
Azapeptides as an Efficient Tool to Improve the Activity of Biologically Effective Peptides



Abstract
:1. Introduction
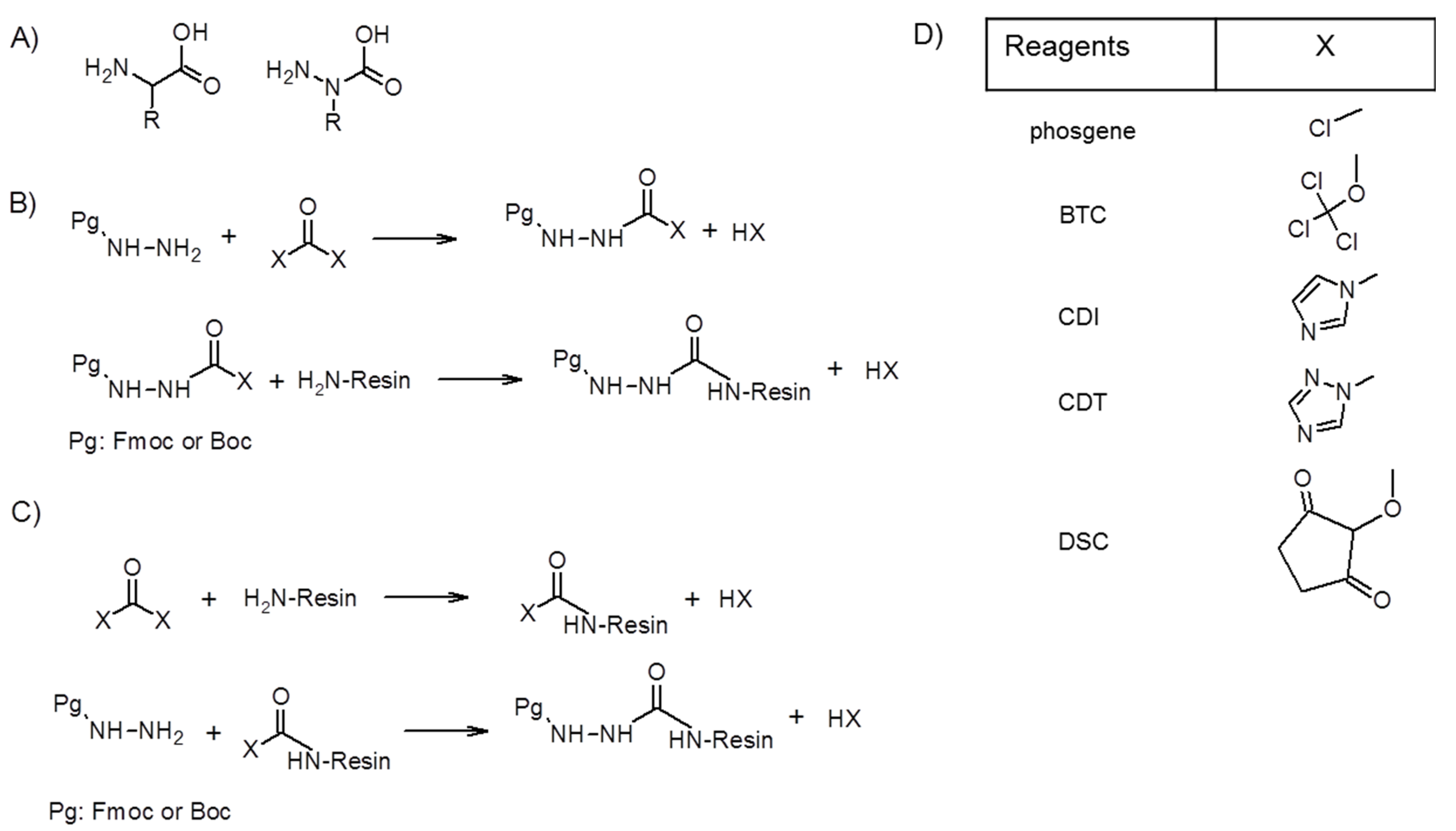
2. Synthesis of Azapeptides
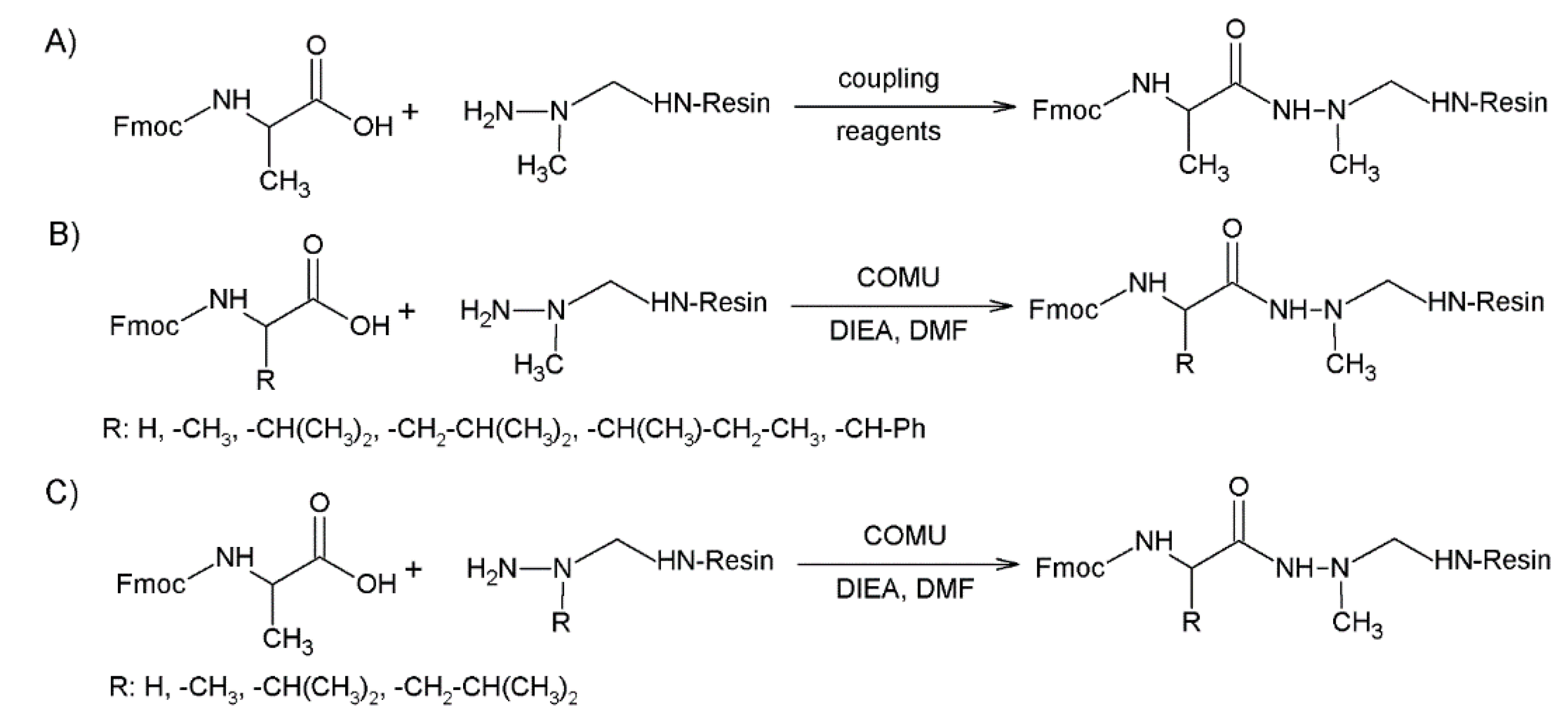
2.1. Incorporation of N-Alkylated Hydrazine Derivatives
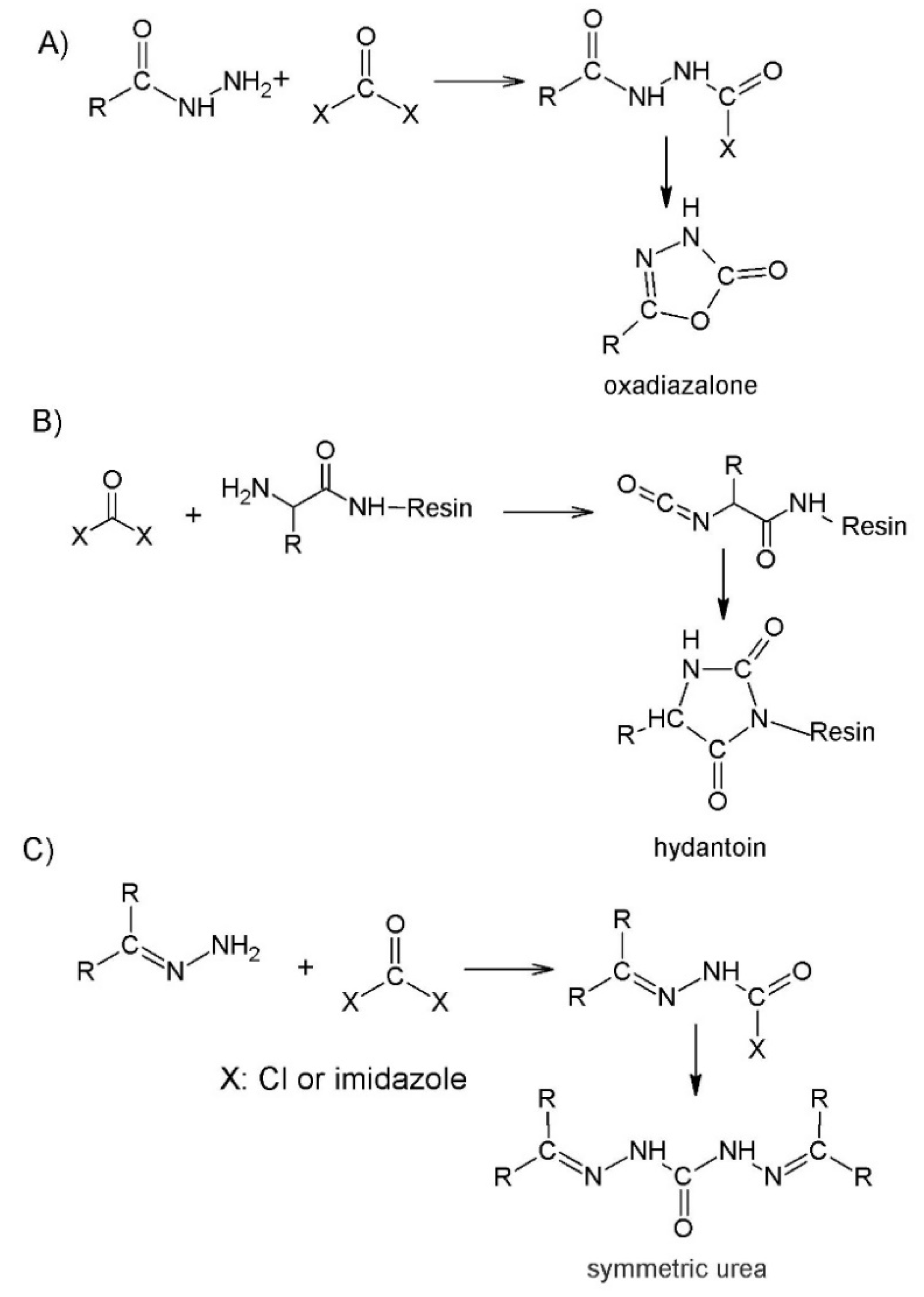
2.2. Submonomer Synthesis of Azapeptides
2.3. Coupling the Next Amino Acid to Azaamino Acid on Resin
3. The Effect of Azaamino Acid Substitution on the Peptide Structure
4. Biological Activity of Azapeptides
4.1. Enzyme Inhibitors
4.2. Receptor Ligands
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide chemistry toolbox—Transforming natural peptides into peptide therapeutics. Bioorg. Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef]
- Avan, I.; Dennis Hall, C.; Katritzky, A.R. Peptidomimetics via modifications of amino acids and peptide bonds. Chem. Soc. Rev. 2014, 43, 3575–3594. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.M.; Cabalteja, C.C.; Horne, W.S. Peptide Backbone Composition and Protease Susceptibility: Impact of Modification Type, Position, and Tandem Substitution. Chembiochem 2016, 17, 712–718. [Google Scholar] [CrossRef]
- Frey, V.; Viaud, J.; Subra, G.; Cauquil, N.; Guichou, J.F.; Casara, P.; Grassy, G.; Chavanieu, A. Structure-activity relationships of Bak derived peptides: Affinity and specificity modulations by amino acid replacement. Eur. J. Med. Chem. 2008, 43, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Hess, H.J.; Moreland, W.T.; Laubach, G.D. N- [2-Isopropyl-3-(-aspartyl-l-arginyl)-carbazoyl]-l-tyrosyl-l-valyl-l-histidyl-l-prolyl-l-phenylalanine, an Isostere of Bovine Angiotensin II. J. Am. Chem. Soc. 1963, 85, 4040–4041. [Google Scholar] [CrossRef]
- Proulx, C.; Sabatino, D.; Hopewell, R.; Spiegel, J.; García Ramos, Y.; Lubell, W.D. Azapeptides and their therapeutic potential. Future Med. Chem. 2011, 3, 1139–1164. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.C.; Carroll, D.L. Reaction of acyl carbazates with proteolytic enzymes. Biochem. Biophys. Res. Commun. 1975, 67, 639–644. [Google Scholar] [CrossRef]
- Magrath, J.; Abeles, R.H. Cysteine Protease Inhibition by Azapeptide Esters. J. Med. Chem. 1992, 35, 4279–4283. [Google Scholar] [CrossRef]
- Powers, J.C.; Boone, R.; Carroll, D.L.; Gupton, B.F.; Kam, C.M.; Nishino, N.; Sakamoto, M.; Tuhy, P.M. Reaction of azapeptides with human leukocyte elastase and porcine pancreatic elastase. New inhibitors and active site titrants. J. Biol. Chem. 1984, 259, 4288–4294. [Google Scholar] [CrossRef]
- Chingle, R.; Mulumba, M.; Chung, N.N.; Nguyen, T.M.D.; Ong, H.; Ballet, S.; Schiller, P.W.; Lubell, W.D. Solid-Phase Azopeptide Diels-Alder Chemistry for Aza-pipecolyl Residue Synthesis to Study Peptide Conformation. J. Org. Chem. 2019, 84, 6006–6016. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Beckmann, A.M.; Dudic, A.; Li, T.; Sellier, R.; Bartz, U.; Gütschow, M. 3-Cyano-3-aza-β-aminoAcid Derivativesas Inhibitors of Human Cysteine Cathepsins. ACS Med. Chem. Lett. 2014, 5, 1076. [Google Scholar] [CrossRef]
- Miyata, K.; Narita, A.; Fujisawa, R.; Roppongi, M.; Ito, S.; Shingo, T.; Oba, T. Synthesis of boronophenylalanine-like aza-amino acids for boron-containing azapeptide precursors. Tetrahedron Lett. 2020, 61, 152585. [Google Scholar] [CrossRef]
- Boeglin, D.; Xiang, Z.; Sorenson, N.B.; Wood, M.S.; Haskell-Luevano, C.; Lubell, W.D. Aza-scanning of the potent melanocortin receptor agonist Ac-His-D-Phe-Arg-Trp-NH. Chem. Biol. Drug Des. 2006, 67, 275–283. [Google Scholar] [CrossRef]
- Chingle, R.; Proulx, C.; Lubell, W.D. Azapeptide Synthesis Methods for Expanding Side-Chain Diversity for Biomedical Applications. Acc. Chem. Res. 2017, 50, 1541–1556. [Google Scholar] [CrossRef]
- Zhang, Y.; Malamakal, R.M.; Chenoweth, D.M. Aza-Glycine Induces Collagen Hyperstability. J. Am. Chem. Soc. 2015, 137, 12422–12425. [Google Scholar] [CrossRef]
- Mcmechen, M.A.; Willis, E.L.; Gourville, P.C.; Proulx, C.; Mosberg, H.; Sawyer, T.; Haskell-Luevano, C. Aza-Amino Acids Disrupt β-Sheet Secondary Structures. Molecules 2019, 24, 1919. [Google Scholar] [CrossRef]
- Kasznel, A.J.; Harris, T.; Porter, N.J.; Zhang, Y.; Chenoweth, D.M. Aza-proline effectively mimics L-proline stereochemistry in triple helical collagen. Chem. Sci. 2019, 10, 6979–6983. [Google Scholar] [CrossRef]
- Galibert, M.; Wartenberg, M.; Lecaille, F.; Saidi, A.; Mavel, S.; Joulin-Giet, A.; Korkmaz, B.; Brömme, D.; Aucagne, V.; Delmas, A.F.; et al. Substrate-derived triazolo- and azapeptides as inhibitors of cathepsins K and S. Eur. J. Med. Chem. 2018, 144, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Melton, S.D.; Brackhahn, E.A.E.; Orlin, S.J.; Jin, P.; Chenoweth, D.M. Rules for the design of aza-glycine stabilized triple-helical collagen peptides. Chem. Sci. 2020, 11, 10638–10646. [Google Scholar] [CrossRef] [PubMed]
- Traoré, M.; Gignac, M.; Doan, N.D.; Hof, F.; Lubell, W.D. Aza-amino acid scanning of chromobox homolog 7 (CBX7) ligands. J. Pept. Sci. 2017, 23, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Ahsanullah; Chingle, R.; Ohm, R.G.; Chauhan, P.S.; Lubell, W.D. Aza-propargylglycine installation by aza-amino acylation: Synthesis and Ala-scan of an azacyclopeptide CD36 modulator. Pept. Sci. 2019, 111, e24102. [Google Scholar] [CrossRef]
- André, F.; Marraud, M.; Tsouloufis, T.; Tzartos, S.J.; Boussard, G. Triphosgene: An efficient carbonylating agent for liquid and solid-phase aza-peptide synthesis. Application to the synthesis of two aza-analogues of the AChR MIR decapeptide. J. Pept. Sci. 1997, 3, 429–441. [Google Scholar] [CrossRef]
- Melton, S.D.; Smith, M.S.; Chenoweth, D.M. Incorporation of Aza-Glycine into Collagen Peptides. J. Org. Chem. 2020, 85, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Shrivastava, R.; Singh, G.; Ali, R.; Sankar Ampapathi, R.; Bhadhuria, S.; Haq, W. AzaGly-Appended Peptidomimetics Structurally Related to PTR6154 as Potential PKB/Akt Inhibitors. Chembiochem 2017, 18, 1061–1065. [Google Scholar] [CrossRef]
- Ahn, I.A.; Kim, S.W.; Ro, S. Solid phase synthesis of azapeptides using an automatic synthesizer. Mol. Divers. 1998, 4, 23–24. [Google Scholar] [CrossRef]
- Jȩdrzejewska, H.; Szumna, A. Peptide-based capsules with chirality-controlled functionalized interiors—Rational design and amplification from dynamic combinatorial libraries. Chem. Sci. 2019, 10, 4412–4421. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.S.; Morley, J.S. Polypeptides. Part XIII. Preparation of α-aza-amino-acid (carbazic acid) derivatives and intermediates for the preparation of α-aza-peptides. J. Chem. Soc. Perkin Trans. 1 1975, 1, 1712–1720. [Google Scholar] [CrossRef]
- Melendez, R.E.; Lubell, W.D. Aza-amino acid scan for rapid identification of secondary structure based on the application of N-Boc-aza1-dipeptides in peptide synthesis. J. Am. Chem. Soc. 2004, 126, 6759–6764. [Google Scholar] [CrossRef] [PubMed]
- Frochot, C.; Vanderesse, R.; Driou, A.; Linden, G.; Marraud, M.; Cung, M.T. A solid-phase synthesis of three aza-, iminoaza- and reduced aza-peptides from the same precursor. Lett. Pept. Sci. 1997, 4, 219–225. [Google Scholar] [CrossRef]
- Boeglin, D.; Lubell, W.D. Aza-amino acid scanning of secondary structure suited for solid-phase peptide synthesis with Fmoc chemistry and aza-amino acids with heteroatomic side chains. J. Comb. Chem. 2005, 7, 864–878. [Google Scholar] [CrossRef]
- Freeman, N.S.; Hurevich, M.; Gilon, C. Synthesis of N’-substituted Ddz-protected hydrazines and their application in solid phase synthesis of aza-peptides. Tetrahedron 2009, 65, 1737–1745. [Google Scholar] [CrossRef]
- Bourguet, C.B.; Sabatino, D.; Lubell, W.D. Benzophenone semicarbazone protection strategy for synthesis of aza-glycine containing aza-peptides. Biopolymers 2008, 90, 824–831. [Google Scholar] [CrossRef]
- Zhang, J.; Mulumba, M.; Ong, H.; Lubell, W.D. Diversity-Oriented Synthesis of Cyclic Azapeptides by A3-Macrocyclization Provides High-Affinity CD36-Modulating Peptidomimetics. Angew. Chemie Int. Ed. 2017, 56, 6284–6288. [Google Scholar] [CrossRef]
- Sabatino, D.; Proulx, C.; Klocek, S.; Bourguet, C.B.; Boeglin, D.; Ong, H.; Lubell, W.D. Exploring side-chain diversity by Submonomer solid-phase aza-peptide synthesis. Org. Lett. 2009, 11, 3650–3653. [Google Scholar] [CrossRef]
- Garcia-Ramos, Y.; Lubell, W.D. Synthesis and alkylation of aza-glycinyl dipeptide building blocks. J. Pept. Sci. 2013, 19, 725–729. [Google Scholar] [CrossRef]
- Chignen Possi, K.; Mulumba, M.; Omri, S.; Garcia-Ramos, Y.; Tahiri, H.; Chemtob, S.; Ong, H.; Lubell, W.D. Influences of Histidine-1 and Azaphenylalanine-4 on the Affinity, Anti-inflammatory, and Antiangiogenic Activities of Azapeptide Cluster of Differentiation 36 Receptor Modulators. J. Med. Chem. 2017, 60, 9263–9274. [Google Scholar] [CrossRef]
- Dai, C.; Ma, J.; Li, M.; Wu, W.; Xia, X.; Zhang, J. Diversity-oriented submonomer synthesis of azapeptides mediated by the Mitsunobu reaction. Org. Chem. Front. 2019, 6, 2529–2533. [Google Scholar] [CrossRef]
- Doan, N.D.; Zhang, J.; Traoré, M.; Kamdem, W.; Lubell, W.D. Solid-phase synthesis of C-terminal azapeptides. J. Pept. Sci. 2015, 21, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Bánóczi, Z.; Tantos, Á.; Farkas, A.; Majer, Z.; Dókus, L.E.; Tompa, P.; Hudecz, F. New m-calpain substrate-based azapeptide inhibitors. J. Pept. Sci. 2013, 19, 370–376. [Google Scholar] [CrossRef]
- Arujõe, M.; Ploom, A.; Mastitski, A.; Järv, J. Comparison of various coupling reagents in solid-phase aza-peptide synthesis. Tetrahedron Lett. 2017, 58, 3421–3425. [Google Scholar] [CrossRef]
- Arujõe, M.; Ploom, A.; Mastitski, A.; Järv, J. Influence of steric effects in solid-phase aza-peptide synthesis. Tetrahedron Lett. 2018, 59, 2010–2013. [Google Scholar] [CrossRef]
- Troska, A.; Arujõe, M.; Mastitski, A.; Järv, J.; Ploom, A. Steric impact of aza-amino acid on solid-phase aza-peptide bond synthesis. Tetrahedron Lett. 2021, 69, 152973. [Google Scholar] [CrossRef]
- André, F.; Vicherat, A.; Boussard, G.; Aubry, A.; Marraud, M. Aza-peptides. III. Experimental structural analysis of aza-alanine and aza-asparagine-containing peptides. J. Pept. Res. 1997, 50, 372–381. [Google Scholar] [CrossRef]
- Benatalah, Z.; Aubry, A.; Boussard, G.; Marraud, M. Evidence for a β-turn in an azadipeptide sequence. Int. J. Pept. Protein Res. 1991, 38, 603–605. [Google Scholar] [CrossRef]
- Adhikary, R.; Zimmermann, J.; Liu, J.; Forrest, R.P.; Janicki, T.D.; Dawson, P.E.; Corcelli, S.A.; Romesberg, F.E. Evidence of an Unusual N–H···N Hydrogen Bond in Proteins. J. Am. Chem. Soc. 2014, 136, 13474–13477. [Google Scholar] [CrossRef]
- Baruah, K.; Sahariah, B.; Sakpal, S.S.; Deka, J.K.R.; Bar, A.K.; Bagchi, S.; Sarma, B.K. Stabilization of Azapeptides by N amide ···H–N amide Hydrogen Bonds. Org. Lett. 2021, 23, 4949–4954. [Google Scholar] [CrossRef]
- Lee, H.J.; Ahn, I.A.; Ro, S.; Choi, K.H.; Choi, Y.S.; Lee, K.B. Role of azaamino acid residue in beta-turn formation and stability in designed peptide. J. Pept. Res. 2000, 56, 35–46. [Google Scholar] [CrossRef]
- El Khabchi, M.; Lahlou, H.; El Adnani, Z.; Mcharfi, M.; Benzakour, M.; Fitri, A.; Benjelloun, A.T. Conformational preferences of Ac-Pro-azaXaa-NHMe (Xaa = Asn, Asp, Ala) and the effect of intramolecular hydrogen bonds on their stability in gas phase and solution. J. Mol. Model. 2021, 27, 368. [Google Scholar] [CrossRef]
- Zhang, Y.; Herling, M.; Chenoweth, D.M. General Solution for Stabilizing Triple Helical Collagen. J. Am. Chem. Soc. 2016, 138, 9751–9754. [Google Scholar] [CrossRef]
- Zhang, Y.; Malamakal, R.M.; Chenoweth, D.M. A Single Stereodynamic Center Modulates the Rate of Self-Assembly in a Biomolecular System. Angew. Chem. Int. Ed. Engl. 2015, 54, 10826–10832. [Google Scholar] [CrossRef]
- Harris, T.; Chenoweth, D.M. Sterics and Stereoelectronics in Aza-Glycine: Impact of Aza-Glycine Preorganization in Triple Helical Collagen. J. Am. Chem. Soc. 2019, 141, 18021–18029. [Google Scholar] [CrossRef]
- Zhou, Z.; Deng, C.; Abbas, C.; Didierjean, C.; Averlant-Petit, M.-C.; Bodiguel, J.; Vanderesse, R.; Jamart-Grégoire, B. Synthesis and Structural Characterization of 2:1 [α/Aza]-oligomers. Eur. J. Org. Chem. 2014, 2014, 7643–7650. [Google Scholar] [CrossRef]
- Ibrahim, M.I.A.; Zhou, Z.; Deng, C.; Didierjean, C.; Vanderesse, R.; Bodiguel, J.; Averlant-Petit, M.C.; Jamart-Grégoire, B. Impact of Cα-Chirality on Supramolecular Self-Assembly in Cyclo-2:1-[α/aza]-Hexamers (d/l-Phe-azaPhe-Ala)2. Europ. J. Org. Chem. 2017, 2017, 4703–4712. [Google Scholar] [CrossRef]
- Tonali, N.; Correia, I.; Lesma, J.; Bernadat, G.; Ongeri, S.; Lequin, O. Introducing sequential aza-amino acids units induces repeated β-turns and helical conformations in peptides. Org. Biomol. Chem. 2020, 18, 3452–3458. [Google Scholar] [CrossRef]
- Danelius, E.; Ohm, R.G.; Ahsanullah; Mulumba, M.; Ong, H.; Chemtob, S.; Erdelyi, M.; Lubell, W.D. Dynamic Chirality in the Mechanism of Action of Allosteric CD36 Modulators of Macrophage-Driven Inflammation. J. Med. Chem. 2019, 62, 11071–11079. [Google Scholar] [CrossRef]
- Wieczerzak, E.; Jankowska, E.; Rodziewicz-Motowidło, S.; Giełdoń, A.; Ła̧Giewka, J.; Grzonka, Z.; Abrahamson, M.; Grubb, A.; Brömme, D. Novel azapeptide inhibitors of cathepsins B and K. Structural background to increased specificity for cathepsin B. J. Pept. Res. 2005, 66, 1–11. [Google Scholar] [CrossRef]
- Corrigan, T.S.; Lotti Diaz, L.M.; Border, S.E.; Ratigan, S.C.; Kasper, K.Q.; Sojka, D.; Fajtova, P.; Caffrey, C.R.; Salvesen, G.S.; McElroy, C.A.; et al. Design, synthesis, and in vitro evaluation of aza-peptide aldehydes and ketones as novel and selective protease inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 1387–1402. [Google Scholar] [CrossRef]
- Löser, R.; Frizler, M.; Schilling, K.; Gütschow, M. Azadipeptide Nitriles: Highly Potent and Proteolytically Stable Inhibitors of Papain-Like Cysteine Proteases. Angew. Chem. Int. Ed. 2008, 47, 4331–4334. [Google Scholar] [CrossRef] [PubMed]
- Jílková, A.; Horn, M.; Fanfrlík, J.; Küppers, J.; Pachl, P.; Řezáčová, P.; Lepšík, M.; Fajtová, P.; Rubešová, P.; Chanová, M.; et al. Azanitrile Inhibitors of the SmCB1 Protease Target Are Lethal to Schistosoma mansoni: Structural and Mechanistic Insights into Chemotype Reactivity. ACS Infect. Dis. 2021, 7, 189–201. [Google Scholar] [CrossRef]
- Breidenbach, J.; Lemke, C.; Pillaiyar, T.; Schäkel, L.; Al Hamwi, G.; Diett, M.; Gedschold, R.; Geiger, N.; Lopez, V.; Mirza, S.; et al. Targeting the Main Protease of SARS-CoV-2: From the Establishment of High Throughput Screening to the Design of Tailored Inhibitors. Angew. Chem. Int. Ed. 2021, 60, 10423–10429. [Google Scholar] [CrossRef]
- Freeman, N.S.; Tal-Gan, Y.; Klein, S.; Levitzki, A.; Gilon, C. Microwave-Assisted Solid-Phase Aza-peptide Synthesis: Aza Scan of a PKB/Akt Inhibitor Using Aza-arginine and Aza-proline Precursors. J. Org. Chem. 2011, 76, 3078–3085. [Google Scholar] [CrossRef]
- Proulx, C.; Picard, É.; Boeglin, D.; Pohankova, P.; Chemtob, S.; Ong, H.; Lubell, W.D. Azapeptide analogues of the growth hormone releasing peptide 6 as cluster of differentiation 36 receptor ligands with reduced affinity for the growth hormone secretagogue receptor 1a. J. Med. Chem. 2012, 55, 6502–6511. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.; Mas-Moruno, C.; Kessler, H.; Lubell, W.D. Cyclic aza-peptide integrin ligand synthesis and biological activity. J. Org. Chem. 2012, 77, 5271–5278. [Google Scholar] [CrossRef]
- Frizler, M.; Lohr, F.; Furtmann, N.; Kläs, J.; Gütschow, M. Structural optimization of azadipeptide nitriles strongly increases association rates and allows the development of selective cathepsin inhibitors. J. Med. Chem. 2011, 54, 396–400. [Google Scholar] [CrossRef]
- Litman, P.; Ohne, O.; Ben-Yaakov, S.; Shemesh-Darvish, L.; Yechezkel, T.; Salitra, Y.; Rubnov, S.; Cohen, I.; Senderowitz, H.; Kidron, D.; et al. A Novel Substrate Mimetic Inhibitor of PKB/Akt Inhibits Prostate Cancer Tumor Growth in Mice by Blocking the PKB Pathway. Biochemistry 2007, 46, 4716–4724. [Google Scholar] [CrossRef]
- Hiromura, M.; Okada, F.; Obata, T.; Auguin, D.; Shibata, T.; Roumestand, C.; Noguchi, M. Inhibition of Akt Kinase Activity by a Peptide Spanning the βA Strand of the Proto-oncogene TCL1. J. Biol. Chem. 2004, 279, 53407–53418. [Google Scholar] [CrossRef] [PubMed]
- Elsawy, M.A.; Tikhonova, I.G.; Martin, L.L.; Walker, B. Smac-derived Aza-peptide As an Aminopeptidase-resistant XIAP BIR3 Antagonist. Protein Pept. Lett. 2015, 22, 836–843. [Google Scholar] [CrossRef]
- Randolph, J.T.; Zhang, X.; Huang, P.P.; Klein, L.L.; Kurtz, K.A.; Konstantinidis, A.K.; He, W.; Kati, W.M.; Kempf, D.J. Synthesis, antiviral activity, and conformational studies of a P3 aza-peptide analog of a potent macrocyclic tripeptide HCV protease inhibitor. Bioorg. Med. Chem. Lett. 2008, 18, 2745–2750. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2022, 118, 115–129. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Angin, Y.; Steinbusch, L.K.M.; Schwenk, R.W.; Luiken, J.J.F.P. CD36 as a target to prevent cardiac lipotoxicity and insulin resistance. Prostaglandins Leukot. Essent. Fat. Acids 2013, 88, 71–77. [Google Scholar] [CrossRef]
- Berlanga-Acosta, J.; Guillen Nieto, G.; Lopez-Mola, E.; Herrera-Martinez, L. Growth hormone releasing peptide-6 (GHRP-6) and other related secretagogue synthetic peptides: A mine of medical potentialities for unmet medical needs. Integr. Mol. Med. 2016, 3, 616–623. [Google Scholar] [CrossRef]
- Proulx, C.; Zhang, J.; Sabatino, D.; Chemtob, S.; Ong, H.; Lubell, W.D. Synthesis and Biomedical Potential of Azapeptide Modulators of the Cluster of Differentiation 36 Receptor (CD36). Biomedicines 2020, 8, 241. [Google Scholar] [CrossRef]
- Huynh, D.N.; Bessi, V.L.; Menard, L.; Piquereau, J.; Proul, C.; Febbraio, M.; Lubell, W.D.; Carpentier, A.C.; Burelle, Y.; Ong, H.; et al. Adiponectin has a pivotal role in the cardioprotective effect of CP-3(iv), a selective CD36 azapeptide ligand, after transient coronary artery occlusion in mice. FASEB J. 2018, 32, 807–818. [Google Scholar] [CrossRef]
- Frégeau, G.; Sarduy, R.; Elimam, H.; Esposito, C.L.; Mellal, K.; Ménard, L.; Leitão da Graça, S.D.; Proulx, C.; Zhang, J.; Febbraio, M.; et al. Atheroprotective and atheroregressive potential of azapeptide derivatives of GHRP-6 as selective CD36 ligands in apolipoprotein E-deficient mice. Atherosclerosis 2020, 307, 52–62. [Google Scholar] [CrossRef]
- Mellal, K.; Omri, S.; Mulumba, M.; Tahiri, H.; Fortin, C.; Dorion, M.F.; Pham, H.; Garcia Ramos, Y.; Zhang, J.; Pundir, S.; et al. Immunometabolic modulation of retinal inflammation by CD36 ligand. Sci. Rep. 2019, 9, 12903. [Google Scholar] [CrossRef]
- Ohm, R.G.; Mulumba, M.; Chingle, R.M.; Ahsanullah; Zhang, J.; Chemtob, S.; Ong, H.; Lubell, W.D. Diversity-Oriented A 3-Macrocyclization for Studying Influences of Ring-Size and Shape of Cyclic Peptides: CD36 Receptor Modulators. J. Med. Chem. 2021, 64, 9365–9380. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Azapeptide | Biological Activity | Reference |
|---|---|---|
| Z-Arg-Leu-His-Agly-Ile-Val-OMe | Cathpsin B-specific inhibitor | [57] |
| Ac-ThrSerLeuAglySerProProProSer-NH2 | M-calpain inhibitor | [40] |
| Z-Leu-Leu-azaLeu-CHO | 20S proteasome inhibitor | [58] |
| Z-Asp-Glu-Val-azaAsp-COMe | Caspase-3 inhibitor | [58] |
| Z-Ile-Glu-Thr-azaAsp-COMe | Caspase-6 inhibitor | [58] |
| Z-Ala-Ala-azaAsn-COBn | Legumain inhibitor | [58] |
| Z-Phe-(N-Me)azaAla-nitril | Cathepsin inhibitor | [59] |
| Z-Phe-(N-Me)azahPhe-nitril | SmCB1 inhibitor | [60] |
| Z-Abu-Tle-Phe-azaGln-nitril | SARS-CoV-2 main protease inhibitor | [61] |
| H-Arg-azaPro-Arg-Nva-Tyr-Dap-Hol-NH2 | PKB/Akt inhibitor | [62] |
| azaGly-Arg-Pro-Arg-Nle-Tyr-Dap-Nle-NH2 | PKB/Akt inhibitor | [25] |
| Ala-D-Trp-Ala-AzaPhe-D-Phe-Lys-NH2 | CD36 ligand | [63] |
| H-His-D-Trp-Ala-aza(4-n-propoxy)Phe-D-Phe-Lys-NH2 | CD36 ligand | [37] |
| c[ArgR-azaGly-AspD-D-Phef-Val] | Integrin ligand | [64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarchoun, K.; Yousef, M.; Bánóczi, Z. Azapeptides as an Efficient Tool to Improve the Activity of Biologically Effective Peptides. Future Pharmacol. 2022, 2, 293-305. https://doi.org/10.3390/futurepharmacol2030020
Tarchoun K, Yousef M, Bánóczi Z. Azapeptides as an Efficient Tool to Improve the Activity of Biologically Effective Peptides. Future Pharmacology. 2022; 2(3):293-305. https://doi.org/10.3390/futurepharmacol2030020
Chicago/Turabian StyleTarchoun, Karima, Mo’ath Yousef, and Zoltán Bánóczi. 2022. "Azapeptides as an Efficient Tool to Improve the Activity of Biologically Effective Peptides" Future Pharmacology 2, no. 3: 293-305. https://doi.org/10.3390/futurepharmacol2030020
APA StyleTarchoun, K., Yousef, M., & Bánóczi, Z. (2022). Azapeptides as an Efficient Tool to Improve the Activity of Biologically Effective Peptides. Future Pharmacology, 2(3), 293-305. https://doi.org/10.3390/futurepharmacol2030020