
Identifying Monomeric Fe Species for Efficient Direct Methane Oxidation to C1 Oxygenates with H2O2 over Fe/MOR Catalysts


Abstract
:1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
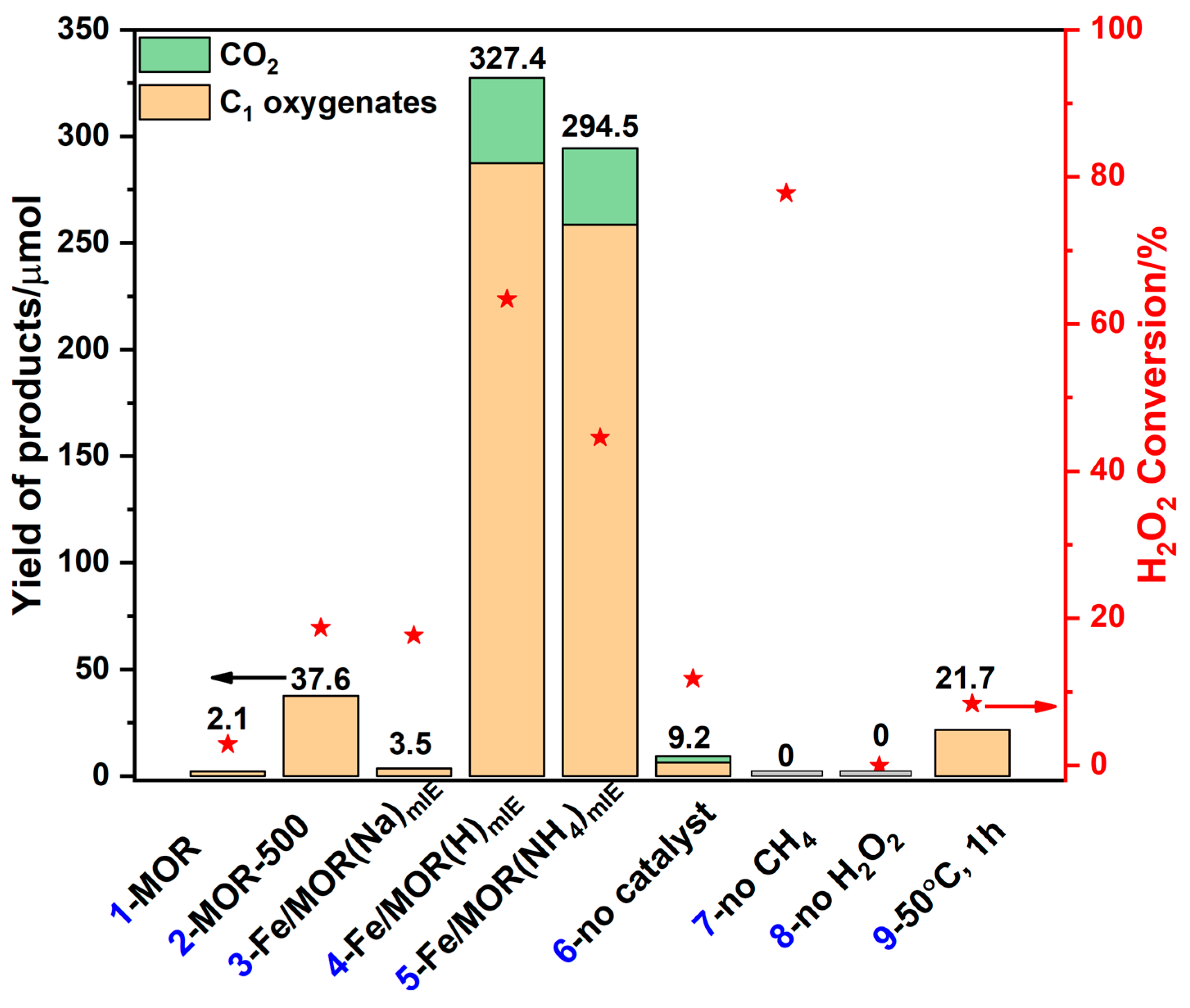
2.1.1. Different Loading Methods
2.1.2. Different Calcination Temperatures
2.1.3. MOR with Different Counter Cations
2.1.4. Different Zeolites Used as Supports
2.1.5. Different Metal Species
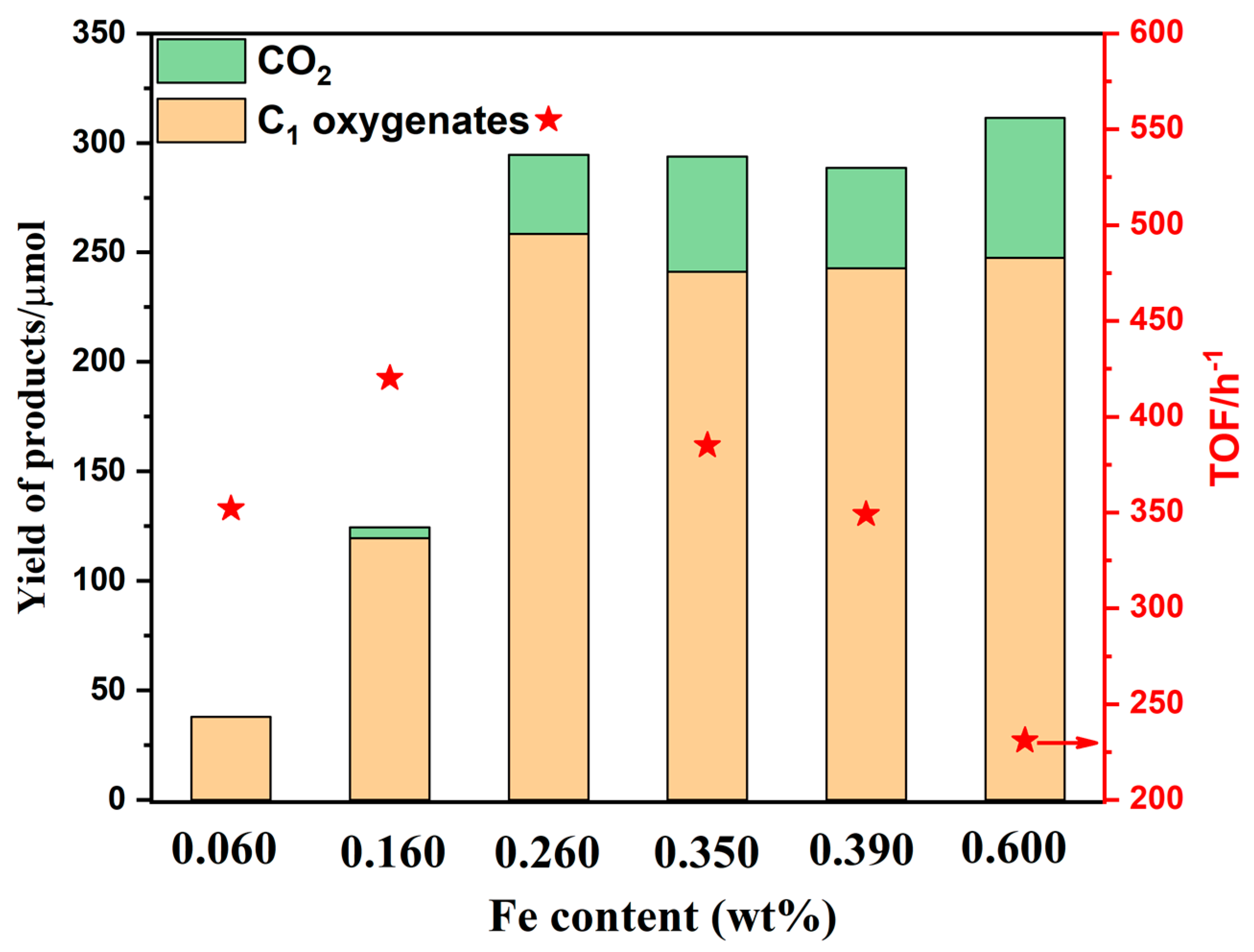
2.1.6. Different Fe Loading Contents
2.2. Experimental Methods
2.2.1. Catalytic Methane Oxidation Test
2.2.2. Analysis of Products
- Gas−phase analysis
- 2.
- Liquid−phase analysis
- 3.
- H2O2 quantification
- 4.
- The calculation of the performance indicators
- (1)
- The selectivity of the product was defined using Equation (1):where n(MeOH) refers to the molar amount of MeOH in the reactor after the reaction obtained by the NMR test.
- (2)
- The conversion of H2O2 was defined using Equation (2) or Equation (3):The peak areas of H2O2 before and after the reaction were obtained by HPLC measurements.The consumed volume of Ce(SO4)2 for reaction solution was obtained by titration.
- (3)
- The utilization ratio of H2O2 was defined using Equation (4):The consumption of H2O2 of the C1 oxygenates in Equation (4) is based on the reaction in Equation (5):
- (4)
- Turnover frequencies (TOFs) were defined as micromoles of C1 oxygenates per micromole of iron and hour (h−1), as shown in Equation (6).
- (5)
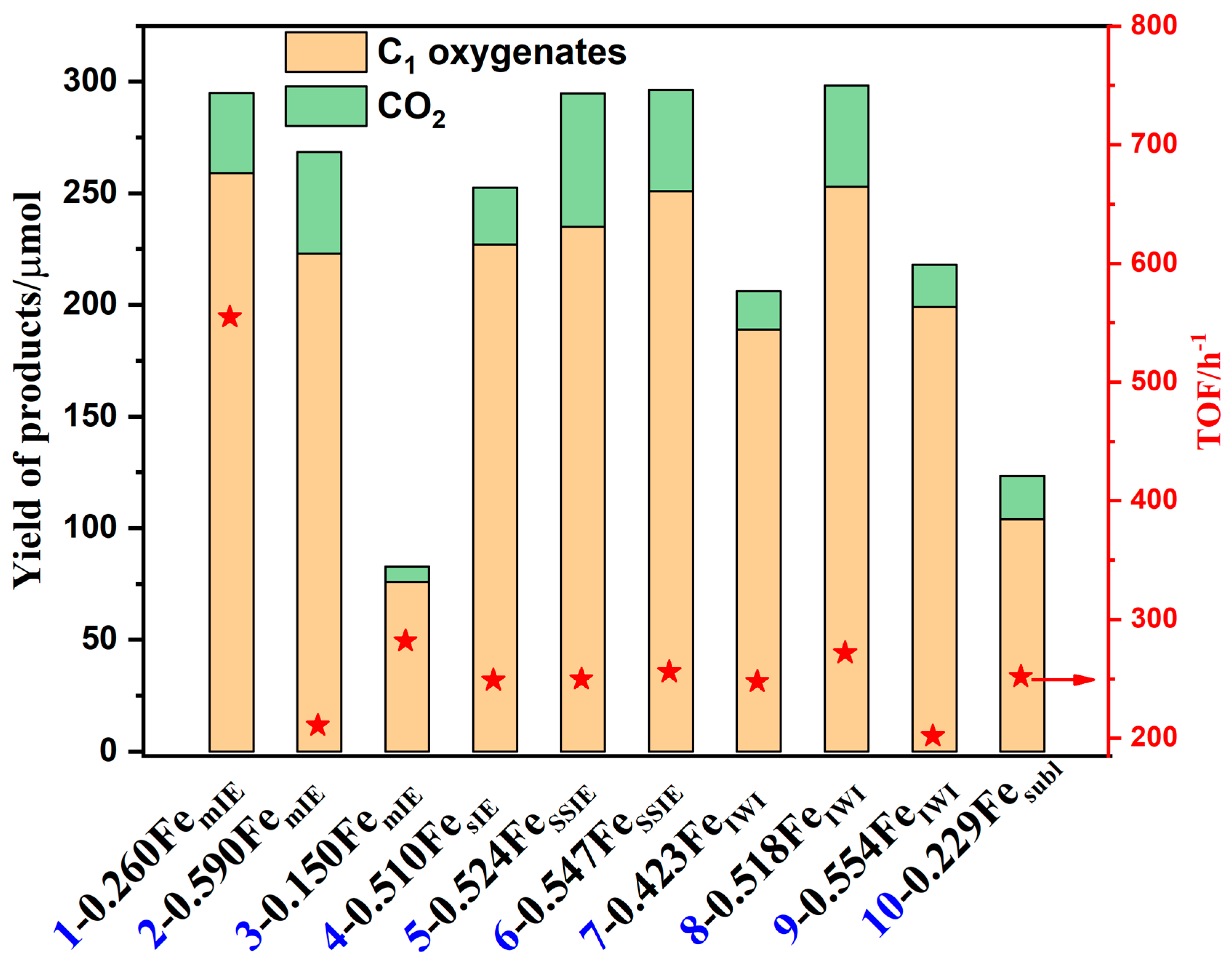
- Reproducibility of the catalytic resultsThe important catalysts in our work were tested repeatedly to ensure reproducibility, especially the 0.260Fe/MOR(NH4)mIE sample. Multiple catalytic experiments using 0.260Fe/MOR(NH4)mIE sample showed that its activity could maintain a TOF of around 555 ± 8 h−1 and a product yield of about 258 ± 5 μmol.
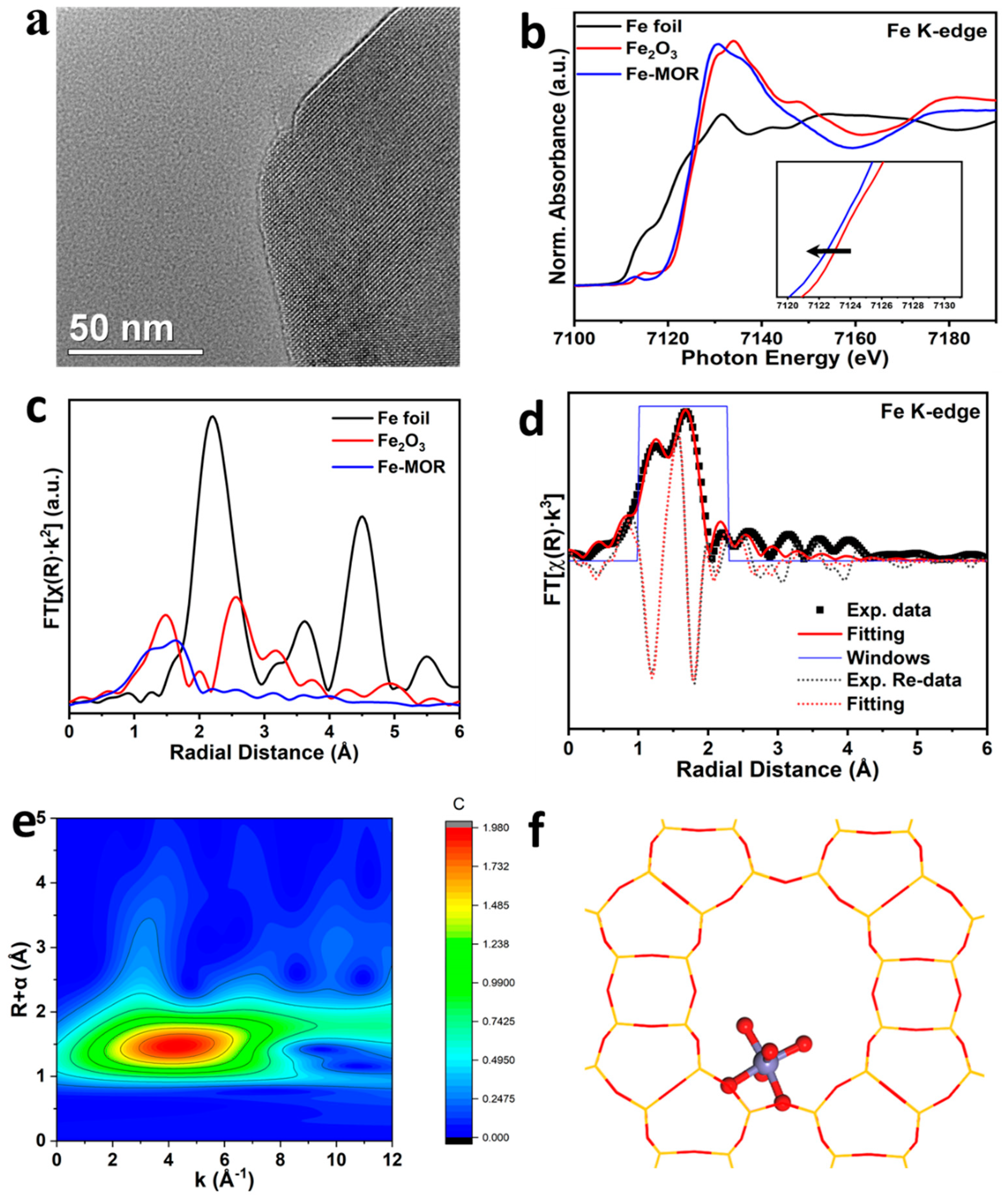
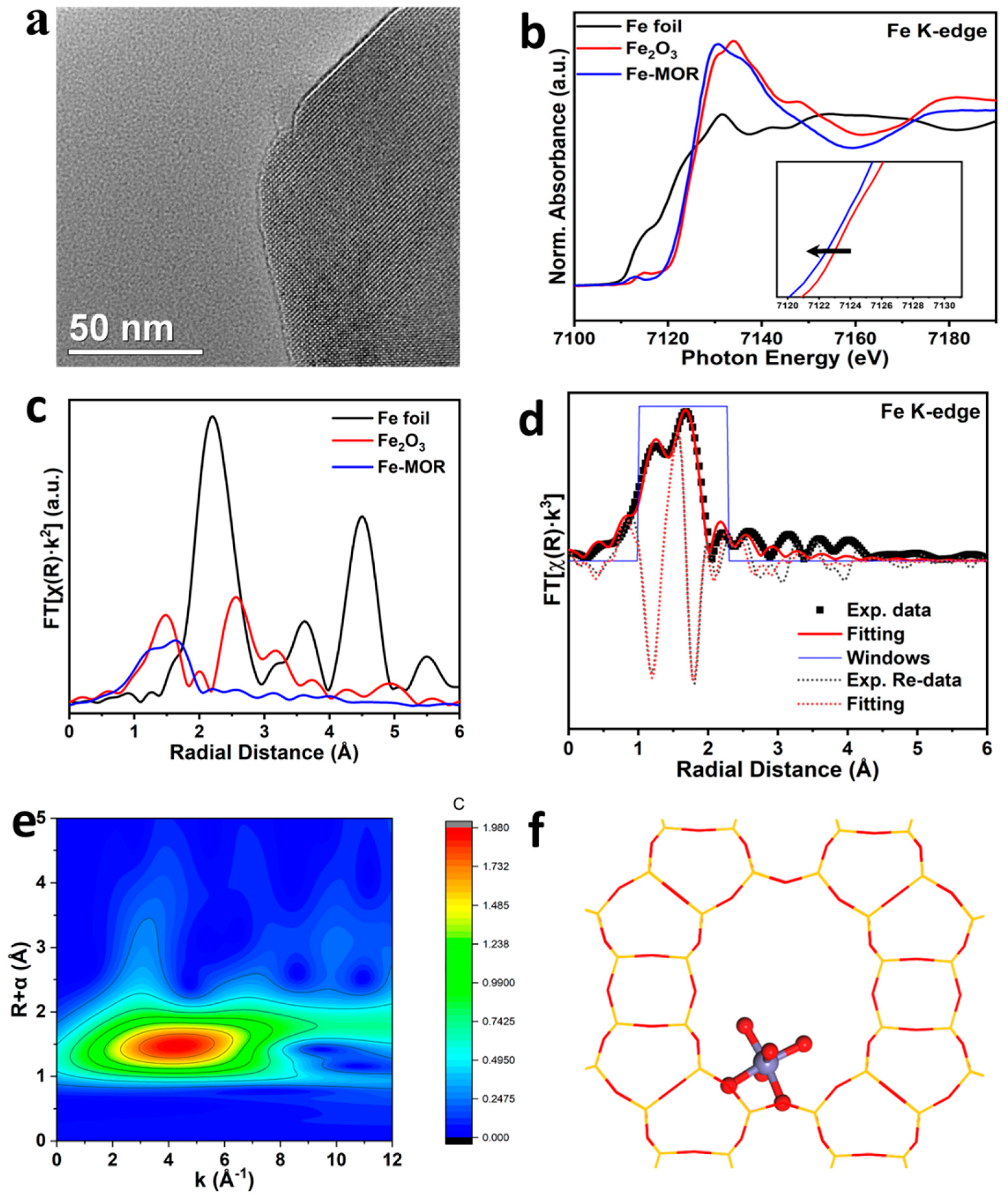
2.3. Characterizations of the Catalysts
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arndtsen, B.A.; Bergman, R.G.; Mobley, T.A.; Peterson, T.H. Selective Intermolecular Carbon−Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution. Accounts Chem. Res. 1995, 28, 154–162. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.M.; Serna, P.; Meyer, R.J.; Dincă, M.; Román−Leshkov, Y. Viewpoint on the Partial Oxidation of Methane to Methanol Using Cu− and Fe−Exchanged Zeolites. ACS Catal. 2018, 8, 8306–8313. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Li, Y.; Peng, J.; Questell-Santiago, Y.M.; Akkiraju, K.; Giordano, L.; Zheng, D.J.; Bagi, S.; Román-Leshkov, Y.; Shao-Horn, Y. Conversion of Methane into Liquid Fuels—Bridging Thermal Catalysis with Electrocatalysis. Adv. Energy Mater. 2020, 10, 2002054. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Guan, N.; Li, L. Methane Activation and Utilization: Current Status and Future Challenges. Energy Technol. 2019, 8, 1900826. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Bols, M.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes. Chem. Rev. 2018, 118, 2718–2768. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; Van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol—A Critical Assessment. Angew. Chem. Int. Ed. 2017, 56, 16464–16483. [Google Scholar] [CrossRef]
- Freakley, S.J.; Dimitratos, N.; Willock, D.J.; Taylor, S.H.; Kiely, C.J.; Hutchings, G.J. Methane Oxidation to Methanol in Water. Acc. Chem. Res. 2021, 54, 2614–2623. [Google Scholar] [CrossRef]
- Newton, M.A.; Knorpp, A.J.; Sushkevich, V.L.; Palagin, D.; Van Bokhoven, J.A. Active sites and mechanisms in the direct conversion of methane to methanol using Cu in zeolitic hosts: A critical examination. Chem. Soc. Rev. 2020, 49, 1449–1486. [Google Scholar] [CrossRef]
- Cui, X.; Li, H.; Wang, Y.; Hu, Y.; Hua, L.; Li, H.; Han, X.; Liu, Q.; Yang, F.; He, L.; et al. Room−Temperature Methane Conversion by Graphene−Confined Single Iron Atoms. Chem 2018, 4, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Hammond, C.; Forde, M.; Rahim, M.H.A.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez−Sanchez, J.A.; Dummer, N.; Murphy, D.; et al. Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by using Copper−Promoted Fe−ZSM−5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Zuo, H.; Xin, Q.; Meynen, V.; Klemm, E. Sensitivity of the selective oxidation of methane over Fe/ZSM−5 zeolites in a micro fixed−bed reactor for the catalyst preparation method. Appl. Catal. A Gen. 2018, 566, 96–103. [Google Scholar] [CrossRef]
- Rahim, M.H.A.; Forde, M.; Jenkins, R.L.; Hammond, C.; He, Q.; Dimitratos, N.; Lopez−Sanchez, J.A.; Carley, A.F.; Taylor, S.; Willock, D.J.; et al. Oxidation of Methane to Methanol with Hydrogen Peroxide Using Supported Gold−Palladium Alloy Nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Kim, T.Y.; Kwon, G.; Yi, J.; Lee, H. Selective Activation of Methane on Single−Atom Catalyst of Rhodium Dispersed on Zirconia for Direct Conversion. J. Am. Chem. Soc. 2017, 139, 17694–17699. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Liu, F.; Huang, B.; Li, F.; Lin, H.; Wu, T.; Sun, M.; Wu, J.; Shao, Q.; Xu, Y.; et al. High−efficiency direct methane conversion to oxygenates on a cerium dioxide nanowires supported rhodium single−atom catalyst. Nat. Commun. 2020, 11, 954. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, T.Y.; Zhao, X.Y.; Zhao, Y.F.; Lv, H.W.; Fang, S.; Wang, X.Q.; Zhou, F.Y.; Xu, Q.; Xu, J.; et al. A Supported Nickel Catalyst Stabilized by a Surface Digging Effect for Efficient Methane Oxidation. Angew. Chem. Int. Ed. 2019, 58, 18388–18393. [Google Scholar] [CrossRef]
- Shen, Q.; Cao, C.; Huang, R.; Zhu, L.; Zhou, X.; Zhang, Q.; Gu, L.; Song, W.-G. Single Chromium Atoms Supported on Titanium Dioxide Nanoparticles for Synergic Catalytic Methane Conversion under Mild Conditions. Angew. Chem. Int. Ed. 2019, 59, 1216–1219. [Google Scholar] [CrossRef]
- Osadchii, D.Y.; Olivos−Suarez, A.I.; Szécsényi, Á.; Li, G.; Nasalevich, M.A.; Dugulan, I.A.; Crespo, P.S.; Hensen, E.J.M.; Veber, S.L.; Fedin, M.V.; et al. Isolated Fe Sites in Metal Organic Frameworks Catalyze the Direct Conversion of Methane to Methanol. ACS Catal. 2018, 8, 5542–5548. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Shi, Y.; Jiang, Y.; Zhang, X.; Long, C.; An, P.; Zhu, Y.; Shao, S.; Yan, Z.; Li, G.; et al. Fe-O Clusters Anchored on Nodes of Metal–Organic Frameworks for Direct Methane Oxidation. Angew. Chem. Int. Ed. 2021, 60, 5811–5815. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Jones, W.; Liu, Y.; He, Q.; Song, W.; Du, P.; Yang, B.; An, H.; Farmer, D.M.; et al. Identifying key mononuclear Fe species for low−temperature methane oxidation. Chem. Sci. 2021, 12, 3152–3160. [Google Scholar] [CrossRef]
- Fang, Z.; Murayama, H.; Zhao, Q.; Liu, B.; Jiang, F.; Xu, Y.; Tokunaga, M.; Liu, X. Selective mild oxidation of methane to methanol or formic acid on Fe–MOR catalysts. Catal. Sci. Technol. 2019, 9, 6946–6956. [Google Scholar] [CrossRef]
- Qi, G.S.; Gatt, J.E.; Yang, R.T. Selective catalytic oxidation (SCO) of ammonia to nitrogen over Fe−exchanged zeolites prepared by sublimation of FeCl3. J. Catal. 2004, 226, 120–128. [Google Scholar] [CrossRef]
- Hammond, C.; Dimitratos, N.; Jenkins, R.L.; Lopez−Sanchez, J.A.; Kondrat, S.A.; ab Rahim, M.H.; Forde, M.M.; Thetford, A.; Taylor, S.H.; Hagen, H.; et al. Elucidation and Evolution of the Active Component within Cu/Fe/ZSM−5 for Catalytic Methane Oxidation: From Synthesis to Catalysis. ACS Catal. 2013, 3, 689–699. [Google Scholar] [CrossRef]
- Zuo, H.; Klemm, E. Selective oxidation of methane with H2O2 over Fe−silicalite−1: An investigation of the influence of crystal sizes, calcination temperatures and acidities. Appl. Catal. A Gen. 2019, 583, 117121. [Google Scholar] [CrossRef]
- Zheng, L.; Zhao, Y.; Tang, K.; Ma, C.; Hong, C.; Han, Y.; Cui, M.; Guo, Z. A new experiment station on beamline 4B7A at Beijing Synchrotron Radiation Facility. Spectrochim. Acta Part B At. Spectrosc. 2014, 101, 1–5. [Google Scholar] [CrossRef]
- Rehr, J.J.; Albers, R.C. Theoretical approaches to X-ray absorption fine structure. Rev. Mod. Phys. 2000, 72, 621–654. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. Athena, Artemis, Hephaestus: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, P.; Luo, Y.; Ma, Z.; Huang, C.; Miao, C.; Yue, Y.; Hua, W.; Gao, Z. Catalytic decomposition of N2O over Fe−ZSM−11 catalysts prepared by different methods: Nature of active Fe species. J. Catal. 2015, 330, 311–322. [Google Scholar] [CrossRef]
- Marturano, P.; Drozdová, L.; Kogelbauer, A.; Prins, R. Fe/ZSM−5 Prepared by Sublimation of FeCl3: The Structure of the Fe Species as Determined by IR, 27Al MAS NMR, and EXAFS Spectroscopy. J. Catal. 2000, 192, 236–247. [Google Scholar] [CrossRef]
- Oda, A.; Aono, K.; Murata, N.; Murata, K.; Yasumoto, M.; Tsunoji, N.; Sawabe, K.; Satsuma, A. Rational design of ZSM−5 zeolite containing a high concentration of single Fe sites capable of catalyzing the partial oxidation of methane with high turnover frequency. Catal. Sci. Technol. 2021, 12, 542–550. [Google Scholar] [CrossRef]
- Zhu, K.; Liang, S.; Cui, X.; Huang, R.; Wan, N.; Hua, L.; Li, H.; Chen, H.; Zhao, Z.; Hou, G.; et al. Highly efficient conversion of methane to formic acid under mild conditions at ZSM−5−confined Fe−sites. Nano Energy 2021, 82, 105718. [Google Scholar] [CrossRef]
- Yu, T.; Su, Y.; Wang, A.; Weckhuysen, B.M.; Luo, W. Efficient Synthesis of Monomeric Fe Species in Zeolite ZSM-5 for the Low-Temperature Oxidation of Methane. ChemCatChem 2021, 13, 2766–2770. [Google Scholar] [CrossRef]
- Kim, M.S.; Park, E.D. Aqueous−phase partial oxidation of methane with H2O2 over Fe−ZSM−5 catalysts prepared from different iron precursors. Microporous Mesoporous Mater. 2021, 324, 111278. [Google Scholar] [CrossRef]
- Hammond, C.; Dimitratos, N.; Lopez−Sanchez, J.A.; Jenkins, R.L.; Whiting, G.; Kondrat, S.A.; ab Rahim, M.H.; Forde, M.M.; Thetford, A.; Hagen, H.; et al. Aqueous−Phase Methane Oxidation over Fe−MFI Zeolites; Promotion through Isomorphous Framework Substitution. ACS Catal. 2013, 3, 1835–1844. [Google Scholar] [CrossRef]
- Schwidder, M.; Kumar, M.S.; Klementiev, K.; Pohl, M.M.; Brückner, A.; Grünert, W. Selective reduction of NO with Fe−ZSM−5 catalysts of low Fe content: I. Relations between active site structure and catalytic performance. J. Catal. 2005, 231, 314–330. [Google Scholar] [CrossRef]
- Santhosh Kumar, M.; Schwidder, M.; Grunert, W.; Bentrup, U.; Brückner, A. Selective reduction of NO with Fe−ZSM−5 catalysts of low Fe content: Part II. Assessing the function of different Fe sites by spectroscopic in situ studies. J. Catal. 2006, 239, 173–186. [Google Scholar] [CrossRef]
- Perez−Ramirez, J.; Kumar, M.S.; Bruckner, A. Reduction of N2O with CO over Fe−MFI zeolites: Influence of the preparation method on the iron species and catalytic behavior. J. Catal. 2004, 223, 13–27. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Vanelderen, P.; Bols, M.; Hallaert, S.D.; Böttger, L.H.; Ungur, L.; Pierloot, K.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. The active site of low−temperature methane hydroxylation in iron−containing zeolites. Nature 2016, 536, 317–321. [Google Scholar] [CrossRef]
- Shahami, M.; Shantz, D.F. Zeolite acidity strongly influences hydrogen peroxide activation and oxygenate selectivity in the partial oxidation of methane over M,Fe−MFI (M: Ga, Al, B) zeolites. Catal. Sci. Technol. 2019, 9, 2945–2951. [Google Scholar] [CrossRef]
- Hammond, C.; Jenkins, R.L.; Dimitratos, N.; Lopez−Sanchez, J.A.; Rahim, M.H.A.; Forde, M.M.; Thetford, A.; Murphy, D.M.; Hagen, H.; Stangland, E.E.; et al. Catalytic and Mechanistic Insights of the Low−Temperature Selective Oxidation of Methane over Cu−Promoted Fe−ZSM−5. Chem. A Eur. J. 2012, 18, 15735–15745. [Google Scholar] [CrossRef]
- Choi, S.H.; Wood, B.R.; Ryder, A.J.A.; Bell, A. X-ray Absorption Fine Structure Characterization of the Local Structure of Fe in Fe−ZSM−5. J. Phys. Chem. B 2003, 107, 11843–11851. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Lin, L.; Chu, S.; Su, Y.; Song, W.; Wang, A.; Weckhuysen, B.M.; Luo, W. Highly Selective Oxidation of Methane into Methanol over Cu−Promoted Monomeric Fe/ZSM−5. ACS Catal. 2021, 11, 6684–6691. [Google Scholar] [CrossRef]
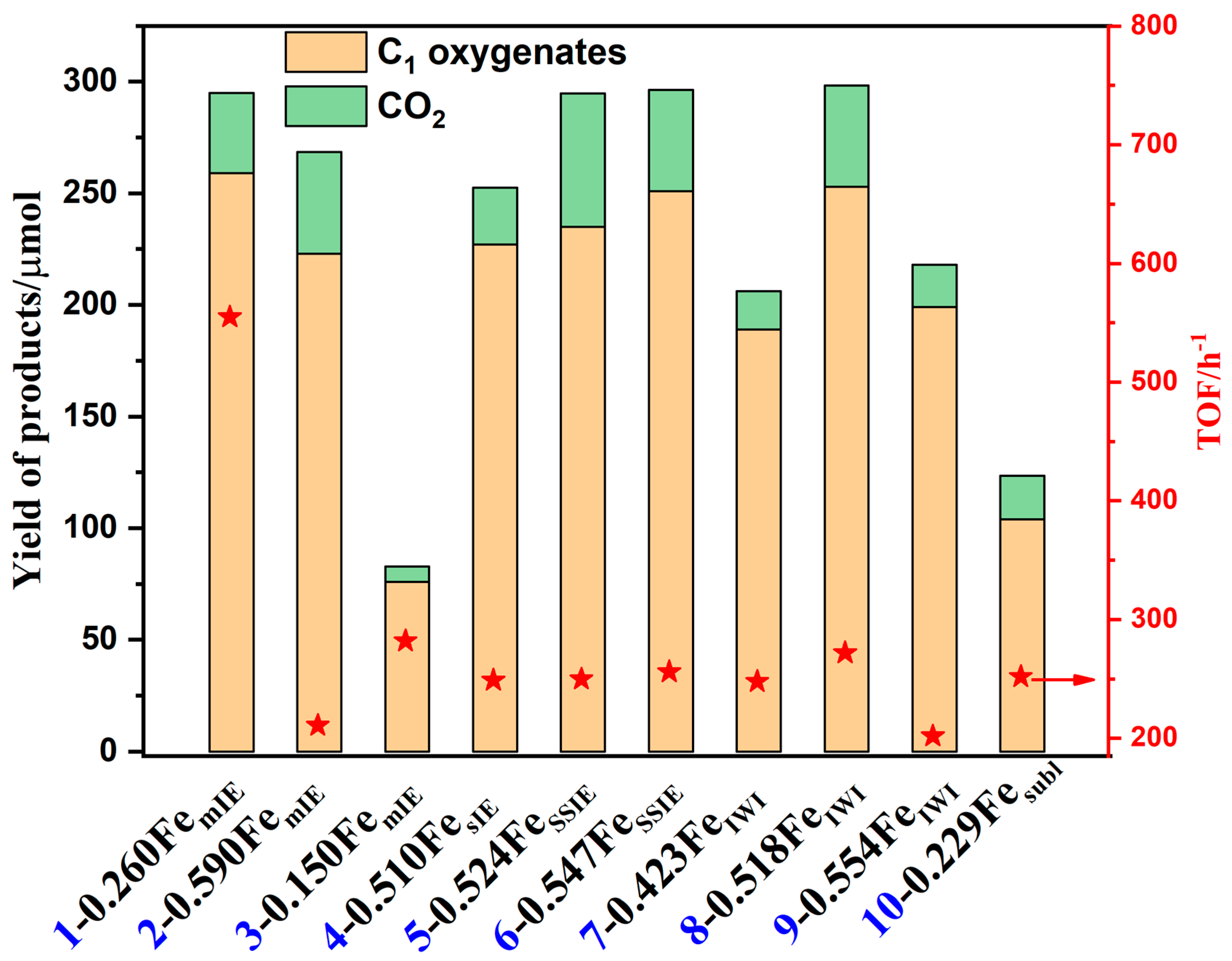
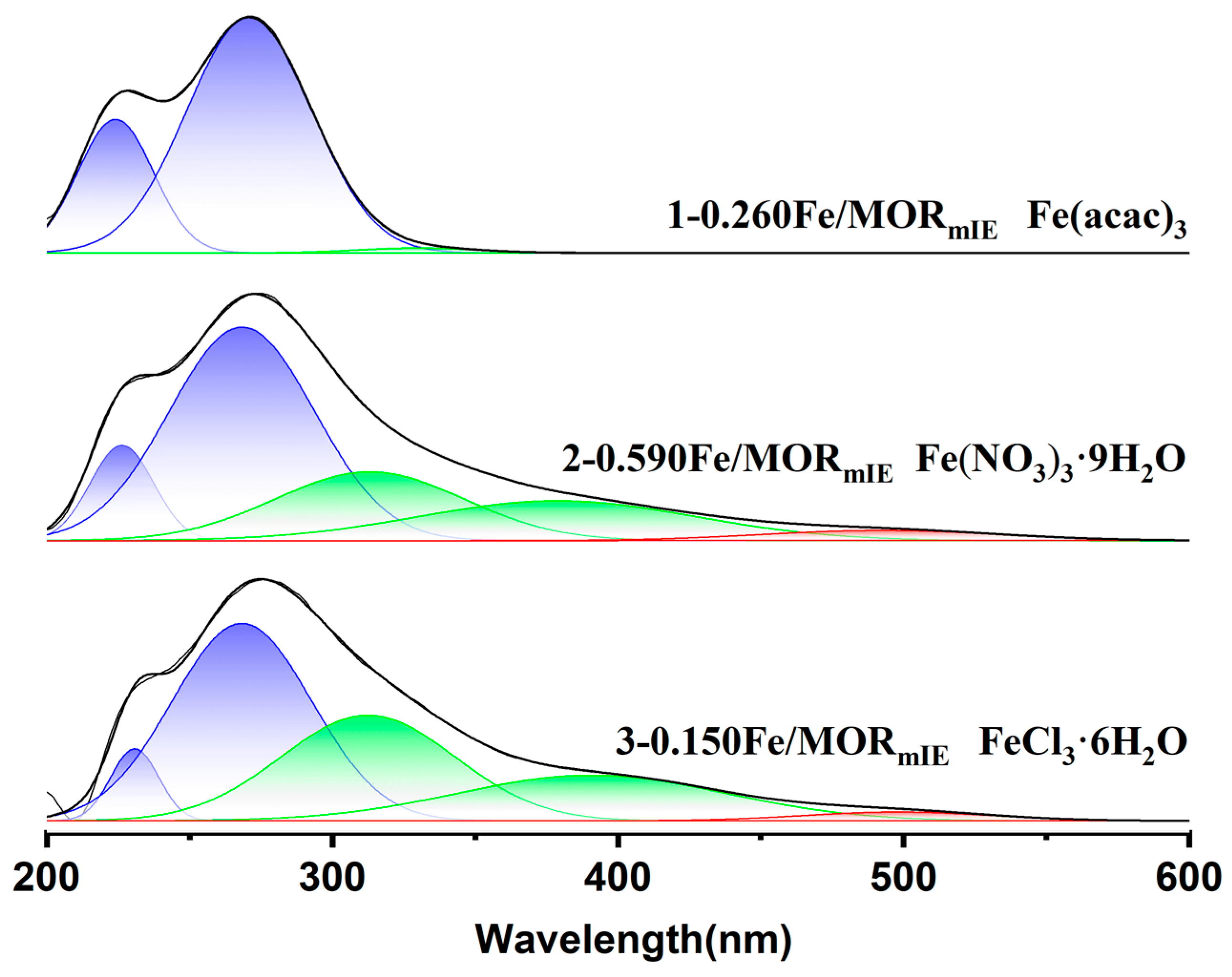
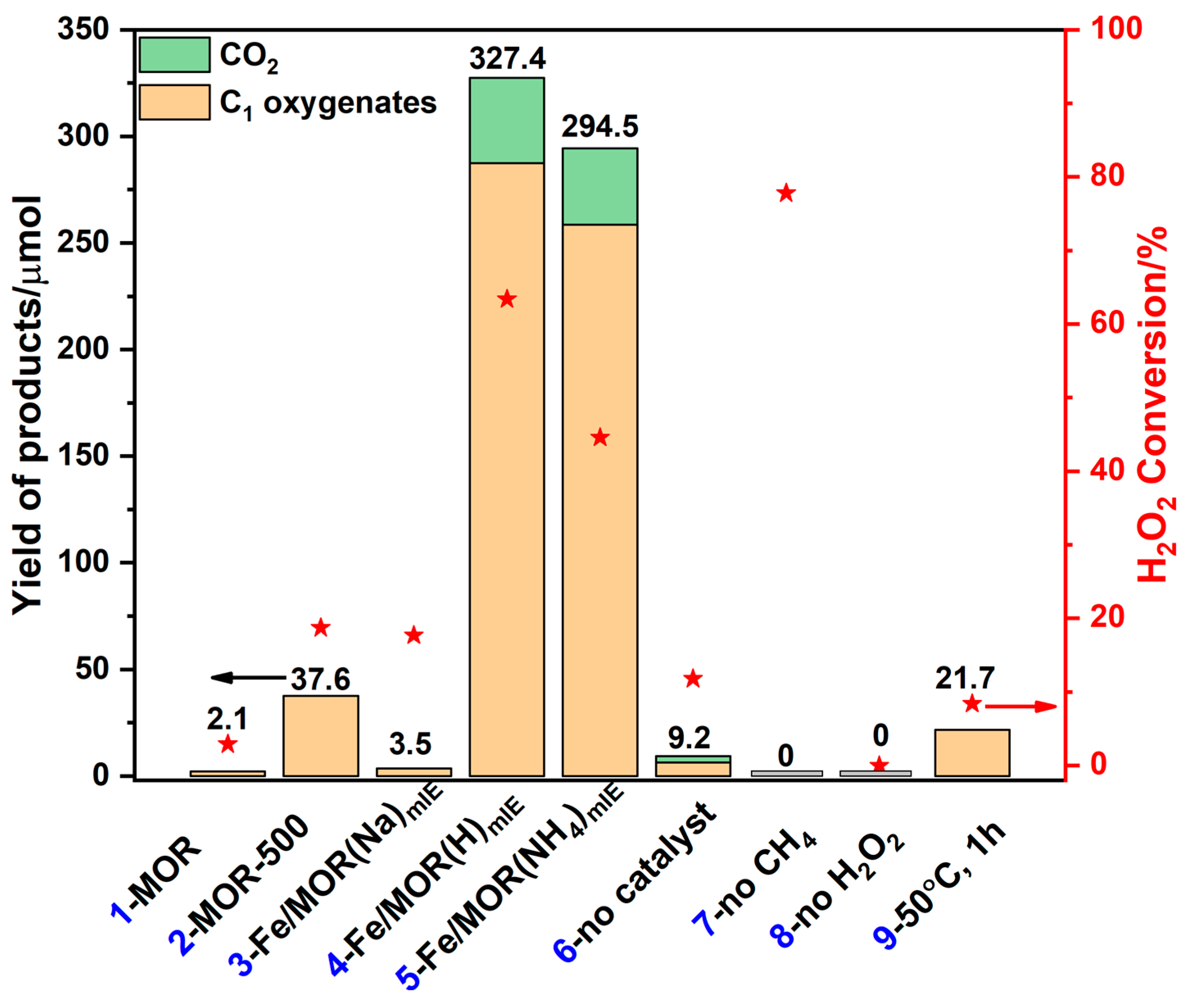
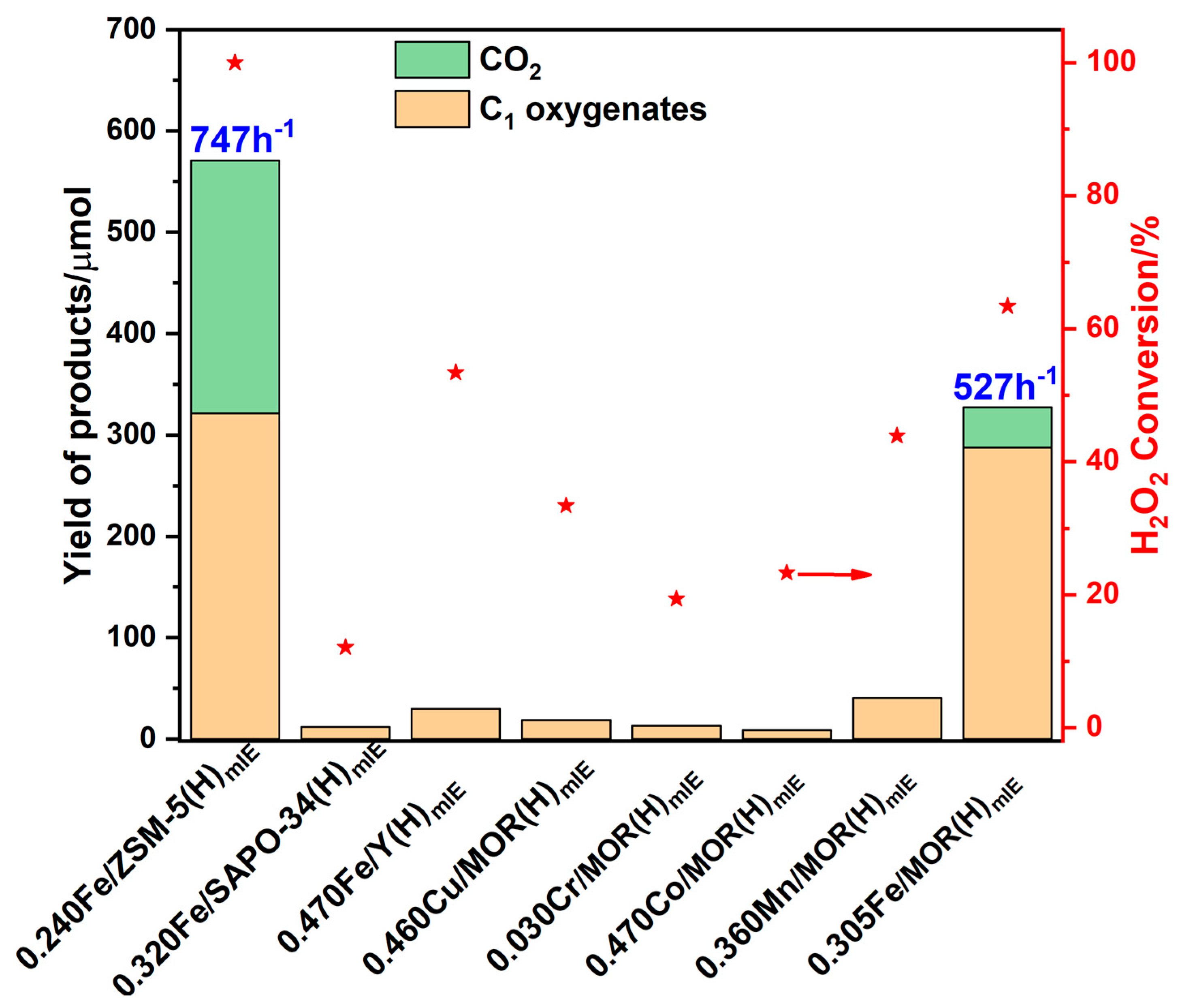


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Relative Contribution of Different Range (%) a | ||
|---|---|---|---|
| I1 (200~300 nm) | I2 (300~400 nm) | I3 (>400 nm) | |
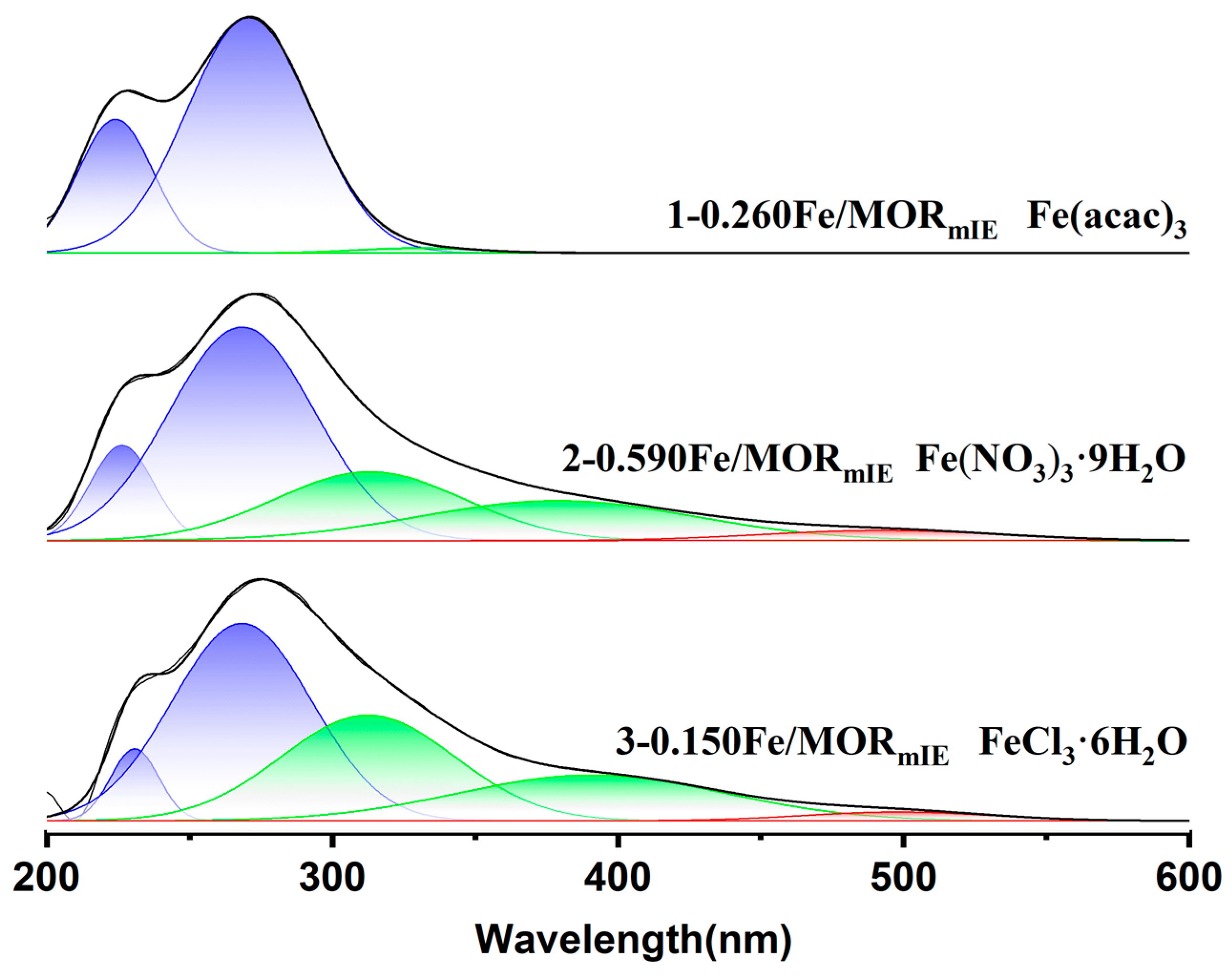
| 1−0.260Fe/MORmIE | 98.7 | 1.3 | 0 |
| 2−0.590Fe/MORmIE | 72.2 | 25.4 | 2.4 |
| 3−0.150Fe/MORmIE | 62.7 | 35.2 | 2.1 |
| Catalysts | Relative Contribution of Different Ranges (%) a | ||
|---|---|---|---|
| I1 (200~300 nm) | I2 (300~400 nm) | I3 (>400 nm) | |
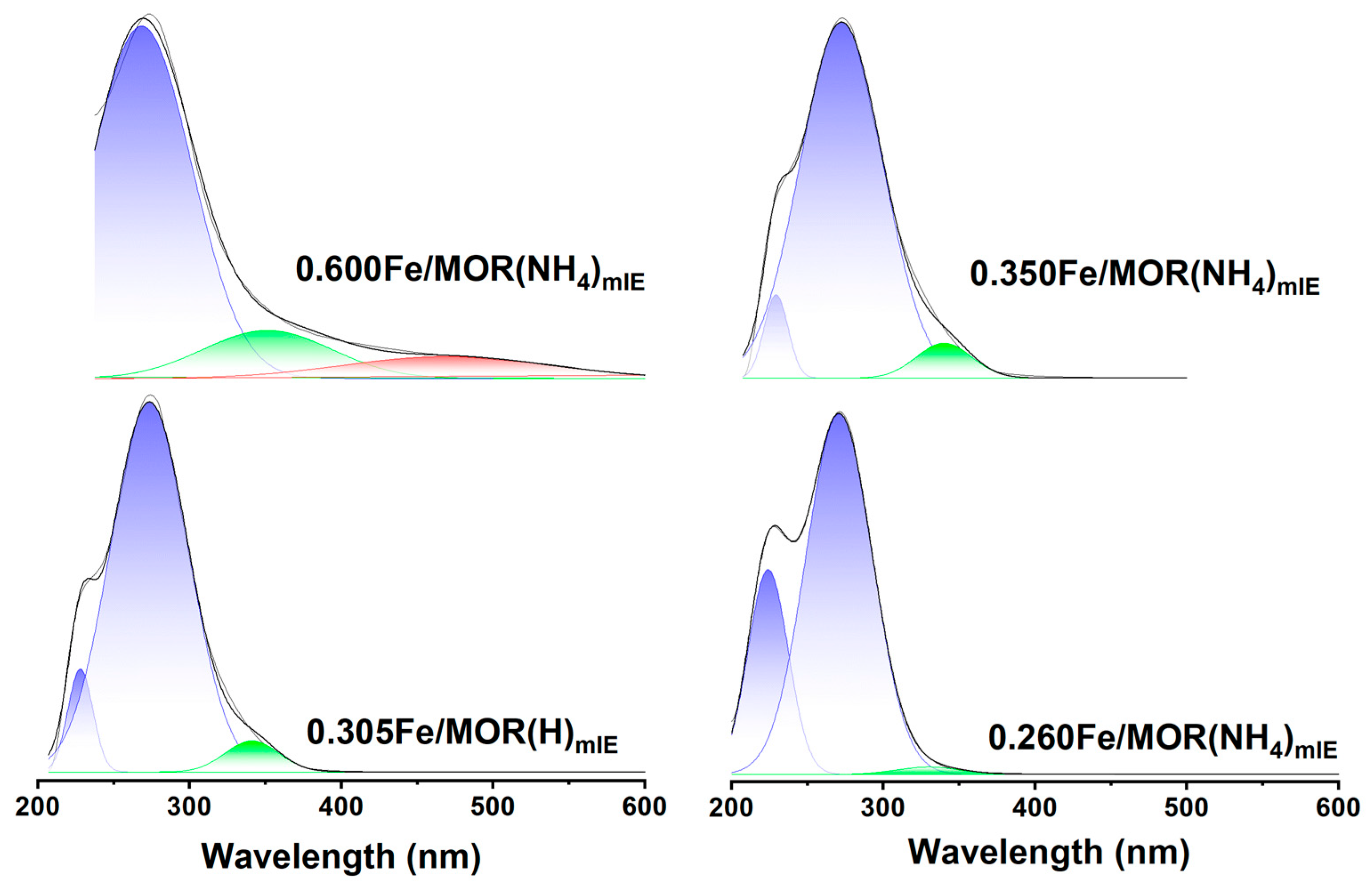
| 0.260Fe/MOR(NH4)mIE | 98.7 | 1.3 | 0 |
| 0.305Fe/MOR(H)mIE | 95.2 | 4.8 | 0 |
| 0.350Fe/MOR(NH4)mIE | 92.6 | 7.4 | 0 |
| 0.600Fe/MOR(NH4)mIE | 83.4 | 11.4 | 5.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, C.; Song, Q.; Merdanoglu, N.; Liu, H.; Klemm, E. Identifying Monomeric Fe Species for Efficient Direct Methane Oxidation to C1 Oxygenates with H2O2 over Fe/MOR Catalysts. Methane 2022, 1, 107-124. https://doi.org/10.3390/methane1020010
Xu C, Song Q, Merdanoglu N, Liu H, Klemm E. Identifying Monomeric Fe Species for Efficient Direct Methane Oxidation to C1 Oxygenates with H2O2 over Fe/MOR Catalysts. Methane. 2022; 1(2):107-124. https://doi.org/10.3390/methane1020010
Chicago/Turabian StyleXu, Caiyun, Qian Song, Nagme Merdanoglu, Hang Liu, and Elias Klemm. 2022. "Identifying Monomeric Fe Species for Efficient Direct Methane Oxidation to C1 Oxygenates with H2O2 over Fe/MOR Catalysts" Methane 1, no. 2: 107-124. https://doi.org/10.3390/methane1020010
APA StyleXu, C., Song, Q., Merdanoglu, N., Liu, H., & Klemm, E. (2022). Identifying Monomeric Fe Species for Efficient Direct Methane Oxidation to C1 Oxygenates with H2O2 over Fe/MOR Catalysts. Methane, 1(2), 107-124. https://doi.org/10.3390/methane1020010