
Chromatin Organization during C. elegans Early Development


Abstract
1. Introduction
2. Histone H3K9 Methylation-Mediated Heterochromatin Formation
3. Other Mechanisms of Heterochromatin Formation
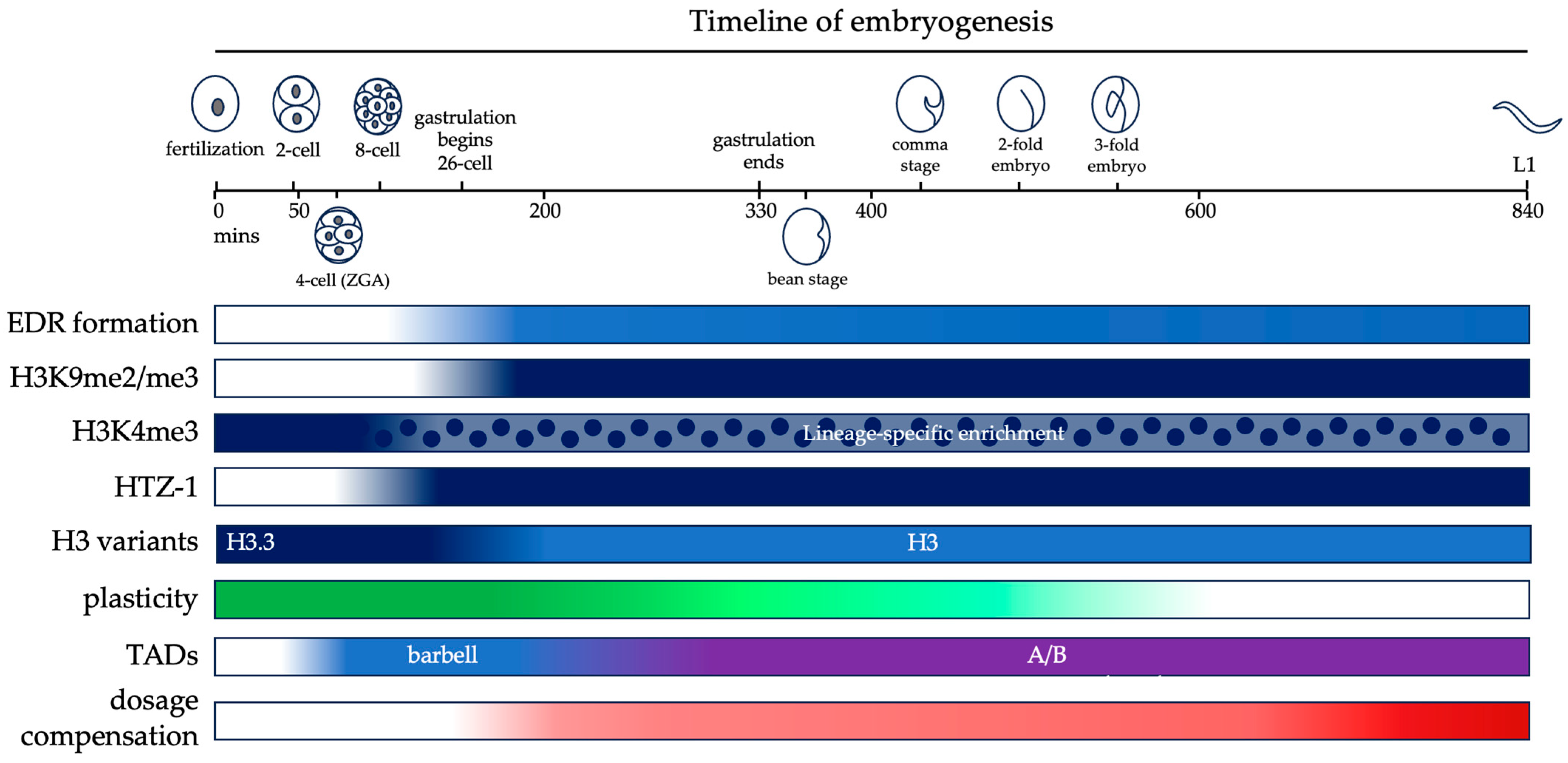
4. Developmental Regulation of Active Chromatin
5. Antagonism between Repressive and Active Histone Marks
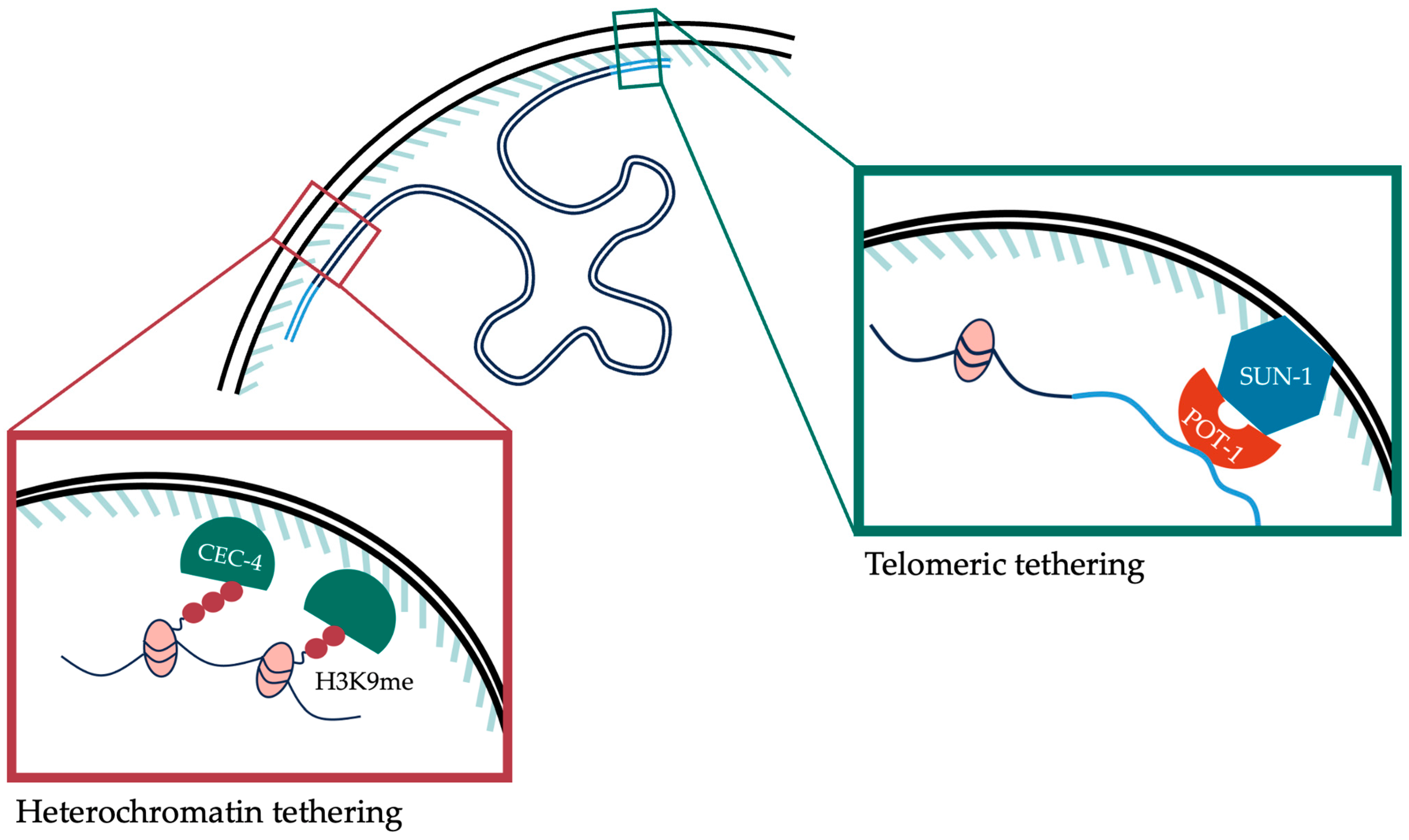
6. Spatial Organization of Chromatin inside the Nucleus
7. Emergence of Topologically Associating Domains (TADs)
8. Epigenetic Modifications and the Loss of Developmental Plasticity
9. Establishment of Dosage Compensation on the X Chromosome
10. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Politz, J.C.R.; Scalzo, D.; Groudine, M. Something Silent This Way Forms: The Functional Organization of the Repressive Nuclear Compartment. Annu. Rev. Cell Dev. Biol. 2013, 29, 241–270. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of Lysine 4 on Histone H3: Intricacy of Writing and Reading a Single Epigenetic Mark. Mol. Cell 2007, 25, 15–30. [Google Scholar] [CrossRef]
- Ahringer, J.; Gasser, S.M. Repressive Chromatin in Caenorhabditis elegans: Establishment, Composition, and Function. Genetics 2018, 208, 491–511. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-Dimensional Folding and Functional Organization Principles of the Drosophila Genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef]
- Gonzalez-Sandoval, A.; Gasser, S.M. On TADs and LADs: Spatial Control Over Gene Expression. Trends Genet. 2016, 32, 485–495. [Google Scholar] [CrossRef]
- Meyer, B.J.; Casson, L.P. Caenorhabditis elegans Compensates for the Difference in X Chromosome Dosage between the Sexes by Regulating Transcript Levels. Cell 1986, 47, 871–881. [Google Scholar] [CrossRef]
- Tadros, W.; Lipshitz, H.D. The Maternal-to-Zygotic Transition: A Play in Two Acts. Dev. Camb. Engl. 2009, 136, 3033–3042. [Google Scholar] [CrossRef]
- Mutlu, B.; Chen, H.-M.; Moresco, J.J.; Orelo, B.D.; Yang, B.; Gaspar, J.M.; Keppler-Ross, S.; Yates, J.R.; Hall, D.H.; Maine, E.M.; et al. Regulated Nuclear Accumulation of a Histone Methyltransferase Times the Onset of Heterochromatin Formation in C. Elegans Embryos. Sci. Adv. 2018, 4, eaat6224. [Google Scholar] [CrossRef]
- Becker, J.S.; Nicetto, D.; Zaret, K.S. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 2016, 32, 29–41. [Google Scholar] [CrossRef]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-Methylated Heterochromatin and Its Functions in Tissue Differentiation and Maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef]
- Montavon, T.; Shukeir, N.; Erikson, G.; Engist, B.; Onishi-Seebacher, M.; Ryan, D.; Musa, Y.; Mittler, G.; Meyer, A.G.; Genoud, C.; et al. Complete Loss of H3K9 Methylation Dissolves Mouse Heterochromatin Organization. Nat. Commun. 2021, 12, 4359. [Google Scholar] [CrossRef]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Bian, Q.; Anderson, E.C.; Yang, Q.; Meyer, B.J. Histone H3K9 Methylation Promotes Formation of Genome Compartments in Caenorhabditis elegans via Chromosome Compaction and Perinuclear Anchoring. Proc. Natl. Acad. Sci. USA 2020, 117, 11459–11470. [Google Scholar] [CrossRef]
- Sawh, A.N.; Shafer, M.E.R.; Su, J.-H.; Zhuang, X.; Wang, S.; Mango, S.E. Lamina-Dependent Stretching and Unconventional Chromosome Compartments in Early C. Elegans Embryos. Mol. Cell 2020, 78, 96–111.e6. [Google Scholar] [CrossRef]
- Towbin, B.D.; González-Aguilera, C.; Sack, R.; Gaidatzis, D.; Kalck, V.; Meister, P.; Askjaer, P.; Gasser, S.M. Step-Wise Methylation of Histone H3K9 Positions Heterochromatin at the Nuclear Periphery. Cell 2012, 150, 934–947. [Google Scholar] [CrossRef]
- Cabianca, D.S.; Muñoz-Jiménez, C.; Kalck, V.; Gaidatzis, D.; Padeken, J.; Seeber, A.; Askjaer, P.; Gasser, S.M. Active Chromatin Marks Drive Spatial Sequestration of Heterochromatin in C. Elegans Nuclei. Nature 2019, 569, 734–739. [Google Scholar] [CrossRef]
- Delaney, C.E.; Methot, S.P.; Guidi, M.; Katic, I.; Gasser, S.M.; Padeken, J. Heterochromatic Foci and Transcriptional Repression by an Unstructured MET-2/SETDB1 Co-Factor LIN-65. J. Cell Biol. 2019, 218, 820–838. [Google Scholar] [CrossRef]
- Delaney, C.E.; Methot, S.P.; Kalck, V.; Seebacher, J.; Hess, D.; Gasser, S.M.; Padeken, J. SETDB1-like MET-2 Promotes Transcriptional Silencing and Development Independently of Its H3K9me-Associated Catalytic Activity. Nat. Struct. Mol. Biol. 2022, 29, 85–96. [Google Scholar] [CrossRef]
- Mutlu, B.; Chen, H.-M.; Gutnik, S.; Hall, D.H.; Keppler-Ross, S.; Mango, S.E. Distinct Functions and Temporal Regulation of Methylated Histone H3 during Early Embryogenesis. Development 2019, 146, dev174516. [Google Scholar] [CrossRef]
- Padeken, J.; Methot, S.; Zeller, P.; Delaney, C.E.; Kalck, V.; Gasser, S.M. Argonaute NRDE-3 and MBT Domain Protein LIN-61 Redundantly Recruit an H3K9me3 HMT to Prevent Embryonic Lethality and Transposon Expression. Genes Dev. 2021, 35, 82–101. [Google Scholar] [CrossRef]
- Zeller, P.; Padeken, J.; van Schendel, R.; Kalck, V.; Tijsterman, M.; Gasser, S.M. Histone H3K9 Methylation Is Dispensable for Caenorhabditis elegans Development but Suppresses RNA:DNA Hybrid-Associated Repeat Instability. Nat. Genet. 2016, 48, 1385–1395. [Google Scholar] [CrossRef]
- Ho, J.W.K.; Jung, Y.L.; Liu, T.; Alver, B.H.; Lee, S.; Ikegami, K.; Sohn, K.-A.; Minoda, A.; Tolstorukov, M.Y.; Appert, A.; et al. Comparative Analysis of Metazoan Chromatin Organization. Nature 2014, 512, 449–452. [Google Scholar] [CrossRef]
- Vandamme, J.; Sidoli, S.; Mariani, L.; Friis, C.; Christensen, J.; Helin, K.; Jensen, O.N.; Salcini, A.E. H3K23me2 Is a New Heterochromatic Mark in Caenorhabditis elegans. Nucleic Acids Res. 2015, 43, 9694–9710. [Google Scholar] [CrossRef]
- Liu, T.; Rechtsteiner, A.; Egelhofer, T.A.; Vielle, A.; Latorre, I.; Cheung, M.-S.; Ercan, S.; Ikegami, K.; Jensen, M.; Kolasinska-Zwierz, P.; et al. Broad Chromosomal Domains of Histone Modification Patterns in C. elegans. Genome Res. 2011, 21, 227–236. [Google Scholar] [CrossRef]
- Garrigues, J.M.; Sidoli, S.; Garcia, B.A.; Strome, S. Defining Heterochromatin in C. Elegans through Genome-Wide Analysis of the Heterochromatin Protein 1 Homolog HPL-2. Genome Res. 2015, 25, 76–88. [Google Scholar] [CrossRef]
- Gaydos, L.J.; Wang, W.; Strome, S. H3K27me and PRC2 Transmit a Memory of Repression across Generations and during Development. Science 2014, 345, 1515–1518. [Google Scholar] [CrossRef]
- Gaydos, L.J.; Rechtsteiner, A.; Egelhofer, T.A.; Carroll, C.R.; Strome, S. Antagonism between MES-4 and Polycomb Repressive Complex 2 Promotes Appropriate Gene Expression in C. Elegans Germ Cells. Cell Rep. 2012, 2, 1169–1177. [Google Scholar] [CrossRef]
- Bender, L.B.; Cao, R.; Zhang, Y.; Strome, S. The MES-2/MES-3/MES-6 Complex and Regulation of Histone H3 Methylation in C. elegans. Curr. Biol. CB 2004, 14, 1639–1643. [Google Scholar] [CrossRef]
- Yuzyuk, T.; Fakhouri, T.H.I.; Kiefer, J.; Mango, S.E. The Polycomb Complex Protein Mes-2/E(z) Promotes the Transition from Developmental Plasticity to Differentiation in C. Elegans Embryos. Dev. Cell 2009, 16, 699–710. [Google Scholar] [CrossRef]
- Vandamme, J.; Lettier, G.; Sidoli, S.; Schiavi, E.D.; Jensen, O.N.; Salcini, A.E. The C. Elegans H3K27 Demethylase UTX-1 Is Essential for Normal Development, Independent of Its Enzymatic Activity. PLoS Genet. 2012, 8, e1002647. [Google Scholar] [CrossRef]
- Käser-Pébernard, S.; Pfefferli, C.; Aschinger, C.; Wicky, C. Fine-Tuning of Chromatin Composition and Polycomb Recruitment by Two Mi2 Homologues during C. Elegans Early Embryonic Development. Epigenetics Chromatin 2016, 9, 1–17. [Google Scholar] [CrossRef]
- Reynolds, N.; Latos, P.; Hynes-Allen, A.; Loos, R.; Leaford, D.; O’Shaughnessy, A.; Mosaku, O.; Signolet, J.; Brennecke, P.; Kalkan, T.; et al. NuRD Suppresses Pluripotency Gene Expression to Promote Transcriptional Heterogeneity and Lineage Commitment. Cell Stem Cell 2012, 10, 583–594. [Google Scholar] [CrossRef]
- O’Shaughnessy-Kirwan, A.; Signolet, J.; Costello, I.; Gharbi, S.; Hendrich, B. Constraint of Gene Expression by the Chromatin Remodelling Protein CHD4 Facilitates Lineage Specification. Development 2015, 142, 2586–2597. [Google Scholar] [CrossRef]
- Hendrich, B.; Guy, J.; Ramsahoye, B.; Wilson, V.A.; Bird, A. Closely Related Proteins MBD2 and MBD3 Play Distinctive but Interacting Roles in Mouse Development. Genes Dev. 2001, 15, 710–723. [Google Scholar] [CrossRef]
- Myers, T.R.; Amendola, P.G.; Lussi, Y.C.; Salcini, A.E. JMJD-1.2 Controls Multiple Histone Post-Translational Modifications in Germ Cells and Protects the Genome from Replication Stress. Sci. Rep. 2018, 8, 3765. [Google Scholar] [CrossRef]
- Jack, A.P.M.; Bussemer, S.; Hahn, M.; Pünzeler, S.; Snyder, M.; Wells, M.; Csankovszki, G.; Solovei, I.; Schotta, G.; Hake, S.B. H3K56me3 Is a Novel, Conserved Heterochromatic Mark That Largely but Not Completely Overlaps with H3K9me3 in Both Regulation and Localization. PLoS ONE 2013, 8, e51765. [Google Scholar] [CrossRef]
- McKittrick, E.; Gafken, P.R.; Ahmad, K.; Henikoff, S. Histone H3.3 Is Enriched in Covalent Modifications Associated with Active Chromatin. Proc. Natl. Acad. Sci. USA 2004, 101, 1525–1530. [Google Scholar] [CrossRef]
- Loyola, A.; Bonaldi, T.; Roche, D.; Imhof, A.; Almouzni, G. PTMs on H3 Variants before Chromatin Assembly Potentiate Their Final Epigenetic State. Mol. Cell 2006, 24, 309–316. [Google Scholar] [CrossRef]
- Hake, S.B.; Garcia, B.A.; Duncan, E.M.; Kauer, M.; Dellaire, G.; Shabanowitz, J.; Bazett-Jones, D.P.; Allis, C.D.; Hunt, D.F. Expression Patterns and Post-Translational Modifications Associated with Mammalian Histone H3 Variants. J. Biol. Chem. 2006, 281, 559–568. [Google Scholar] [CrossRef]
- Kreher, J.; Takasaki, T.; Cockrum, C.; Sidoli, S.; Garcia, B.A.; Jensen, O.N.; Strome, S. Distinct Roles of Two Histone Methyltransferases in Transmitting H3K36me3-Based Epigenetic Memory Across Generations in Caenorhabditis elegans. Genetics 2018, 210, 969–982. [Google Scholar] [CrossRef]
- Gleason, R.J.; Guo, Y.; Semancik, C.S.; Ow, C.; Lakshminarayanan, G.; Chen, X. Developmentally Programmed Histone H3 Expression Regulates Cellular Plasticity at the Parental-to-Early Embryo Transition. Sci. Adv. 2023, 9, eadh0411. [Google Scholar] [CrossRef]
- Ishiuchi, T.; Abe, S.; Inoue, K.; Yeung, W.K.A.; Miki, Y.; Ogura, A.; Sasaki, H. Reprogramming of the Histone H3.3 Landscape in the Early Mouse Embryo. Nat. Struct. Mol. Biol. 2021, 28, 38–49. [Google Scholar] [CrossRef]
- Jedrusik, M.A.; Schulze, E. A Single Histone H1 Isoform (H1.1) Is Essential for Chromatin Silencing and Germline Development in Caenorhabditis elegans. Development 2001, 128, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Jedrusik, M.A.; Schulze, E. Linker Histone HIS-24 (H1.1) Cytoplasmic Retention Promotes Germ Line Development and Influences Histone H3 Methylation in Caenorhabditis elegans. Mol. Cell. Biol. 2007, 27, 2229–2239. [Google Scholar] [CrossRef]
- Jedrusik-Bode, M. Histone H1 and Heterochromatin Protein 1 (HP1) Regulate Specific Gene Expression and Not Global Transcription. Worm 2013, 2, e23703. [Google Scholar] [CrossRef][Green Version]
- Arico, J.K.; Katz, D.J.; van der Vlag, J.; Kelly, W.G. Epigenetic Patterns Maintained in Early Caenorhabditis elegans Embryos Can Be Established by Gene Activity in the Parental Germ Cells. PLoS Genet. 2011, 7, e1001391. [Google Scholar] [CrossRef]
- Samson, M.; Jow, M.M.; Wong, C.C.L.; Fitzpatrick, C.; Aslanian, A.; Saucedo, I.; Estrada, R.; Ito, T.; Park, S.R.; Iii, J.R.Y.; et al. The Specification and Global Reprogramming of Histone Epigenetic Marks during Gamete Formation and Early Embryo Development in C. elegans. PLoS Genet. 2014, 10, e1004588. [Google Scholar] [CrossRef]
- Rechtsteiner, A.; Ercan, S.; Takasaki, T.; Phippen, T.M.; Egelhofer, T.A.; Wang, W.; Kimura, H.; Lieb, J.D.; Strome, S. The Histone H3K36 Methyltransferase MES-4 Acts Epigenetically to Transmit the Memory of Germline Gene Expression to Progeny. PLoS Genet. 2010, 6, e1001091. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Fisher, K.; Poulin, G.B. Lineage Specific Trimethylation of H3 on Lysine 4 during C. Elegans Early Embryogenesis. Dev. Biol. 2011, 355, 227–238. [Google Scholar] [CrossRef]
- Reuben, M.; Lin, R. Germline X Chromosomes Exhibit Contrasting Patterns of Histone H3 Methylation in Caenorhabditis elegans. Dev. Biol. 2002, 245, 71–82. [Google Scholar] [CrossRef][Green Version]
- Kelly, W.G.; Schaner, C.E.; Dernburg, A.F.; Lee, M.-H.; Kim, S.K.; Villeneuve, A.M.; Reinke, V. X-Chromosome Silencing in the Germline of C. elegans. Development 2002, 129, 479–492. [Google Scholar] [CrossRef]
- Riddle, D.L.; Blumenthal, T.; Meyer, B.J.; Priess, J.R. Specification of Cell Fates in the AB Lineage. In C. elegans II, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Fisher, K.; Southall, S.M.; Wilson, J.R.; Poulin, G.B. Methylation and Demethylation Activities of a C. Elegans MLL-like Complex Attenuate RAS Signalling. Dev. Biol. 2010, 341, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 Are Histone H3K27 Demethylases Involved in HOX Gene Regulation and Development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Bedet, C.; Robert, V.J.P.; Simonet, T.; Dunkelbarger, S.; Rakotomalala, C.; Soete, G.; Korswagen, H.C.; Strome, S.; Palladino, F. Caenorhabditis elegans Chromatin-Associated Proteins SET-2 and ASH-2 Are Differentially Required for Histone H3 Lys 4 Methylation in Embryos and Adult Germ Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8305–8310. [Google Scholar] [CrossRef] [PubMed]
- Andersen, E.C.; Horvitz, H.R. Two C. Elegans Histone Methyltransferases Repress Lin-3EGF Transcription to Inhibit Vulval Development. Development 2007, 134, 2991–2999. [Google Scholar] [CrossRef] [PubMed]
- Whittle, C.M.; McClinic, K.N.; Ercan, S.; Zhang, X.; Green, R.D.; Kelly, W.G.; Lieb, J.D. The Genomic Distribution and Function of Histone Variant HTZ-1 during C. Elegans Embryogenesis. PLoS Genet. 2008, 4, e1000187. [Google Scholar] [CrossRef] [PubMed]
- Borcherds, W.; Bremer, A.; Borgia, M.B.; Mittag, T. How Do Intrinsically Disordered Protein Regions Encode a Driving Force for Liquid–Liquid Phase Separation? Curr. Opin. Struct. Biol. 2021, 67, 41–50. [Google Scholar] [CrossRef]
- Saito, M.; Hess, D.; Eglinger, J.; Fritsch, A.W.; Kreysing, M.; Weinert, B.T.; Choudhary, C.; Matthias, P. Acetylation of Intrinsically Disordered Regions Regulates Phase Separation. Nat. Chem. Biol. 2019, 15, 51–61. [Google Scholar] [CrossRef]
- Methot, S.P.; Padeken, J.; Brancati, G.; Zeller, P.; Delaney, C.E.; Gaidatzis, D.; Kohler, H.; van Oudenaarden, A.; Großhans, H.; Gasser, S.M. H3K9me Selectively Blocks Transcription Factor Activity and Ensures Differentiated Tissue Integrity. Nat. Cell Biol. 2021, 23, 1163–1175. [Google Scholar] [CrossRef]
- Towbin, B.D.; Meister, P.; Pike, B.L.; Gasser, S.M. Repetitive Transgenes in C. Elegans Accumulate Heterochromatic Marks and Are Sequestered at the Nuclear Envelope in a Copy-Number- and Lamin-Dependent Manner. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 555–565. [Google Scholar] [CrossRef]
- Snyder, M.J.; Lau, A.C.; Brouhard, E.A.; Davis, M.B.; Jiang, J.; Sifuentes, M.H.; Csankovszki, G. Anchoring of Heterochromatin to the Nuclear Lamina Reinforces Dosage Compensation-Mediated Gene Repression. PLoS Genet. 2016, 12, e1006341. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sandoval, A.; Towbin, B.D.; Kalck, V.; Cabianca, D.S.; Gaidatzis, D.; Hauer, M.H.; Geng, L.; Wang, L.; Yang, T.; Wang, X.; et al. Perinuclear Anchoring of H3K9-Methylated Chromatin Stabilizes Induced Cell Fate in C. Elegans Embryos. Cell 2015, 163, 1333–1347. [Google Scholar] [CrossRef]
- Ikegami, K.; Egelhofer, T.A.; Strome, S.; Lieb, J.D. Caenorhabditis elegans Chromosome Arms Are Anchored to the Nuclear Membrane via Discontinuous Association with LEM-2. Genome Biol. 2010, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.; Kranz, A.-L.; Su, A.; Winterkorn, L.H.; Albritton, S.E.; Ercan, S. Developmental Dynamics of X-Chromosome Dosage Compensation by the DCC and H4K20me1 in C. elegans. PLoS Genet. 2015, 11, e1005698. [Google Scholar] [CrossRef]
- Vielle, A.; Lang, J.; Dong, Y.; Ercan, S.; Kotwaliwale, C.; Rechtsteiner, A.; Appert, A.; Chen, Q.B.; Dose, A.; Egelhofer, T.; et al. H4K20me1 Contributes to Downregulation of X-Linked Genes for C. Elegans Dosage Compensation. PLoS Genet. 2012, 8, e1002933. [Google Scholar] [CrossRef]
- Brejc, K.; Bian, Q.; Uzawa, S.; Wheeler, B.S.; Anderson, E.C.; King, D.S.; Kranzusch, P.J.; Preston, C.G.; Meyer, B.J. Dynamic Control of X Chromosome Conformation and Repression by a Histone H4K20 Demethylase. Cell 2017, 171, 85–102.e23. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.B.; Snyder, M.J.; Custer, L.M.; Csankovszki, G. Caenorhabditis elegans Dosage Compensation Regulates Histone H4 Chromatin State on X Chromosomes. Mol. Cell. Biol. 2012, 32, 1710–1719. [Google Scholar] [CrossRef]
- Custer, L.M.; Snyder, M.J.; Flegel, K.; Csankovszki, G. The Onset of C. Elegans Dosage Compensation Is Linked to the Loss of Developmental Plasticity. Dev. Biol. 2014, 385, 279–290. [Google Scholar] [CrossRef]
- Manzo, S.G.; Dauban, L.; van Steensel, B. Lamina-Associated Domains: Tethers and Looseners. Curr. Opin. Cell Biol. 2022, 74, 80–87. [Google Scholar] [CrossRef]
- Poleshko, A.; Smith, C.L.; Nguyen, S.C.; Sivaramakrishnan, P.; Wong, K.G.; Murray, J.I.; Lakadamyali, M.; Joyce, E.F.; Jain, R.; Epstein, J.A. H3K9me2 Orchestrates Inheritance of Spatial Positioning of Peripheral Heterochromatin through Mitosis. eLife 2019, 8, e49278. [Google Scholar] [CrossRef] [PubMed]
- See, K.; Kiseleva, A.A.; Smith, C.L.; Liu, F.; Li, J.; Poleshko, A.; Epstein, J.A. Histone Methyltransferase Activity Programs Nuclear Peripheral Genome Positioning. Dev. Biol. 2020, 466, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Takano-Maruyama, M.; Pereira-Smith, O.M.; Gaufo, G.O.; Tominaga, K. MRG15, a Component of HAT and HDAC Complexes, Is Essential for Proliferation and Differentiation of Neural Precursor Cells. J. Neurosci. Res. 2009, 87, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Pardo, P.S.; Leung, J.K.; Lucchesi, J.C.; Pereira-Smith, O.M. MRG15, a Novel Chromodomain Protein, Is Present in Two Distinct Multiprotein Complexes Involved in Transcriptional Activation*. J. Biol. Chem. 2002, 277, 50860–50866. [Google Scholar] [CrossRef]
- Hajduskova, M.; Baytek, G.; Kolundzic, E.; Gosdschan, A.; Kazmierczak, M.; Ofenbauer, A.; Beato del Rosal, M.L.; Herzog, S.; ul Fatima, N.; Mertins, P.; et al. MRG-1/MRG15 Is a Barrier for Germ Cell to Neuron Reprogramming in Caenorhabditis elegans. Genetics 2019, 211, 121–139. [Google Scholar] [CrossRef]
- Shi, Y.; Mello, C. A CBP/P300 Homolog Specifies Multiple Differentiation Pathways in Caenorhabditis elegans. Genes Dev. 1998, 12, 943–955. [Google Scholar] [CrossRef]
- Ferreira, H.C.; Towbin, B.D.; Jegou, T.; Gasser, S.M. The Shelterin Protein POT-1 Anchors Caenorhabditis elegans Telomeres through SUN-1 at the Nuclear Periphery. J. Cell Biol. 2013, 203, 727–735. [Google Scholar] [CrossRef]
- Ferreira, H.C.; Luke, B.; Schober, H.; Kalck, V.; Lingner, J.; Gasser, S.M. The PIAS Homologue Siz2 Regulates Perinuclear Telomere Position and Telomerase Activity in Budding Yeast. Nat. Cell Biol. 2011, 13, 867–874. [Google Scholar] [CrossRef]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandão, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-Nucleus Hi-C Reveals Unique Chromatin Reorganization at Oocyte-to-Zygote Transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef]
- Cardozo Gizzi, A.M.; Cattoni, D.I.; Nollmann, M. TADs or No TADS: Lessons from Single-Cell Imaging of Chromosome Architecture. J. Mol. Biol. 2020, 432, 682–693. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D Structures of Individual Mammalian Genomes Studied by Single-Cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two Independent Modes of Chromatin Organization Revealed by Cohesin Removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Goloborodko, A.; Valton, A.-L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930–944.e22. [Google Scholar] [CrossRef] [PubMed]
- Heger, P.; Marin, B.; Schierenberg, E. Loss of the Insulator Protein CTCF during Nematode Evolution. BMC Mol. Biol. 2009, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Crane, E.; Bian, Q.; McCord, R.P.; Lajoie, B.R.; Wheeler, B.S.; Ralston, E.J.; Uzawa, S.; Dekker, J.; Meyer, B.J. Condensin-Driven Remodelling of X Chromosome Topology during Dosage Compensation. Nature 2015, 523, 240–244. [Google Scholar] [CrossRef]
- Lambert, J.; Lloret-Fernández, C.; Laplane, L.; Poole, R.J.; Jarriault, S. Chapter Five—On the Origins and Conceptual Frameworks of Natural Plasticity—Lessons from Single-Cell Models in C. elegans. In Current Topics in Developmental Biology; Jarriault, S., Podbilewicz, B., Eds.; Nematode Models of Development and Disease; Academic Press: Cambridge, MA, USA, 2021; Volume 144, pp. 111–159. [Google Scholar]
- Wood, W.B. Evidence from Reversal of Handedness in C. Elegans Embryos for Early Cell Interactions Determining Cell Fates. Nature 1991, 349, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Fukushige, T.; McGhee, J.D.; Rothman, J.H. Reprogramming of Early Embryonic Blastomeres into Endodermal Progenitors by a Caenorhabditis elegans GATA Factor. Genes Dev. 1998, 12, 3809–3814. [Google Scholar] [CrossRef]
- Fukushige, T.; Krause, M. The Myogenic Potency of HLH-1 Reveals Wide-Spread Developmental Plasticity in Early C. Elegans Embryos. Development 2005, 132, 1795–1805. [Google Scholar] [CrossRef]
- Djabrayan, N.J.-V.; Dudley, N.R.; Sommermann, E.M.; Rothman, J.H. Essential Role for Notch Signaling in Restricting Developmental Plasticity. Genes Dev. 2012, 26, 2386–2391. [Google Scholar] [CrossRef]
- Bucher, E.A.; Seydoux, G. Gastrulation in the Nematode Caenorhabditis elegans. Semin. Dev. Biol. 1994, 5, 121–130. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Liu, J.; Qi, J.; Wei, B.; Yang, J.; Liang, H.; Chen, Y.; Chen, J.; Wu, Y.; et al. H3K9 Methylation Is a Barrier during Somatic Cell Reprogramming into iPSCs. Nat. Genet. 2013, 45, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and Impediments of the Pluripotency Reprogramming Factors’ Initial Engagement with the Genome. Cell 2012, 151, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.E.; Kang, Y.-K.; Beppu, H.; Lei, H.; Li, E. Histone H3-K9 Methyltransferase ESET Is Essential for Early Development. Mol. Cell. Biol. 2004, 24, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a Histone Methyltransferase Plays a Dominant Role in Euchromatic Histone H3 Lysine 9 Methylation and Is Essential for Early Embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.F.; Piccolo, F.M.; Tsubouchi, T.; Sauer, S.; Ryan, N.K.; Bruno, L.; Landeira, D.; Santos, J.; Banito, A.; Gil, J.; et al. ESCs Require PRC2 to Direct the Successful Reprogramming of Differentiated Cells toward Pluripotency. Cell Stem Cell 2010, 6, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.R.; Chuang, P.-T.; Meyer, B.J. DPY-30, a Nuclear Protein Essential Early in Embryogenesis for Caenorhabditis elegans Dosage Compensation. Development 1995, 121, 3323–3334. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R.; Jow, M.M.; Meyer, B.J. The T-Box Transcription Factor SEA-1 Is an Autosomal Element of the X:A Signal That Determines C. Elegans Sex. Dev. Cell 2005, 9, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Carmi, I.; Kopczynski, J.B.; Meyer, B.J. The Nuclear Hormone Receptor SEX-1 Is an X-Chromosome Signal That Determines Nematode Sex. Nature 1998, 396, 168–173. [Google Scholar] [CrossRef]
- Carmi, I.; Meyer, B.J. The Primary Sex Determination Signal of Caenorhabditis elegans. Genetics 1999, 152, 999–1015. [Google Scholar] [CrossRef]
- Nicoll, M.; Akerib, C.C.; Meyer, B.J. X-Chromosome-Counting Mechanisms That Determine Nematode Sex. Nature 1997, 388, 200–204. [Google Scholar] [CrossRef]
- Farboud, B.; Nix, P.; Jow, M.M.; Gladden, J.M.; Meyer, B.J. Molecular Antagonism between X-Chromosome and Autosome Signals Determines Nematode Sex. Genes Dev. 2013, 27, 1159–1178. [Google Scholar] [CrossRef]
- Gladden, J.M.; Meyer, B.J. A ONECUT Homeodomain Protein Communicates X Chromosome Dose to Specify Caenorhabditis elegans Sexual Fate by Repressing a Sex Switch Gene. Genetics 2007, 177, 1621–1637. [Google Scholar] [CrossRef] [PubMed]
- Farboud, B.; Novak, C.S.; Nicoll, M.; Quiogue, A.; Meyer, B.J. Dose-Dependent Action of the RNA Binding Protein FOX-1 to Relay X-Chromosome Number and Determine C. Elegans Sex. eLife 2020, 9, e62963. [Google Scholar] [CrossRef]
- Rhind, N.R.; Miller, L.M.; Kopczynski, J.B.; Meyer, B.J. Xol-1 Acts as an Early Switch in the C. Elegans Male/Hermaphrodite Decision. Cell 1995, 80, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.C.; Nabeshima, K.; Csankovszki, G. The C. Elegans Dosage Compensation Complex Mediates Interphase X Chromosome Compaction. Epigenetics Chromatin 2014, 7, 31. [Google Scholar] [CrossRef]
- Csankovszki, G.; Collette, K.; Spahl, K.; Carey, J.; Snyder, M.; Petty, E.; Patel, U.; Tabuchi, T.; Liu, H.; McLeod, I.; et al. Three Distinct Condensin Complexes Control C. Elegans Chromosome Dynamics. Curr. Biol. 2009, 19, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.-T.; Albertson, D.G.; Meyer, B.J. DPY-27: A Chromosome Condensation Protein Homolog That Regulates C. Elegans Dosage Compensation through Association with the X Chromosome. Cell 1994, 79, 459–474. [Google Scholar] [CrossRef]
- Csankovszki, G.; McDonel, P.; Meyer, B.J. Recruitment and Spreading of the C. Elegans Dosage Compensation Complex Along X Chromosomes. Science 2004, 303, 1182–1185. [Google Scholar] [CrossRef]
- Ercan, S.; Dick, L.L.; Lieb, J.D. The C. Elegans Dosage Compensation Complex Propagates Dynamically and Independently of X Chromosome Sequence. Curr. Biol. CB 2009, 19, 1777–1787. [Google Scholar] [CrossRef]
- Blauwkamp, T.A.; Csankovszki, G. Two Classes of Dosage Compensation Complex Binding Elements along Caenorhabditis elegans X Chromosomes. Mol. Cell. Biol. 2009, 29, 2023–2031. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, J.; Jimenez, D.S.; Ragipani, B.; Zhang, B.; Street, L.A.; Kramer, M.; Albritton, S.E.; Winterkorn, L.H.; Morao, A.K.; Ercan, S. Condensin DC Loads and Spreads from Recruitment Sites to Create Loop-Anchored TADs in C. elegans. eLife 2022, 11, e68745. [Google Scholar] [CrossRef] [PubMed]
- Kruesi, W.S.; Core, L.J.; Waters, C.T.; Lis, J.T.; Meyer, B.J. Condensin Controls Recruitment of RNA Polymerase II to Achieve Nematode X-Chromosome Dosage Compensation. eLife 2013, 2, e00808. [Google Scholar] [CrossRef]
- Anderson, E.C.; Frankino, P.A.; Higuchi-Sanabria, R.; Yang, Q.; Bian, Q.; Podshivalova, K.; Shin, A.; Kenyon, C.; Dillin, A.; Meyer, B.J. X Chromosome Domain Architecture Regulates Caenorhabditis elegans Lifespan but Not Dosage Compensation. Dev. Cell 2019, 51, 192–207.e6. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.B.; Jash, E.; Chawla, B.; Haines, R.A.; Tushman, L.E.; Troll, R.; Csankovszki, G. Dual Roles for Nuclear RNAi Argonautes in Caenorhabditis elegans Dosage Compensation. Genetics 2022, 221, iyac033. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Histone Variant | Histone Marks | Active/Repressive | Localization and Function | Ref |
|---|---|---|---|---|
| H3 | H3K9me2/me3, H3K27me2/me3 | Repressive | Canonical histone H3 that favors the deposition of repressive histone marks. Depleted from chromatin in early embryos. Transcribed in embryos from 2-cell stage and accumulates on chromatin throughout early embryogenesis on all cells except P-lineage cells. Promotes termination of developmental plasticity. | [39,42] |
| H3.3 | H3K4me3, H3K36me2 | Active | Histone H3 variant that favors the deposition of active histone marks. Inherited from germ cells in early embryos in 2-cell to 50-cell stage. Depleted from chromatin during embryo development, except in P-lineage cells. | [38,39,41,42] |
| H1.1 | H3K9me2 | Repressive | Linker histone H1 that promotes the accumulation of repressive histone marks. Rapidly translocated from the cytoplasm into the nucleus after fertilization. Associated with chromatin in all embryonic cells except Z2 and Z3 PGCs. Promotes the silencing of heterochromatic loci. | [44,45] |
| HTZ-1 | Active | Histone H2A variant enriched upstream of transcribed genes required for development where it influences PolII engagement. Incorporated into chromatin starting at 4-cell stage and required for appropriate embryonic development. | [58] |
| Histone Modification | Histone Methyl/Acetyl Transferase | Histone Demethylase/Deacetylase | Active/Repressive | Function | Ref |
|---|---|---|---|---|---|
| H3K9me2 | met-2 | jmjd-1.2 | Repressive | Dynamically increases during gastrulation. Repressive mark enriched on heterochromatin. High levels of H3K9me2 promotes developmental plasticity. | [9,14,19,20,25,36,61] |
| H3K9me3 | set-25 | Repressive | Dynamically increases during gastrulation. Repressive mark enriched on heterochromatin. Linked to heterochromatin formation, tethering of the chromatin to nuclear periphery, TAD formation and establishment of dosage compensation on the hermaphrodite X chromosome. | [14,16,18,20,21,25,62,63,64] | |
| H3K27me3 | mes-2 | utx-1, jmjd-3.1, jmjd-3.2, jmjd-3.3 | Repressive | Inherited from maternal and paternal germ cells and dynamically enriched in embryonic cells in a lineage-specific manner. Repressive mark enriched on heterochromatin and LADs. Promotes termination of developmental plasticity. | [25,27,28,29,30,62,65] |
| H3K23me2 | set-32 | jmjd-1.2 | Repressive | Repressive mark enriched on heterochromatin. | [24,25,36] |
| H4K20me1/me2 | set-4 deposits H4K20me2 | dpy-21 converts H4K20me2 to H4K20me1 on the X chromosome | Repressive | H4K20me1 is selectively enriched on the X chromosome by dpy-21. Repressive mark required for the establishment of dosage compensation in hermaphrodite embryos. | [66,67,68,69,70] |
| H3K4me2 | ash-2, set-16 | Active | Active mark inherited from paternal and maternal germ cells, generally enriched over gene bodies where it permits transcription at genomic loci. Enriched uniformly in all cells in early embryos, enriched specifically on PGCs in late embryos. | [2,47,50,51,52,54,56,57] | |
| H3K4me3 | ash-2, set-2, set-16 | Active | Active mark inherited from paternal and maternal germ cells, generally enriched at TSS where it permits transcription at genomic loci. Enriched uniformly in all cells in early embryos. Enriched in a lineage-specific manner starting at the eight-cell stage. | [2,49,50,51,52,54,57] | |
| H3K36me3 | mes-4, met-1 | Active | Active mark enriched on euchromatin. Required for expression of germline-specific genes in early embryos. | [17,28] | |
| H3K9ac | Active | Active mark excluded from heterochromatin by MET-2 nuclear foci. | [19] | ||
| H3K27ac | Active | Active mark excluded from heterochromatin by MET-2 nuclear foci. | [19] | ||
| H4K16ac | Active | Active mark inherited from maternal germ cells and selectively depleted from the X chromosome. | [69,70] | ||
| H3K56me3 | Repressive | Repressive mark enriched on heterochromatin. | [37] | ||
| H3K79me2 | Active | Active mark inherited from paternal and maternal germ cells. Depleted on chromatin in 1–4 cell embryos, then enriched after 16-cell stage. | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jash, E.; Csankovszki, G. Chromatin Organization during C. elegans Early Development. DNA 2024, 4, 64-83. https://doi.org/10.3390/dna4010004
Jash E, Csankovszki G. Chromatin Organization during C. elegans Early Development. DNA. 2024; 4(1):64-83. https://doi.org/10.3390/dna4010004
Chicago/Turabian StyleJash, Eshna, and Györgyi Csankovszki. 2024. "Chromatin Organization during C. elegans Early Development" DNA 4, no. 1: 64-83. https://doi.org/10.3390/dna4010004
APA StyleJash, E., & Csankovszki, G. (2024). Chromatin Organization during C. elegans Early Development. DNA, 4(1), 64-83. https://doi.org/10.3390/dna4010004