

An Update of Clinical, Epidemiological, and Psychosocial Features in Gamma-Sarcoglycanopathy




Abstract
1. Introduction
2. Evolution of the LGMD Concept
3. Epidemiology and Psychosocial Consequences of LGMDs
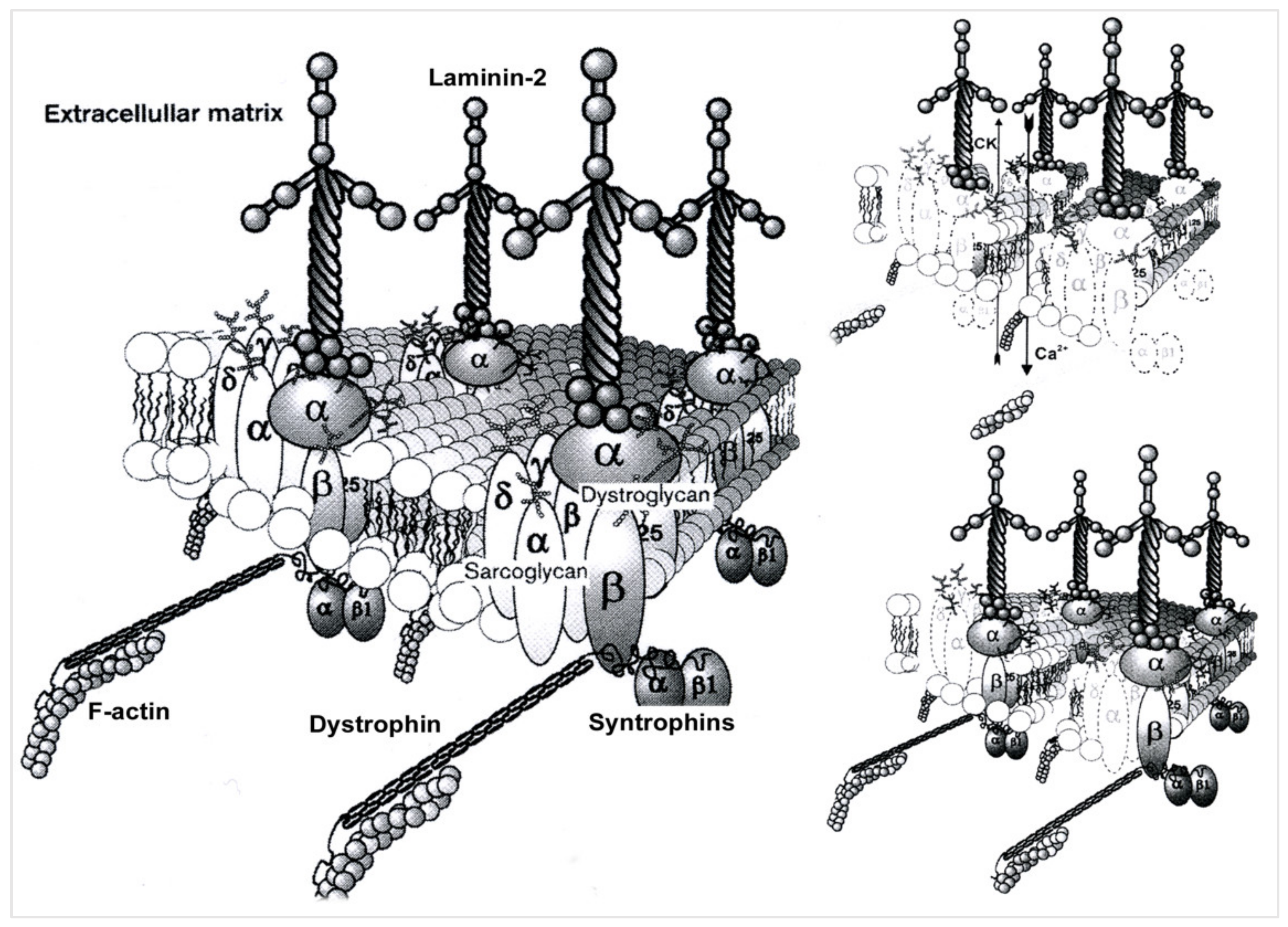
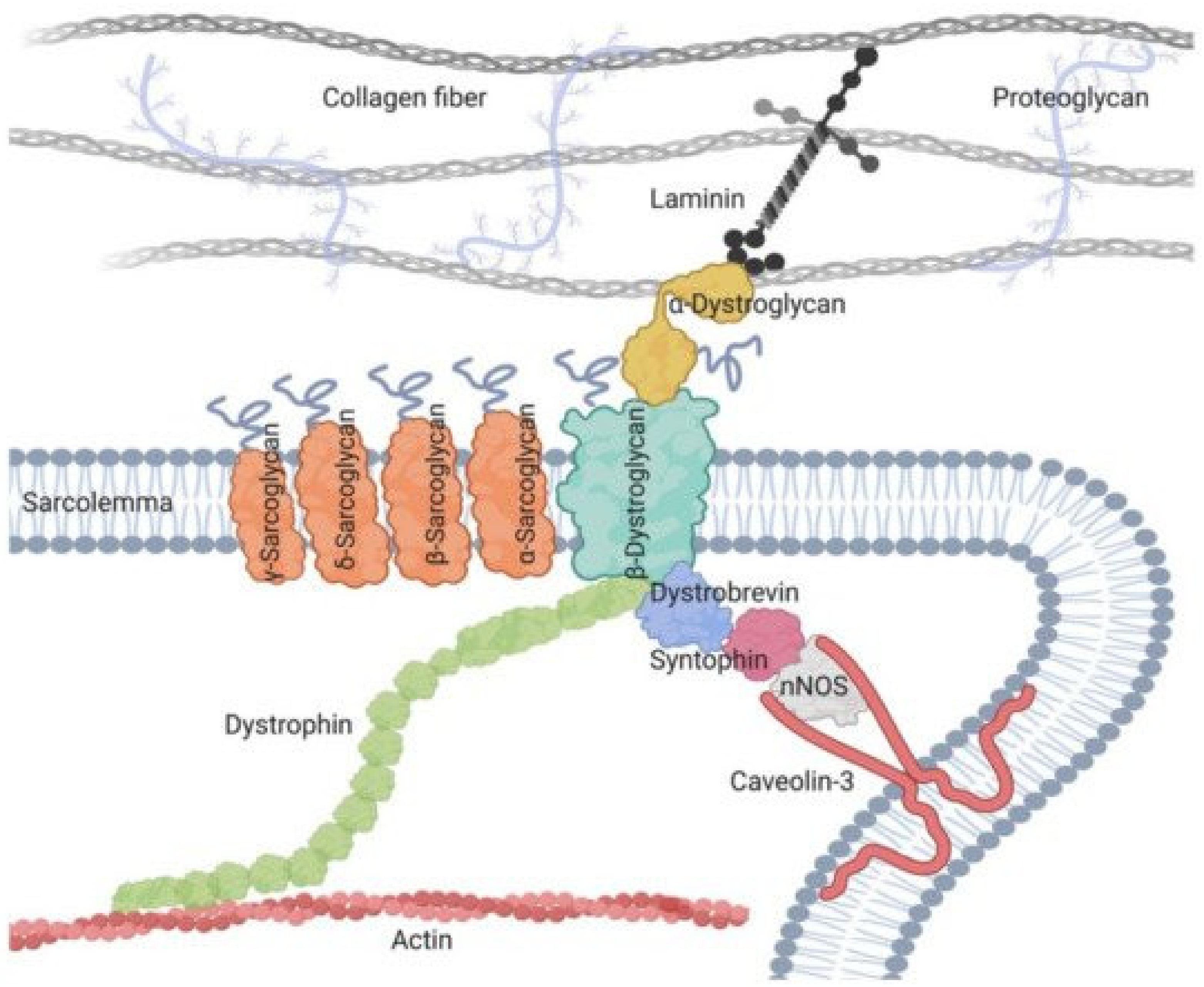
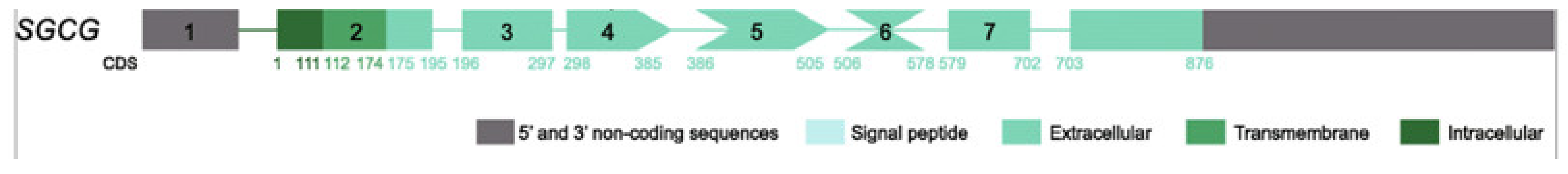
4. Pathophysiological Data for LGMDR5
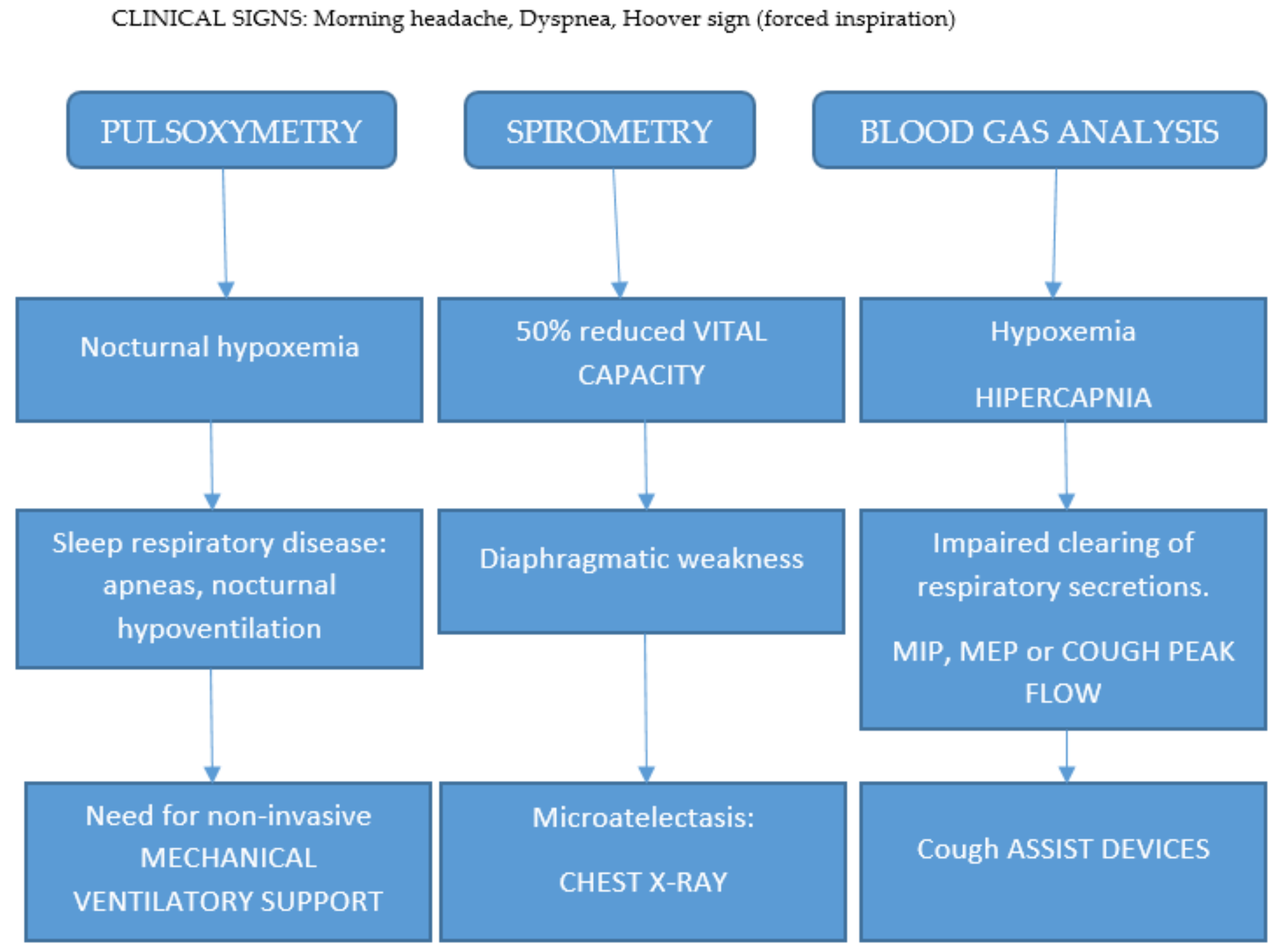
5. Diagnostic Approach
6. Therapeutic Perspectives
7. Socioeconomic and Quality of Life Consideration in the Case Report and Comparison between Italian and Tunisian Patients
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ben Othmane, K.; Ben Hamida, M.; Pericak-Vance, M.A.; Ben Hamida, C.; Blel, S.; Carter, S.C.; Bowcock, A.M.; Petruhkin, K.; Gilliam, T.C.; Roses, A.D.; et al. Linkage of Tunisian autosomal recessive Duchenne—Like muscular dystrophy to the pericentromeric region of chromosome 13q. Nat. Genet. 1992, 2, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, H. Epidemiology of muscular dystrophies in the Mediterranean area. Acta Myol. 2013, 32, 138. [Google Scholar]
- Noguchi, S.; McNally, E.M.; Othmane, K.B.; Hagiwara, Y.; Mizuno, Y.; Yoshida, M.; Yamamoto, H.; Bönnemann, C.G.; Gussoni, E.; Denton, P.H.; et al. Mutations in the dystrophin-associated protein γ-sarcoglycan in chromosome 13 muscular dystrophy. Science 1995, 270, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Kefi, M.; Amouri, R.; Driss, A.; Hamida, C.B.; Hamidaa, M.B.; Kunkel, L.M.; Hentati, F. Phenotype and sarcoglycan expression in Tunisian LGMD 2C patients sharing the same del521-T mutation. Neuromuscul. Disord. 2003, 13, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C. LGMD. Identification, description and classification. Acta Myol. 2020, 39, 207. [Google Scholar] [CrossRef]
- Okizuka, Y.; Takeshima, Y.; Itoh, K.; Zhang, Z.; Awano, H.; Maruyama, K.; Kumagai, T.; Yagi, M.; Matsuo, M. Low incidence of limb-girdle muscular dystrophy type2C revealed by a mutation study in Japanese patients clinically diagnosed as DMD. BMC Med. Genet. 2010, 11, 49. [Google Scholar] [CrossRef]
- Elloumi-Zghal, H.; Bouhamed, H.C. Genetics and genomic medicine in Tunisia. Mol. Genet. Genom. Med. 2018, 6, 134. [Google Scholar] [CrossRef]
- Bönnemann, C.G.; Wong, J.; Hamida, C.B.; Hamida, M.B.; Hentati, F.; Kunkel, L.M. LGMD 2E in Tunisia is caused by a homozygous missense mutation in β-sarcoglycan exon 3. Neuromuscul. Disord. 1998, 8, 193–197. [Google Scholar] [CrossRef]
- Driss, A.; Amouri, R.; Hamida, C.B.; Souilem, S.; Gouider-Khouja, N.; Hamida, M.B.; Hentati, F. A new locus for autosomal recessive limb-girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13. 3. Neuromuscul. Disord. 2000, 10, 240–246. [Google Scholar] [CrossRef]
- Rekik, S.; Sakka, S.; Ben Romdhan, S.; Farhat, N.; Baba Amer, Y.; Lehkim, L.; Authier, F.J.; Mhiri, C. Novel missense CAPN3 mutation responsible for adult-onset limb girdle muscular dystrophy with calves hypertrophy. J. Mol. Neurosci. 2019, 69, 563–569. [Google Scholar] [CrossRef]
- Ben Hamida, M.; Fardeau, M.; Attia, N. Severe childhood muscular dystrophy affecting both sexes and frequent in Tunisia. Muscle Nerve 1983, 6, 469–480. [Google Scholar] [CrossRef]
- Tsubata, S.; Bowles, K.R.; Vatta, M.; Zintz, C.; Titus, J.; Muhonen, L.; Bowles, N.E.; Towbin, J.A. Mutations in the human δ-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J. Clin. Invest. 2000, 106, 655–662. [Google Scholar] [CrossRef]
- Beckmann, J.S. β-sarcoglycane: Une protéine du complexe dystrophine glycoprotéines est responsable d’une forme récessive de dystrophie musculaire. Méd. Sci. 1995, 11, 1732–1738. Available online: https://www.ipubli.inserm.fr/bitstream/handle/10608/2376/1995_12_1732.pdf?sequence=1. (accessed on 14 October 2022).
- Bushby, K. Diagnostic criteria for the limb-girdle muscular dystrophies: Report of the ENMC Consortium on Limb-Girdle Dystrophies. Neuromuscul. Disord. 1995, 1, 71–74. [Google Scholar] [CrossRef]
- Straub, V.; Murphy, A.; Udd, B.; LGMD workshop study group. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef]
- Duggan, D.J.; Gorospe, J.R.; Fanin, M.; Hoffman, E.P.; Angelini, C. Mutations in the sarcoglycan genes in patients with myopathy. N. Engl. J. Med. 1997, 336, 618–625. [Google Scholar] [CrossRef]
- Cukierman, L. La Banque des Données des Mutations du Gene DMD UMD-DMD France. Available online: www.umd.be/DMD/W_DMD/gene_FR.html (accessed on 21 October 2022).
- Bulakh, M.V.; Ryzhkova, O.P.; Polyakov, A.V. Sarcoglycanopathies: Clinical, Molecular and Genetic Characteristics, Epidemiology, Diagnostics and Treatment Options. Russ. J. Genet. 2018, 54, 129–144. [Google Scholar] [CrossRef]
- Du Montcel, S.T.; Clot, F.; Vidailhet, M.; Roze, E.; Damier, P.; Jedynak, C.P.; Camuzat, A.; Lagueny, A.; Vercueil, L.; Doummar, D.; et al. Epsilon sarcoglycan mutations and phenotype in French patients with myoclonic syndromes. J. Med. Genet. 2006, 43, 394–400. [Google Scholar] [CrossRef]
- Angelini, C.; Fanin, M. Pathogenesis, clinical features and diagnosis of sarcoglycanopathies. Expert. Opin. Orphan Drugs 2016, 4, 1239–1251. [Google Scholar] [CrossRef]
- Guimarães-Costa, R.; Fernández-Eulate, G.; Wahbi, K.; Leturcq, F.; Malfatti, E.; Behin, A.; Leonard-Louis, S.; Desguerre, I.; Barnerias, C.; Nougues, M.C.; et al. Clinical correlations and long-term follow-up in 100 patients with sarcoglycanopathies. Eur. J. Neurol. 2021, 28, 660–669. [Google Scholar] [CrossRef]
- Xie, Z.; Hou, Y.; Yu, M.; Liu, Y.; Fan, Y.; Zhang, W.; Wang, Z.; Xiong, H.; Yuan, Y. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet J. Rare Dis. 2019, 14, 1–13. Available online: https://ojrd.biomedcentral.com/articles/10.1186/s13023-019-1021-9 (accessed on 3 November 2022). [CrossRef] [PubMed]
- Liu, W.; Pajusalu, S.; Lake, N.J.; Zhou, G.; Ioannidis, N.; Mittal, P.; Johnson, N.E.; Weihl, C.C.; Williams, B.A.; Albrecht, D.E.; et al. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet. Med. 2019, 21, 2512–2520. [Google Scholar] [CrossRef] [PubMed]
- Nallamilli, B.R.R.; Chakravorty, S.; Kesari, A.; Tanner, A.; Ankala, A.; Schneider, T.; da Silva, C.; Beadling, R.; Alexander, J.J.; Askree, S.H.; et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann. Clin. Transit. Neurol. 2018, 5, 1574–1587. [Google Scholar] [CrossRef] [PubMed]
- Sobrido, M.; Cacheiro, P.; Carracedo, A.; Bertram, L. Databases for neurogenetics: Introduction, overview, and challenges. Hum. Mutat. 2012, 33, 1311–1314. [Google Scholar] [CrossRef]
- World Medical Association. World Medical Association Declaration of Helsinki-Ethical Principles for Medical Research Involving Human Subjects. Bull. World Health Organ. 2001, 79, 373–374. [Google Scholar]
- Winckler, P.B.; da Silva, A.M.S.; Coimbra-Neto, A.R.; Carvalho, E.; Cavalcanti, E.B.U.; Sobreira, C.F.R.; Marrone, C.D.; Machado-Costa, M.C.; Carvalho, A.A.S.; Feio, R.H.F.; et al. Clinicogenetic lessons from 370 patients with autosomal recessive limb-girdle muscular dystrophy. Clin. Genet. 2019, 96, 341–353. [Google Scholar] [CrossRef]
- Leiden Muscular Dystrophy Pages. Available online: https://www.dmd.nl/ (accessed on 3 November 2022).
- Vainzof, M.; Souza, L.S.; Gurgel-Giannetti, J.; Zatz, M. Sarcoglycanopathies: An update. Neuromuscul. Disord. 2021, 31, 1021–1027. [Google Scholar] [CrossRef]
- Magri, F.; Nigro, V.; Angelini, C.; Mongini, T.; Mora, M.; Moroni, I.; Toscano, A.; D’angelo, M.G.; Tomelleri, G.; Siciliano, G.; et al. The italian limb girdle muscular dystrophy registry: Relative frequency, clinical features, and differential diagnosis. Muscle Nerve 2017, 55, 55–68. [Google Scholar] [CrossRef]
- Nalini, A.; Gayathri, N.; Thaha, F.; Das, S.; Shylashree, S. Sarcoglycanopathy: Clinical and histochemical characteristics in 66 patients. Neurol. India 2010, 58, 691–696. [Google Scholar] [CrossRef]
- Auteur, M.; Validateur, U.J.A.; Validateur, L.S.; Validateur, B.M. Avancées dans les Myopathies des Ceintures; Avancées de la Recherche: Garches, France, 2019. [Google Scholar]
- Kalaydjieva, L.; Gresham, D.; Calafell, F. Genetic studies of the Roma (Gypsies): A review. BMC Med. Genet. 2001, 2, 5. [Google Scholar] [CrossRef]
- OMIM Data Base.608896 SARCOGLYCAN,GAMMA; SGCG. Available online: https://www.omim.org/entry/608896 (accessed on 13 October 2022).
- Tarakci, H.; Berger, J. The sarcoglycan complex in skeletal muscle. Front. Biosci.-Landmark 2016, 21, 744–756. [Google Scholar]
- Sandona, D.; Betto, R. Sarcoglycanopathies: Molecular pathogenesis and therapeutic prospects. Expert Rev. Mol. Med. 2009, 11, E28. [Google Scholar] [CrossRef]
- Hamida, M.B.; Miladi, N.; Turki, I.; Zaiem, H. Duchenne muscular dystrophy in Tunisia: A clinical and morphological study of 77 cases. J. Neurol. Sci. 1992, 107, 60–64. [Google Scholar] [CrossRef]
- Mercuri, E.; Pichiecchio, A.; Allsop, J.; Messina, S.; Pane, M.; Muntoni, F. Muscle MRI in inherited neuromuscular disorders: Past, present, and future. J. Magn. Reson. Imaging 2007, 25, 433–440. [Google Scholar] [CrossRef]
- Recenti, M.; Ricciardi, C.; Edmunds, K.; Jacob, D.; Gambacorta, M.; Gargiulo, P. Testing soft tissue radiodensity parameters interplay with age and self-reported physical activity. Eur. J. Transl. Myol. 2021, 31, 9929. [Google Scholar] [CrossRef]
- Recenti, M.; Ricciardi, C.; Edmunds, K.; Gislason, M.K.; Gargiulo, P. Machine learning predictive system based upon radiodensitometric distributions from mid-thigh CT images. Eur. J. Transl. Myol. 2020, 30, 8892. [Google Scholar] [CrossRef]
- Gargiulo, P.; Helgason, T.; Ramon, C.; Jónsson, H.; Carraro, U., Jr. CT and MRI Assessment and Characterization Using Segmentation and 3D Modeling Techniques: Applications to Muscle, Bone and Brain. Eur. J. Transl. Myol. 2014, 24, 3298. [Google Scholar] [CrossRef]
- Chabbi, N. Duchenne, de la Mutation a la Reparation, Part II. Scholarpersonnal Communication, YouTube. Available online: https://www.youtube.com/watch?v=W_7vMNWJaH0 (accessed on 14 November 2022).
- Herson, S.; Hentati, F.; Rigolet, A.; Behin, A.; Romero, N.B.; Leturcq, F.; Laforêt, P.; Maisonobe, T.; Amouri, R.; Haddad, H.; et al. A phase I trial of adeno-associated virus serotype 1-γ-sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C. Brain 2012, 135, 483–492. [Google Scholar] [CrossRef]
- Israeli, D.; Cosette, J.; Corre, G.; Amor, F.; Poupiot, J.; Stockholm, D.; Montus, M.; Gjata, B.; Richard, I. An AAV-SGCG dose-response study in a γ-sarcoglycanopathy mouse model in the context of mechanical stress. Mol. Ther.-Methods Clin. Dev. 2019, 13, 494–502. [Google Scholar] [CrossRef]
- Escobar, H.; Krause, A.; Keiper, S.; Kieshauer, J.; Müthel, S.; de Paredes, M.G.; Metzler, E.; Kühn, R.; Heyd, F.; Spuler, S. Base editing repairs an SGCA mutation in human primary muscle stem cells. JCI Insight 2021, 6, e145994. [Google Scholar] [CrossRef]
- Koronyo, Y.; Biggs, D.; Barron, E.; Boyer, D.S.; Pearlman, J.A.; Au, W.J.; Kile, S.J.; Blanco, A.; Fuchs, D.T.; Ashfaq, A.; et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight 2017, 2, e93621. [Google Scholar] [CrossRef]
- Bogdanovich, S.; McNally, E.M.; Khurana, T.S. Myostatin blockade improves function but not histopathology in a murine model of limb-girdle muscular dystrophy 2C. Muscle Nerve 2008, 37, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Soheili, T.; Gicquel, E.; Poupiot, J.; N’Guyen, L.; Le Roy, F.; Bartoli, M.; Richard, I. Rescue of sarcoglycan mutations by inhibition of endoplasmic reticulum quality control is associated with minimal structural modifications. Hum. Mutat. 2012, 33, 429–439. [Google Scholar] [CrossRef]
- Fanin, M.; Hoffman, E.P.; Angelini, C.; Pegoraro, E. Private β-and γ-sarcoglycan gene mutations: Evidence of a founder effect in Northern Italy. Hum. Mutat. 2000, 16, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Kovalchick, L.V.; Bates, K.; Statland, J.; Weihl, C.; Kang, P.B.; Lowes, L.P.; Mozaffar, T.; Straub, V.; Wicklund, M.; Heatwole, C.; et al. Patient reported quality of life in limb girdle muscular dystrophy. Neuromuscul. Disord. 2022, 32, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Peric, M.; Peric, S.; Stevanovic, J.; Milovanovic, S.; Basta, I.; Nikolic, A.; Kacar, A.; Rakocevic-Stojanovic, V. Quality of life in adult patients with limb–girdle muscular dystrophies. Acta Neurol. Belg. 2018, 118, 243–250. [Google Scholar] [CrossRef]
- Sansone, V.A.; Panzeri, M.; Montanari, M.; Apolone, G.; Gandossini, S.; Rose, M.R.; Politano, L.; Solimene, C.; Siciliano, G.; Volpi, L.; et al. Italian validation of INQoL, a quality of life questionnaire for adults with muscle diseases. Eur. J. Neurol. 2010, 17, 1178–1187. [Google Scholar] [CrossRef]
- Rodríguez, A.A.; Martínez, Ó.; Amayra, I.; López-Paz, J.F.; Al-Rashaida, M.; Lázaro, E.; Caballero, P.; Pérez, M.; Berrocoso, S.; García, M.; et al. Diseases Costs and Impact of the Caring Role on Informal Carers of Children with Neuromuscular Disease. Int. J. Environ. Res. Public Health 2021, 18, 2991. [Google Scholar] [CrossRef]
- Pegoraro, V.; Angelini, C. Circulating miR-206 as a biomarker for patients affected by severe limb girdle muscle dystrophies. Genes 2021, 12, 85. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| New Name | Old Name | Locus | Gene | Protein |
|---|---|---|---|---|
| LGMDR3 | LGMD2D | 17q21 | SGCA | α-sarcoglycan |
| LGMDR4 | LGMD2E | 4q12 | SGCB | β-sarcoglycan |
| LGMDR5 | LGMD2C | 13q12 | SGCG | λ-sarcoglycan |
| LGMDR6 | LGMD2F | 5q33 | SGCD | δ-sarcoglycan |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chabbi, N.; Angelini, C.; Rodriguez, A.A. An Update of Clinical, Epidemiological, and Psychosocial Features in Gamma-Sarcoglycanopathy. Muscles 2023, 2, 164-176. https://doi.org/10.3390/muscles2020012
Chabbi N, Angelini C, Rodriguez AA. An Update of Clinical, Epidemiological, and Psychosocial Features in Gamma-Sarcoglycanopathy. Muscles. 2023; 2(2):164-176. https://doi.org/10.3390/muscles2020012
Chicago/Turabian StyleChabbi, Naoufel, Corrado Angelini, and Alicia Aurora Rodriguez. 2023. "An Update of Clinical, Epidemiological, and Psychosocial Features in Gamma-Sarcoglycanopathy" Muscles 2, no. 2: 164-176. https://doi.org/10.3390/muscles2020012
APA StyleChabbi, N., Angelini, C., & Rodriguez, A. A. (2023). An Update of Clinical, Epidemiological, and Psychosocial Features in Gamma-Sarcoglycanopathy. Muscles, 2(2), 164-176. https://doi.org/10.3390/muscles2020012