 14. [Google Scholar] [CrossRef]
- Brower, J.V.; Lim, C.H.; Han, C.; Hankowski, K.E.; Hamazaki, T.; Terada, N. Differential CpG island methylation of murine adenine nucleotide translocase genes. Biochim. Biophys. Acta 2009, 1789, 198–203. [Google Scholar] [CrossRef]
- Stepien, G.; Torroni, A.; Chung, A.B.; Hodge, J.A.; Wallace, D.C. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Biol. Chem. 1992, 267, 14592–14597. [Google Scholar] [CrossRef]
- Flierl, A.; Schriner, S.E.; Hancock, S.; Coskun, P.E.; Wallace, D.C. The mitochondrial adenine nucleotide transporters in myogenesis. Free Radic. Biol. Med. 2022, 188, 312–327. [Google Scholar] [CrossRef]
- James, G.; Chen, X.; Diwan, A.; Hodges, P.W. Fat infiltration in the multifidus muscle is related to inflammatory cytokine expression in the muscle and epidural adipose tissue in individuals undergoing surgery for intervertebral disc herniation. Eur. Spine J. 2021, 30, 837–845. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, T.J.; Li, Y.; Gao, Y. Mesenchymal Stem Cells Decrease M1/M2 Ratio and Alleviate Inflammation to Improve Limb Ischemia in Mice. Med. Sci. Monit. 2020, 26, e923287. [Google Scholar] [CrossRef]
- Yan, M.; Yang, Y.; Zhou, Y.; Yu, C.; Li, R.; Gong, W.; Zheng, J. Interleukin-7 aggravates myocardial ischaemia/reperfusion injury by regulating macrophage infiltration and polarization. J. Cell Mol. Med. 2021, 25, 9939–9952. [Google Scholar] [CrossRef]
- Xing, Y.; Pan, S.; Zhu, L.; Cui, Q.; Tang, Z.; Liu, Z.; Liu, F. Advanced Glycation End Products Induce Atherosclerosis via RAGE/TLR4 Signaling Mediated-M1 Macrophage Polarization-Dependent Vascular Smooth Muscle Cell Phenotypic Conversion. Oxid. Med. Cell Longev. 2022, 2022, 9763377. [Google Scholar] [CrossRef]
- Zogbi, C.; Oliveira, N.C.; Levy, D.; Bydlowski, S.P.; Bassaneze, V.; Neri, E.A.; Krieger, J.E. Beneficial effects of IL-4 and IL-6 on rat neonatal target cardiac cells. Sci. Rep. 2020, 10, 12350. [Google Scholar] [CrossRef]
- Zhu, H.; Qu, X.; Zhang, C.; Yu, Y. Interleukin-10 promotes proliferation of vascular smooth muscle cells by inhibiting inflammation in rabbit abdominal aortic aneurysm. Int. J. Clin. Exp. Pathol. 2019, 12, 1260–1271. [Google Scholar]
- Deng, B.; Wehling-Henricks, M.; Villalta, S.A.; Wang, Y.; Tidball, J.G. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol. 2012, 189, 3669–3680. [Google Scholar] [CrossRef]
- Han, H.; Li, M.; Liu, H.; Li, H. Electroacupuncture regulates inflammation, collagen deposition and macrophage function in skeletal muscle through the TGF-beta1/Smad3/p38/ERK1/2 pathway. Exp. Ther Med. 2021, 22, 1457. [Google Scholar] [CrossRef]
- Lin, S.; Zhou, Z.; Zhao, H.; Xu, C.; Guo, Y.; Gao, S.; Mei, X.; Tian, H. TNF promotes M1 polarization through mitochondrial metabolism in injured spinal cord. Free Radic. Biol. Med. 2021, 172, 622–632. [Google Scholar] [CrossRef]
- Tobin, S.W.; Alibhai, F.J.; Wlodarek, L.; Yeganeh, A.; Millar, S.; Wu, J.; Li, S.H.; Weisel, R.D.; Li, R.K. Delineating the relationship between immune system aging and myogenesis in muscle repair. Aging Cell 2021, 20, e13312. [Google Scholar] [CrossRef]
- Garcia, I.; Calderon, F.; la Torre, P.; Vallier, S.S.; Rodriguez, C.; Agarwala, D.; Keniry, M.; Innis-Whitehouse, W.; Gilkerson, R. Mitochondrial OPA1 cleavage is reversibly activated by differentiation of H9c2 cardiomyoblasts. Mitochondrion 2021, 57, 88–96. [Google Scholar] [CrossRef]
- Gilkerson, R.; De La Torre, P.; St Vallier, S. Mitochondrial OMA1 and OPA1 as Gatekeepers of Organellar Structure/Function and Cellular Stress Response. Front. Cell Dev. Biol. 2021, 9, 626117. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; Wade, S.; Triolo, M.; Girgis, J.; Chwastek, D.; Larrigan, S.; Feige, P.; Fujita, R.; Crist, C.; Rudnicki, M.A.; et al. The mitochondrial protein OPA1 regulates the quiescent state of adult muscle stem cells. Cell Stem Cell 2022, 29, 1315–1332.e9. [Google Scholar] [CrossRef] [PubMed]
- Luo, N.; Yue, F.; Jia, Z.; Chen, J.; Deng, Q.; Zhao, Y.; Kuang, S. Reduced electron transport chain complex I protein abundance and function in Mfn2-deficient myogenic progenitors lead to oxidative stress and mitochondria swelling. FASEB J. 2021, 35, e21426. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jia, Y.; Zhao, J.; Lesner, N.P.; Menezes, C.J.; Shelton, S.D.; Venigalla, S.S.K.; Xu, J.; Cai, C.; Mishra, P. A Mitofusin 2—Hif1alpha axis sets a maturation checkpoint in regenerating skeletal muscle. J. Clin. Investig. 2022, 132, e161638. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, X.; Yu, J.; Tian, Y.; Yang, C.; Chen, Y.; Chen, H.; Ge, H. LonP1 regulates mitochondrial network remodeling through the PINK1/Parkin pathway during myoblast differentiation. Am. J. Physiol. Cell Physiol. 2020, 319, C1020–C1028. [Google Scholar] [CrossRef]
- Esteca, M.V.; Severino, M.B.; Silvestre, J.G.; Palmeira Dos Santos, G.; Tamborlin, L.; Luchessi, A.D.; Moriscot, A.S.; Gustafsson, A.B.; Baptista, I.L. Loss of Parkin Results in Altered Muscle Stem Cell Differentiation during Regeneration. Int. J. Mol. Sci. 2020, 21, 8007. [Google Scholar] [CrossRef]
- Zhang, J.; Hua, C.; Zhang, Y.; Wei, P.; Tu, Y.; Wei, T. KAP1-associated transcriptional inhibitory complex regulates C2C12 myoblasts differentiation and mitochondrial biogenesis via miR-133a repression. Cell Death Dis. 2020, 11, 732. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Alway, S.E.; Pereira, S.L.; Edens, N.K.; Hao, Y.; Bennett, B.T. β-Hydroxy-β-methylbutyrate (HMB) enhances the proliferation of satellite cells in fast muscles of aged rats during recovery from disuse atrophy. Exp. Gerontol. 2013, 48, 973–984. [Google Scholar] [CrossRef]
- Bennett, B.T.; Mohamed, J.S.; Alway, S.E. The Effects of Calcium-beta-Hydroxy-beta-Methylbutyrate on Aging-Associated Apoptotic Signaling and Muscle Mass and Function in Unloaded but Nonatrophied Extensor Digitorum Longus Muscles of Aged Rats. Oxid. Med. Cell Longev. 2020, 2020, 3938672. [Google Scholar] [CrossRef]
- Takahashi, H.; Suzuki, Y.; Mohamed, J.S.; Gotoh, T.; Pereira, S.L.; Alway, S.E. Epigallocatechin-3-gallate increases autophagy signaling in resting and unloaded plantaris muscles but selectively suppresses autophagy protein abundance in reloaded muscles of aged rats. Exp. Gerontol. 2017, 92, 56–66. [Google Scholar] [CrossRef]
- Li, P.; Liu, A.; Xiong, W.; Lin, H.; Xiao, W.; Huang, J.; Zhang, S.; Liu, Z. Catechins enhance skeletal muscle performance. Crit Rev. Food Sci. Nutr. 2020, 60, 515–528. [Google Scholar] [CrossRef]
- Bose, C.; Alves, I.; Singh, P.; Palade, P.T.; Carvalho, E.; Borsheim, E.; Jun, S.R.; Cheema, A.; Boerma, M.; Awasthi, S.; et al. Sulforaphane prevents age-associated cardiac and muscular dysfunction through Nrf2 signaling. Aging Cell 2020, 19, e13261. [Google Scholar] [CrossRef]
- Seldeen, K.L.; Shahini, A.; Thiyagarajan, R.; Redae, Y.; Leiker, M.; Rajabian, N.; Dynka, A.; Andreadis, S.T.; Troen, B.R. Short-term nicotinamide riboside treatment improves muscle quality and function in mice and increases cellular energetics and differentiating capacity of myogenic progenitors. Nutrition 2021, 87–88, 111189. [Google Scholar] [CrossRef]
- Roman, W.; Pinheiro, H.; Pimentel, M.R.; Segales, J.; Oliveira, L.M.; Garcia-Dominguez, E.; Gomez-Cabrera, M.C.; Serrano, A.L.; Gomes, E.R.; Munoz-Canoves, P. Muscle repair after physiological damage relies on nuclear migration for cellular reconstruction. Science 2021, 374, 355–359. [Google Scholar] [CrossRef]
- Debattisti, V.; Horn, A.; Singh, R.; Seifert, E.L.; Hogarth, M.W.; Mazala, D.A.; Huang, K.T.; Horvath, R.; Jaiswal, J.K.; Hajnoczky, G. Dysregulation of Mitochondrial Ca(2+) Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell Rep. 2019, 29, 1274–1286.e1276. [Google Scholar] [CrossRef]
- Oliveira, A.N.; Richards, B.J.; Hood, D.A. Measurement of Protein Import Capacity of Skeletal Muscle Mitochondria. J. Vis. Exp. 2022, 179, e63055. [Google Scholar] [CrossRef]
- Hendrickse, P.W.; Venckunas, T.; Platkevicius, J.; Kairaitis, R.; Kamandulis, S.; Snieckus, A.; Stasiulis, A.; Vitkiene, J.; Subocius, A.; Degens, H. Endurance training-induced increase in muscle oxidative capacity without loss of muscle mass in younger and older resistance-trained men. Eur. J. Appl. Physiol. 2021, 121, 3161–3172. [Google Scholar] [CrossRef] [PubMed]
- Hendrickse, P.W.; Krusnauskas, R.; Hodson-Tole, E.; Venckunas, T.; Degens, H. Regular endurance exercise of overloaded muscle of young and old male mice does not attenuate hypertrophy and improves fatigue resistance. Geroscience 2021, 43, 741–757. [Google Scholar] [CrossRef] [PubMed]
- Hendrickse, P.; Degens, H. The role of the microcirculation in muscle function and plasticity. J. Muscle Res. Cell Motil. 2019, 40, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Slavin, M.B.; Kumari, R.; Hood, D.A. ATF5 is a regulator of exercise-induced mitochondrial quality control in skeletal muscle. Mol. Metab 2022, 66, 101623. [Google Scholar] [CrossRef]
- Memme, J.M.; Erlich, A.T.; Phukan, G.; Hood, D.A. Exercise and mitochondrial health. J. Physiol. 2021, 599, 803–817. [Google Scholar] [CrossRef]
- Porter, C.; Reidy, P.T.; Bhattarai, N.; Sidossis, L.S.; Rasmussen, B.B. Resistance Exercise Training Alters Mitochondrial Function in Human Skeletal Muscle. Med. Sci. Sports Exerc. 2015, 47, 1922–1931. [Google Scholar] [CrossRef]
- Andrade-Souza, V.A.; Ghiarone, T.; Sansonio, A.; Santos Silva, K.A.; Tomazini, F.; Arcoverde, L.; Fyfe, J.; Perri, E.; Saner, N.; Kuang, J.; et al. Exercise twice-a-day potentiates markers of mitochondrial biogenesis in men. FASEB J. 2020, 34, 1602–1619. [Google Scholar] [CrossRef]
- Mathew, S.J.; Hansen, J.M.; Merrell, A.J.; Murphy, M.M.; Lawson, J.A.; Hutcheson, D.A.; Hansen, M.S.; Angus-Hill, M.; Kardon, G. Connective tissue fibroblasts and Tcf4 regulate myogenesis. Development 2011, 138, 371–384. [Google Scholar] [CrossRef]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.J.; Hutcheson, D.A.; Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138, 3625–3637. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef]
- Dulac, M.; Leduc-Gaudet, J.P.; Cefis, M.; Ayoub, M.B.; Reynaud, O.; Shams, A.; Moamer, A.; Nery Ferreira, M.F.; Hussain, S.N.; Gouspillou, G. Regulation of muscle and mitochondrial health by the mitochondrial fission protein Drp1 in aged mice. J. Physiol. 2021, 599, 4045–4063. [Google Scholar] [CrossRef]
- Dulac, M.; Leduc-Gaudet, J.P.; Reynaud, O.; Ayoub, M.B.; Guerin, A.; Finkelchtein, M.; Hussain, S.N.; Gouspillou, G. Drp1 knockdown induces severe muscle atrophy and remodelling, mitochondrial dysfunction, autophagy impairment and denervation. J. Physiol. 2020, 598, 3691–3710. [Google Scholar] [CrossRef]
- Maezawa, T.; Tanaka, M.; Kanazashi, M.; Maeshige, N.; Kondo, H.; Ishihara, A.; Fujino, H. Astaxanthin supplementation attenuates immobilization-induced skeletal muscle fibrosis via suppression of oxidative stress. J. Physiol. Sci. 2017, 67, 603–611. [Google Scholar] [CrossRef]
- McCully, J.D.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef]
- Preble, J.M.; Pacak, C.A.; Kondo, H.; MacKay, A.A.; Cowan, D.B.; McCully, J.D. Rapid isolation and purification of mitochondria for transplantation by tissue dissociation and differential filtration. J. Vis. Exp. 2014, 2014, e51682. [Google Scholar] [CrossRef]
- Ali Pour, P.; Kenney, M.C.; Kheradvar, A. Bioenergetics Consequences of Mitochondrial Transplantation in Cardiomyocytes. J. Am. Heart Assoc. 2020, 9, e014501. [Google Scholar] [CrossRef]
- Khan, M.M.; Paez, H.G.; Pitzer, C.R.; Alway, S.E. The Therapeutic Potential of Mitochondria Transplantation Therapy in Neurodegenerative and Neurovascular Disorders. Curr. Neuropharmacol. 2022; in press. [Google Scholar] [CrossRef]
- Orfany, A.; Arriola, C.G.; Doulamis, I.P.; Guariento, A.; Ramirez-Barbieri, G.; Moskowitzova, K.; Shin, B.; Blitzer, D.; Rogers, C.; Del Nido, P.J.; et al. Mitochondrial transplantation ameliorates acute limb ischemia. J. Vasc. Surg. 2020, 71, 1014–1026. [Google Scholar] [CrossRef]
- Paliwal, S.; Chaudhuri, R.; Agrawal, A.; Mohanty, S. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J. Biomed. Sci. 2018, 25, 31. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, D.; Abe, J.; Takeda, A.; Harashima, H.; Yamada, Y. Transplantation of MITO cells, mitochondria activated cardiac progenitor cells, to the ischemic myocardium of mouse enhances the therapeutic effect. Sci. Rep. 2022, 12, 4344. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wei, L.; Tian, K.; Xu, M.; Chen, X.; Chen, F.; Zha, D.; Xue, T. NRG1/ErbB2 axis regulated mitochondrial function and antioxidant enzymes of neural stem cells in the cochlear nucleus partially through PGC-1alpha. Neurosci. Lett. 2023, 792, 136942. [Google Scholar] [CrossRef] [PubMed]
- Sainz de la Maza, D.; Hof-Michel, S.; Phillimore, L.; Bokel, C.; Amoyel, M. Cell-cycle exit and stem cell differentiation are coupled through regulation of mitochondrial activity in the Drosophila testis. Cell Rep. 2022, 39, 110774. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; Yao, Y.; Zhao, T.; Chen, Y.Y.; Shen, Y.L.; Wang, L.L.; Zhu, Y. Stem cell-derived mitochondria transplantation: A novel strategy and the challenges for the treatment of tissue injury. Stem Cell Res. Ther. 2018, 9, 106. [Google Scholar] [CrossRef]
- Kim, Y.S.; Yoon, J.W.; Kim, D.; Choi, S.; Kim, H.K.; Youm, J.B.; Han, J.; Heo, S.C.; Hyun, S.A.; Seo, J.W.; et al. Tomatidine-stimulated maturation of human embryonic stem cell-derived cardiomyocytes for modeling mitochondrial dysfunction. Exp. Mol. Med. 2022, 54, 493–502. [Google Scholar] [CrossRef]







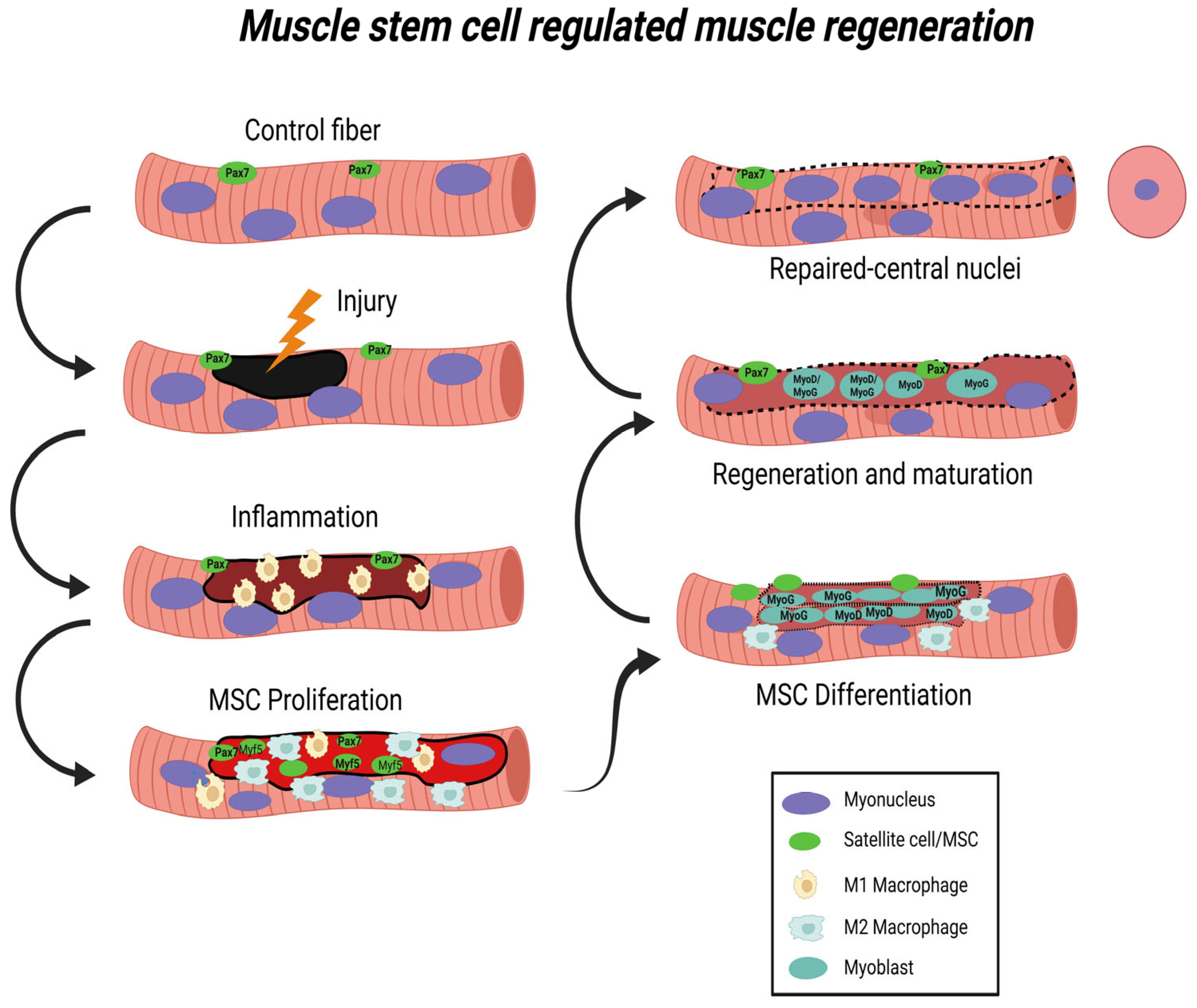{kind=link}
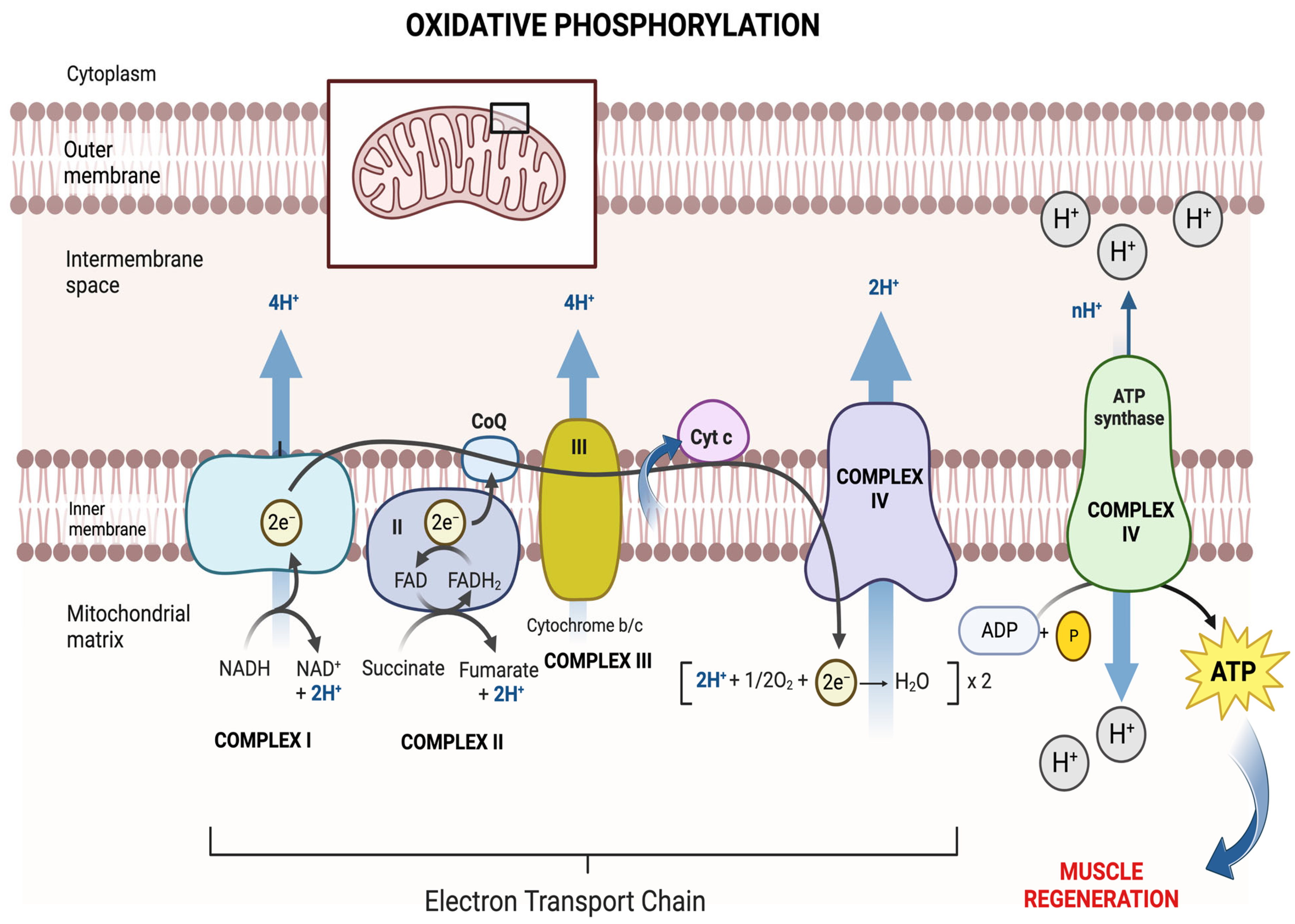{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TLR9 | Inflammation, muscle atrophy, increase cytokines (IL-6) | [54,175,176,180,181,182,183] |
| MFN1/2 loss | Atrophy, inflammation | [54,176,177,178] |
| Parkin1 loss | Increase in cytokines (cytokines IL-6, IFNβ1, IL-12, IL-13, XXCL1, CCL2, CCL4) | [187] |
| Pink1 loss | Increase in immune genes, and cytokines IL-6, IFNβ1, TNFα, IL-1β CCL2, IL-12(p70), IL-13, IL-17, CXCL1, CCL4 | [187,200,201] |
| cGAS-STING | Increase type I Interferon | [187,210,211,212,213,214] |
| Mitophagy/mitochondria stress | NRLP3, mtDAMPs (mtDNA, TFAM, cardiolipin) | [177,188,189,190,191] |
| Condition | mRNA Increases | Protein Increases | Functional Increases | References |
|---|---|---|---|---|
| Eccentric exercise | PCG1α, TFAM | SDHa, CKmt2, ANT1 | mitochondria biogenesis and function | [342] |
| Ischemic injury | TBC1D15 | TBC1D15 | mitochondrial homeostasis | [343] |
| CTX injury | Nmrk2 | differentiation of myoblasts | [344] | |
| Ischemic injury | GJA1-20k | mitochondrial size, recruits actin to mitochondria (decreased ROS) | [345] | |
| Myogenesis/repair | PERM1, PGC1α | mitochondria biogenesis and mitochondrial respiratory function | [346,347,348] | |
| Aging, denervation injury, ischemia | Bax, ATF6, GRP-78, caspase 3 | Bax, caspase 3, caspase 9, cytochrome c | apoptosis and atrophy | [13,148,273,349] |
| CTX muscle repair | SIRT1, p53 | mitochondria size, Complex III activity | [59] | |
| CTX muscle repair | SIRT1, p53, SOD1, CAT | mitochondria size, Complex I, III, ATPase activity | [350] | |
| CTX and freezing muscle repair | PGC1β, PRC, NRF-1, NRF-2, TFAM, ERR, Drp1 | Mitochondrial fission, fusion and biogenesis | [351,352] | |
| CTX muscle repair | Drp1, ULK1, BNIP3, and MAP1LC3-II | Mitochondrial fission, fusion, and biogenesis | [353,354] | |
| Freezing muscle repair | Drp1 BNIP3, Pink1, Parkin | Mitochondrial fission, mitophagy | [88,355] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alway, S.E.; Paez, H.G.; Pitzer, C.R. The Role of Mitochondria in Mediation of Skeletal Muscle Repair. Muscles 2023, 2, 119-163. https://doi.org/10.3390/muscles2020011
Alway SE, Paez HG, Pitzer CR. The Role of Mitochondria in Mediation of Skeletal Muscle Repair. Muscles. 2023; 2(2):119-163. https://doi.org/10.3390/muscles2020011
Chicago/Turabian StyleAlway, Stephen E., Hector G. Paez, and Christopher R. Pitzer. 2023. "The Role of Mitochondria in Mediation of Skeletal Muscle Repair" Muscles 2, no. 2: 119-163. https://doi.org/10.3390/muscles2020011
APA StyleAlway, S. E., Paez, H. G., & Pitzer, C. R. (2023). The Role of Mitochondria in Mediation of Skeletal Muscle Repair. Muscles, 2(2), 119-163. https://doi.org/10.3390/muscles2020011