

Computational Strategies to Enhance Cell-Free Protein Synthesis Efficiency


Abstract
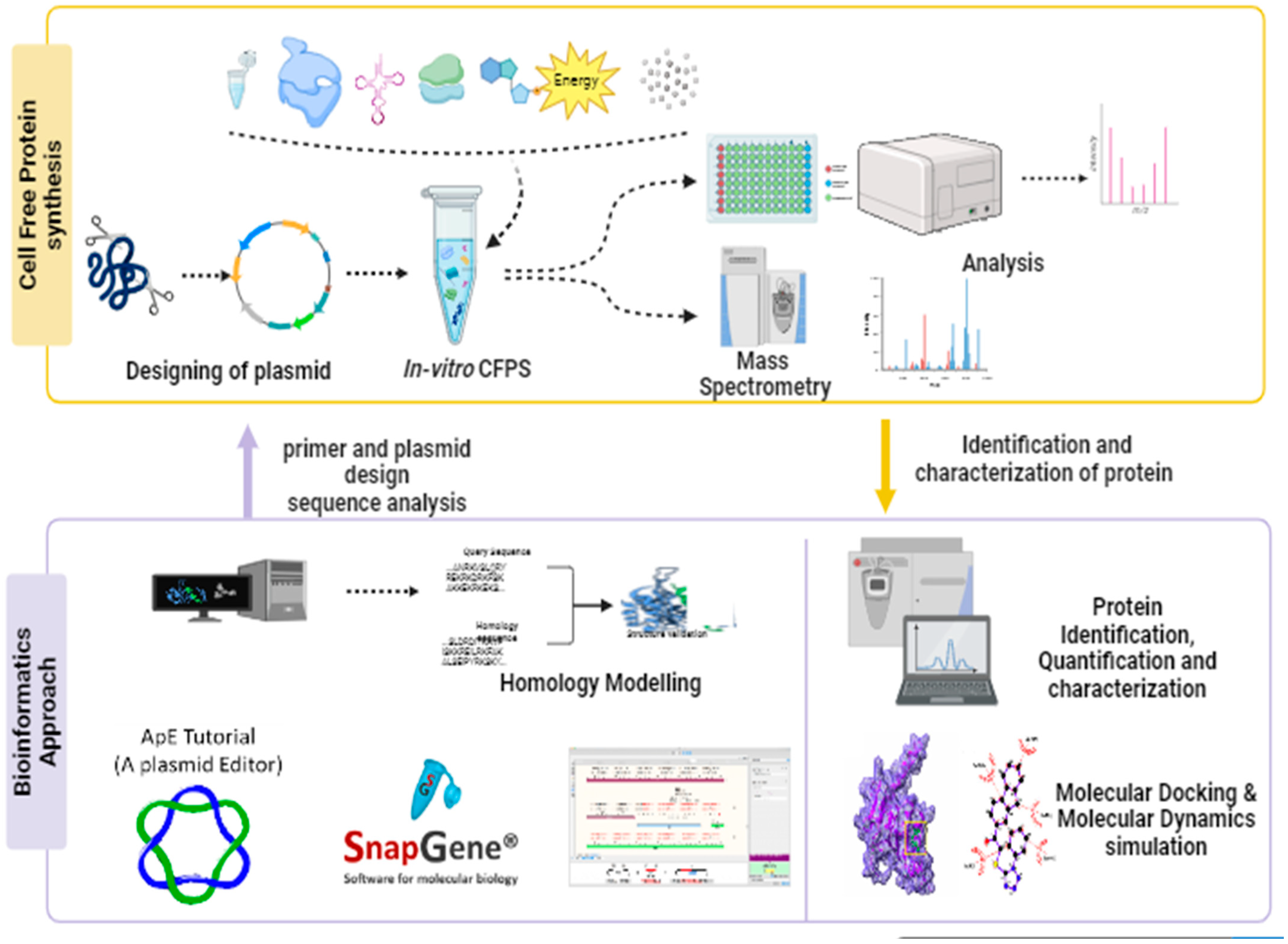
1. Introduction
2. Computational Modeling of CFPS Systems
2.1. Mathematical Models of CFPS Reactions
2.2. Simulation-Based Approaches to Predict Protein Synthesis Kinetics
2.3. Optimization Algorithms for Improving CFPS System Performance
3. Designing DNA Templates for Enhanced Protein Synthesis
3.1. Codon Optimization Strategies
3.2. mRNA Secondary Structure Prediction Tools
3.3. Regulatory Element Engineering for Transcriptional Control
4. Engineering Cell-Free Transcription and Translation Machinery
4.1. Rational Design of Cell-Free Expression Systems
4.2. Computational Tools for Optimizing Translation Initiation and Elongation
5. Predictive Modeling of Metabolic Pathways and Energy Utilization
5.1. Metabolic Flux Analysis in CFPS Systems
5.2. Predicting Substrate Availability and Utilization
5.3. Optimal Resource Allocation Strategies for Efficient Protein Synthesis
6. Machine Learning and Artificial Intelligence Approaches
6.1. Neural Network Models for CFPS Optimization
6.2. Deep Learning Algorithms for Protein Synthesis Prediction
6.3. Reinforcement Learning for Adaptive Control of CFPS Systems
7. Case Studies and Applications
7.1. Essential Bioinformatics Tools
7.2. Examples of Successful CFPS Optimization Using Computational Methods
7.3. Actual vs. Virtual Experiment
7.4. Applications in Synthetic Biology, Biotechnology, and Pharmaceuticals
8. Challenges and Limitations of Current Computational Approaches
9. Future Perspectives and Emerging Trends
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kelwick, R.; Webb, A.J.; Macdonald, J.T.; Freemont, P.S. Development of a bacillus subtilis cell-free transcriptiontranslation system. In Proceedings of the IET Conference Publications, London, UK, 13–15 December 2016. [Google Scholar] [CrossRef]
- Silverman, A.D.; Karim, A.S.; Jewett, M.C. Cell-free gene expression: An expanded repertoire of applications. Nat. Rev. Genet. 2020, 21, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, A.; Iyappan, K.; Kang, T.J. Multiple Gene Expression in Cell-Free Protein Synthesis Systems for Reconstructing Bacteriophages and Metabolic Pathways. Microorganisms 2022, 10, 2477. [Google Scholar] [CrossRef]
- Jewett, M.C.; Swartz, J.R. Substrate replenishment extends protein synthesis with an in vitro translation system designed to mimic the cytoplasm. Biotechnol. Bioeng. 2004, 87, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Zemella, A.; Thoring, L.; Hoffmeister, C.; Kubick, S. Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems. ChemBioChem 2015, 16, 2420–2431. [Google Scholar] [CrossRef] [PubMed]
- Garenne, D.; Thompson, S.; Brisson, A.; Khakimzhan, A.; Noireaux, V. The all-E. coliTXTL toolbox 3.0: New capabilities of a cell-free synthetic biology platform. Synth. Biol. 2021, 6, ysab017. [Google Scholar] [CrossRef] [PubMed]
- Hodgman, C.E.; Jewett, M.C. Cell-free synthetic biology: Thinking outside the cell. Metab. Eng. 2012, 14, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Pardee, K.; Green, A.A.; Ferrante, T.; Cameron, D.E.; Daleykeyser, A.; Yin, P.; Collins, J.J. Paper-based synthetic gene networks. Cell 2014, 159, 940–954. [Google Scholar] [CrossRef]
- Tian, R.; Wang, M.; Shi, J.; Qin, X.; Guo, H.; Jia, X.; Li, J.; Liu, L.; Du, G.; Chen, J.; et al. Cell-free synthesis system-assisted pathway bottleneck diagnosis and engineering in Bacillus subtilis. Synth. Syst. Biotechnol. 2020, 5, 131–136. [Google Scholar] [CrossRef]
- Hong, S.H.; Kwon, Y.C.; Martin, R.W.; Des Soye, B.J.; De Paz, A.M.; Swonger, K.N.; Ntai, I.; Kelleher, N.L.; Jewett, M.C. Improving cell-free protein synthesis through genome engineering of Escherichia coli lacking release factor 1. ChemBioChem 2015, 16, 844–853. [Google Scholar] [CrossRef]
- Chappell, J.; Jensen, K.; Freemont, P.S. Validation of an entirely in vitro approach for rapid prototyping of DNA regulatory elements for synthetic biology. Nucleic Acids Res. 2013, 41, 3471–3481. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, H.; Liu, Q.; Gani, R. Chemical product design—Recent advances and perspectives. Curr. Opin. Chem. Eng. 2020, 27, 22–34. [Google Scholar] [CrossRef]
- Caschera, F.; Karim, A.S.; Gazzola, G.; D’Aquino, A.E.; Packard, N.H.; Jewett, M.C. High-Throughput Optimization Cycle of a Cell-Free Ribosome Assembly and Protein Synthesis System. ACS Synth. Biol. 2018, 7, 2841–2853. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Noireaux, V. An E. coli cell-free expression toolbox: Application to synthetic gene circuits and artificial cells. ACS Synth. Biol. 2012, 1, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.S.; Dudley, Q.M.; Juminaga, A.; Yuan, Y.; Crowe, S.A.; Heggestad, J.T.; Garg, S.; Abdalla, T.; Grubbe, W.S.; Rasor, B.J.; et al. In vitro prototyping and rapid optimization of biosynthetic enzymes for cell design. Nat. Chem. Biol. 2020, 16, 912–919. [Google Scholar] [CrossRef]
- Oakes, B.L.; Nadler, D.C.; Flamholz, A.; Fellmann, C.; Staahl, B.T.; Doudna, J.A.; Savage, D.F. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat. Biotechnol. 2016, 34, 646–651. [Google Scholar] [CrossRef]
- Sun, Z.Z.; Hayes, C.A.; Shin, J.; Caschera, F.; Murray, R.M.; Noireaux, V. Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. J. Vis. Exp. 2013, 79, e50762. [Google Scholar] [CrossRef]
- Kazuta, Y.; Matsuura, T.; Ichihashi, N.; Yomo, T. Synthesis of milligram quantities of proteins using a reconstituted in vitro protein synthesis system. J. Biosci. Bioeng. 2014, 118, 554–557. [Google Scholar] [CrossRef]
- Garamella, J.; Marshall, R.; Rustad, M.; Noireaux, V. The All E. coli TX-TL Toolbox 2.0: A Platform for Cell-Free Synthetic Biology. ACS Synth. Biol. 2016, 5, 344–355. [Google Scholar] [CrossRef]
- Jewett, M.C.; Swartz, J.R. Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol. Bioeng. 2004, 86, 19–26. [Google Scholar] [CrossRef]
- Salis, H.M.; Mirsky, E.A.; Voigt, C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009, 27, 946–950. [Google Scholar] [CrossRef]
- Angov, E.; Hillier, C.J.; Kincaid, R.L.; Lyon, J.A. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS ONE 2008, 3, e2189. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Hoener, C.; Siederdissen, Z.; Tafer, H.; Flamm, C.; Höner Zu Siederdissen, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0 Algorithms for Molecular Biology ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6. [Google Scholar] [CrossRef]
- Rhodius, V.A.; Segall-Shapiro, T.H.; Sharon, B.D.; Ghodasara, A.; Orlova, E.; Tabakh, H.; Burkhardt, D.H.; Clancy, K.; Peterson, T.C.; Gross, C.A.; et al. Design of orthogonal genetic switches based on a crosstalk map of σs, anti-σs, and promoters. Mol. Syst. Biol. 2013, 9, 702. [Google Scholar] [CrossRef]
- Tokmakov, A.A.; Kurotani, A.; Shirouzu, M.; Fukami, Y.; Yokoyama, S. Bioinformatics analysis and optimization of cell-free protein synthesis. Methods Mol. Biol. 2014, 1118, 17–33. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Springer Protocols Handbooks; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005. [Google Scholar] [CrossRef]
- Kurotani, A.; Takagi, T.; Toyama, M.; Shirouzu, M.; Yokoyama, S.; Fukami, Y.; Tokmakov, A.A. Comprehensive bioinformatics analysis of cell-free protein synthesis: Identification of multiple protein properties that correlate with successful expression. FASEB J. 2010, 24, 1095–1104. [Google Scholar] [CrossRef]
- Kumar Goshisht, M. Machine Learning and Deep Learning in Synthetic Biology: Key Architectures, Applications, and Challenges. ACS Omega 2024, 9, 9921–9945. [Google Scholar] [CrossRef]
- Ferro, D.; Franchi, N.; Mangano, V.; Bakiu, R.; Cammarata, M.; Parrinello, N.; Santovito, G.; Ballarin, L. Characterization and metal-induced gene transcription of two new copper zinc superoxide dismutases in the solitary ascidian Ciona intestinalis. Aquat. Toxicol. 2013, 140–141, 369–379. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Hirokawa, T.; Boon-Chieng, S.; Mitaku, S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 1998, 14, 378–379. [Google Scholar] [CrossRef]
- Fereig, R.M.; Abdelbaky, H.H. Comparative study on Toxoplasma gondii dense granule protein 7, peroxiredoxin 1 and 3 based on bioinformatic analysis tools. Ger. J. Microbiol. 2022, 2, 30–38. [Google Scholar] [CrossRef]
- Bryson, K.; McGuffin, L.J.; Marsden, R.L.; Ward, J.J.; Sodhi, J.S.; Jones, D.T. Protein structure prediction servers at University College London. Nucleic Acids Res. 2005, 33, W36–W38. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A.; Van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Ohara, O. DomCut: Prediction of inter-domain linker regions in amino acid sequences. Bioinformatics 2003, 19, 673–674. [Google Scholar] [CrossRef]
- Tokmakov, A.A. Identification of multiple physicochemical and structural properties associated with soluble expression of eukaryotic proteins in cell-free bacterial extracts. Front. Microbiol. 2014, 5, 295. [Google Scholar] [CrossRef]
- Cheng, J.; Saigo, H.; Baldi, P. Large-scale prediction of disulphide bridges using kernel methods, two-dimensional recursive neural networks, and weighted graph matching. Proteins Struct. Funct. Genet. 2006, 62, 617–629. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Šali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Kuszewski, J.J.; Tjandra, N.; Clore, G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003, 160, 65–73. [Google Scholar] [CrossRef]
- Qin, S.; Hicks, A.; Dey, S.; Prasad, R.; Zhou, H.X. ReSMAP: Web Server for Predicting Residue-Specific Membrane-Association Propensities of Intrinsically Disordered Proteins. Membranes 2022, 12, 773. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chan, C.Y.; Lawrence, C.E. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004, 32, W135–W141. [Google Scholar] [CrossRef] [PubMed]
- Kaledhonkar, S.; Fu, Z.; Caban, K.; Li, W.; Chen, B.; Sun, M.; Gonzalez, R.L.; Frank, J. Late steps in bacterial translation initiation visualized using time-resolved cryo-EM. Nature 2019, 570, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.; Perez, J.G.; Carlson, E.D.; Ntai, I.; Isaacs, F.J.; Kelleher, N.L.; Jewett, M.C. Translation system engineering in Escherichia coli enhances non-canonical amino acid incorporation into proteins. Biotechnol. Bioeng. 2017, 114, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Deley Cox, V.E.; Cole, M.F.; Gaucher, E.A. Incorporation of Modified Amino Acids by Engineered Elongation Factors with Expanded Substrate Capabilities. ACS Synth. Biol. 2019, 8, 287–296. [Google Scholar] [CrossRef]
- Opgenorth, P.H.; Korman, T.P.; Bowie, J.U. A synthetic biochemistry module for production of bio-based chemicals from glucose. Nat. Chem. Biol. 2016, 12, 393–395. [Google Scholar] [CrossRef]
- Rollin, J.A.; Tam, T.K.; Zhang, Y.H.P. New biotechnology paradigm: Cell-free biosystems for biomanufacturing. Green Chem. 2013, 15, 1708–1719. [Google Scholar] [CrossRef]
- Crown, S.B.; Long, C.P.; Antoniewicz, M.R. Optimal tracers for parallel labeling experiments and 13C metabolic flux analysis: A new precision and synergy scoring system. Metab. Eng. 2016, 38, 10–18. [Google Scholar] [CrossRef]
- Becker, S.A.; Feist, A.M.; Mo, M.L.; Hannum, G.; Palsson, B.; Herrgard, M.J. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox. Nat. Protoc. 2007, 2, 727–738. [Google Scholar] [CrossRef]
- Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef]
- Martin, J.P.; Rasor, B.J.; DeBonis, J.; Karim, A.S.; Jewett, M.C.; Tyo, K.E.J.; Broadbelt, L.J. A dynamic kinetic model captures cell-free metabolism for improved butanol production. Metab. Eng. 2023, 76, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Hanly, T.J.; Henson, M.A. Dynamic flux balance modeling of microbial co-cultures for efficient batch fermentation of glucose and xylose mixtures. Biotechnol. Bioeng. 2011, 108, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, Y.H.; Schmidt-Dannert, C.; Lee, P.C. Redesign, reconstruction, and directed extension of the brevibacterium linens C40 carotenoid pathway in escherichia coli. Appl. Environ. Microbiol. 2010, 76, 5199–5206. [Google Scholar] [CrossRef] [PubMed]
- Robles-Rodriguez, C.E.; Steur, E. Flux balance analysis-based ranking for model order reduction of biochemical networks. IFAC-PapersOnLine 2021, 54, 556–561. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Ranganathan, S.; Suthers, P.F.; Maranas, C.D. OptForce: An optimization procedure for identifying all genetic manipulations leading to targeted overproductions. PLoS Comput. Biol. 2010, 6, e1000744. [Google Scholar] [CrossRef]
- Wang, F.S.; Wu, W.H. Computer-aided design for genetic modulation to improve biofuel production. In Process Systems Engineering for Biofuels Development; Bonilla-Petriciolet, A., Rangaiah, G.P., Eds.; Wiley: Hoboken, NJ, USA, 2020; pp. 173–189. [Google Scholar] [CrossRef]
- Brookwell, A.; Oza, J.P.; Caschera, F. Biotechnology Applications of Cell-Free Expression Systems. Life 2021, 11, 1367. [Google Scholar] [CrossRef]
- Lim, H.J.; Kim, D.M. Cell-free metabolic engineering: Recent developments and future prospects. Methods Protoc. 2019, 2, 33. [Google Scholar] [CrossRef]
- Duran-Villalobos, C.A.; Ogonah, O.; Melinek, B.; Bracewell, D.G.; Hallam, T.; Lennox, B. Multivariate statistical data analysis of cell-free protein synthesis toward monitoring and control. AIChE J. 2021, 67, e17257. [Google Scholar] [CrossRef]
- Wang, J.; Cao, H.; Zhang, J.Z.H.; Qi, Y. Computational Protein Design with Deep Learning Neural Networks. Sci. Rep. 2018, 8, 6349. [Google Scholar] [CrossRef]
- Caschera, F.; Bedau, M.A.; Buchanan, A.; Cawse, J.; de Lucrezia, D.; Gazzola, G.; Hanczyc, M.M.; Packard, N.H. Coping with complexity: Machine learning optimization of cell-free protein synthesis. Biotechnol. Bioeng. 2011, 108, 2218–2228. [Google Scholar] [CrossRef] [PubMed]
- Pandi, A.; Adam, D.; Zare, A.; Trinh, V.T.; Schaefer, S.L.; Burt, M.; Klabunde, B.; Bobkova, E.; Kushwaha, M.; Foroughijabbari, Y.; et al. Cell-free biosynthesis combined with deep learning accelerates de novo-development of antimicrobial peptides. Nat. Commun. 2023, 14, 7197. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Green, A.A.; Silver, P.A.; Collins, J.J.; Yin, P. Toehold switches: De-novo-designed regulators of gene expression. Cell 2014, 159, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Mahajan, S.P.; Sulam, J.; Gray, J.J. Deep Learning in Protein Structural Modeling and Design. Patterns 2020, 1, 100142. [Google Scholar] [CrossRef]
- Kouba, P.; Kohout, P.; Haddadi, F.; Bushuiev, A.; Samusevich, R.; Sedlar, J.; Damborsky, J.; Pluskal, T.; Sivic, J.; Mazurenko, S. Machine Learning-Guided Protein Engineering. ACS Catal. 2023, 13, 13863–13895. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, J.; Ding, X.; Lin, J.; Jiang, H.; Zhou, H.; Lu, Y. Exploration of the Tolerance Ability of a Cell-Free Biosynthesis System to Toxic Substances. Appl. Biochem. Biotechnol. 2019, 189, 1096–1107. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, X.; Wang, T.; Guo, W.; Lu, Y. Development and comparison of cell-free protein synthesis systems derived from typical bacterial chassis. Bioresour. Bioprocess. 2021, 8, 58. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, S.; Kim, D.M. Translational Detection of Indole by Complementary Cell-free Protein Synthesis Assay. Front. Bioeng. Biotechnol. 2022, 10, 900162. [Google Scholar] [CrossRef]
- Jin, X.; Hong, S.H. Cell-free protein synthesis for producing ‘difficult-to-express’ proteins. Biochem. Eng. J. 2018, 138, 156–164. [Google Scholar] [CrossRef]
- Caschera, F.; Noireaux, V. Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription-translation system. Biochimie 2014, 99, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.S.; Jewett, M.C. A cell-free framework for rapid biosynthetic pathway prototyping and enzyme discovery. Metab. Eng. 2016, 36, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.Z.; Yeung, E.; Hayes, C.A.; Noireaux, V.; Murray, R.M. Linear DNA for rapid prototyping of synthetic biological circuits in an escherichia coli based TX-TL cell-free system. ACS Synth. Biol. 2014, 3, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Caschera, F. Bacterial cell-free expression technology to in vitro systems engineering and optimization. Synth. Syst. Biotechnol. 2017, 2, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Vo, P.N.L.; Na, D. Advancement of metabolic engineering assisted by synthetic biology. Catalysts 2018, 8, 619. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, D.M. Recent advances in development of cell-free protein synthesis systems for fast and efficient production of recombinant proteins. FEMS Microbiol. Lett. 2018, 365, fny174. [Google Scholar] [CrossRef]
- Zhou, L.; Ma, Y.; Wang, K.; Chen, T.; Huang, Y.; Liu, L.; Li, Y.; Sun, J.; Hu, Y.; Li, T.; et al. Omics-guided bacterial engineering of Escherichia coli ER2566 for recombinant protein expression. Appl. Microbiol. Biotechnol. 2023, 107, 853–865. [Google Scholar] [CrossRef]
- Falgenhauer, E.; Von Schönberg, S.; Meng, C.; Mückl, A.; Vogele, K.; Emslander, Q.; Ludwig, C.; Simmel, F.C. Evaluation of an E. coli Cell Extract Prepared by Lysozyme-Assisted Sonication via Gene Expression, Phage Assembly and Proteomics. Chembiochem 2021, 22, 2805–2813. [Google Scholar] [CrossRef]
- Lin, X.; Zhou, C.; Wang, T.; Huang, X.; Chen, J.; Li, Z.; Zhang, J.; Lu, Y. CO2-elevated cell-free protein synthesis. Synth. Syst. Biotechnol. 2022, 7, 911–917. [Google Scholar] [CrossRef]

{kind=link}
| Plasmid Editing and Design Software | Description | Application to CFPS |
|---|---|---|
| SnapGene (Version 7.0) | A versatile molecular biology software for DNA and plasmid sequence analysis, visualization, and annotation. | SnapGene can be used to design and analyze plasmids containing DNA templates for CFPS reactions. Researchers can annotate DNA sequences with relevant features for CFPS, such as promoter regions, coding sequences, and regulatory elements. |
| Geneious (Version 2024.0.2) | Offers tools for plasmid construction, sequence alignment, primer design, and molecular cloning. | Geneious can facilitate the design of plasmids containing genes of interest for CFPS experiments. It provides features for sequence alignment to ensure accurate cloning and primer design for the PCR amplification of DNA templates. |
| Vector NTI (Version 11.5.3) | A comprehensive suite for plasmid design, analysis, and management. | Vector NTI enables the design and analysis of plasmids optimized for CFPS applications. It allows researchers to manipulate DNA sequences, predict restriction enzyme digestion patterns, and manage plasmid libraries efficiently. |
| ApE (A Plasmid Editor) (Version 2.0.45) | Simple and efficient software for DNA sequence visualization, editing, and analysis. | ApE is useful for visualizing and editing plasmid sequences intended for CFPS experiments. It allows researchers to annotate features relevant to CFPS, such as start and stop codons, ribosome binding sites, and protein tags. |
| Benchling (Version 2023.4) | Cloud-based molecular biology platform with tools for plasmid design, cloning, and sequence analysis. | Benchling provides collaborative tools for designing and sharing plasmids optimized for CFPS. It offers features for sequence editing, primer design, and virtual cloning simulations to streamline the design process for CFPS experiments. |
| Feature | Tool/Server | Description | Application | Reference |
|---|---|---|---|---|
| Physicochemical parameters (pI, charge, hydrophobicity) | ProtParam (http://web.expasy.org/protparam/) (accessed on 15 February 2024) | Calculates various physicochemical properties of protein sequences. | Protein function prediction, protein–protein interaction studies, and drug design. | [27] |
| Solvent accessibility | ACCpro 4.0 (http://scratch.proteomics.ics.uci.edu/explanation.html) (accessed on 15 February 2024) | Predicts how accessible each amino acid residue is to solvent. | Understanding protein–protein interactions, protein folding, and stability. | [26,28] |
| Signal sequences | SignalP (http://www.cbs.dtu.dk/services/SignalP/) (accessed on 15 February 2024) | Predicts the presence of signal peptides, which target proteins for secretion from cells/identifies signal sequences for protein export from the cell. | Predicting protein localization, understanding protein targeting pathways, and designing recombinant proteins for expression in different systems. | [26,29,30,31] |
| Transmembrane domains | TM: http://bp.nuap.nagoya-u.ac.jp/sosui/sosuisignal/ (accessed on 15 February 2024) | Predicts the presence and location of transmembrane domains, which anchor proteins to membranes | Identifying membrane proteins, studying protein–lipid interactions. and predicting their topology | [28,32,33] |
| PEST sequences (protein degradation) | PESTfind http://emboss.bioinformatics.nl/cgi-bin/emboss/pestfind (accessed on 15 February 2024) | Predicts the presence of PEST regions, which are often rich in proline, glutamic acid, serine, and threonine, and T associated with rapid protein degradation. | Investigating protein stability and turnover and predicting protein half-life or regulatory roles involved in signal transduction or cell cycle control. | [34,35] |
| Coiled-coil regions (protein–protein interaction) | pepCoil (https://www.bioinformatics.nl/cgi-bin/emboss/pepcoil.) (accessed on 15 February 2024) | Identifies regions that can form helical bundles involved in protein–protein interactions. | Studying protein dimerization or oligomerization, designing protein–protein interaction inhibitors. | [26,28,36] |
| Interdomain linkers | DomCut http://www.bork.embl.de/_suyama/domcut/ (accessed on 15 February 2024) | Identifies flexible linker regions between protein domains. | Understanding protein domain movement and function, protein engineering. | [37,38] |
| S-S bonds | Dipro https://download.igb.uci.edu/bridge.html (accessed on 15 February 2024) | Predicts the formation of disulfide bonds between cysteine residues. | Understanding protein folding and stability, protein engineering. | [39] |
| Secondary Structure Prediction | Mfold http://unafold.rna.albany.edu/?q=mfold (accessed on 15 February 2024) | Mfold is a web server that predicts RNA and DNA secondary structures using energy minimization algorithms based on thermodynamic parameters. | Designing DNA templates with optimized secondary structures to enhance protein synthesis in CFPS systems. | [40] |
| Secondary Structure Prediction | RNAstructure https://rna.urmc.rochester.edu/RNAstructureWeb/Servers/Predict1/Predict1.html (accessed on 15 February 2024) | RNA structure is a software package for predicting RNA secondary structures, offering advanced features including base pairing probabilities and free energy calculations. | Predicting stable RNA secondary structures for optimized mRNA templates in CFPS, potentially improving translational efficiency. | [23] |
| Homology Modeling | MODELLER https://salilab.org/modeller/ (accessed on 15 February 2024) | Predicts the three-dimensional structure of a protein based on the alignment of its sequence to known protein structures (templates). | Predicting protein structures when experimental structures are unavailable, facilitating structure-based studies of proteins, protein engineering, and drug design. | [41] |
| Homology Modeling | XPLOR-NIH https://nmr.cit.nih.gov/xplor-nih/ (accessed on 15 February 2024) | A software suite for computational structural biology, which includes modules for molecular dynamics simulations, energy minimization, and homology modeling based on experimental restraints. | Integrating experimental data, such as NMR spectroscopy or electron microscopy, into homology modeling to refine protein structures and generate accurate models for functional studies. | [42] |
| Predictor of residue-Specific Membrane-Association Propensities of IDPs | ReSMAP https://pipe.rcc.fsu.edu/ReSMAPidp/ (accessed on 15 February 2024) | Predicts the Residue-Specific Membrane-Association Propensities of intrinsically disordered proteins using a sequence-based partition function. | Identifying the residue-wise membrane interaction propensity of intrinsically disordered proteins | [43] |
| Bioinformatics Tools | Description | Application to CFPS |
|---|---|---|
| BLAST (Basic Local Alignment Search Tool) | A widely used tool for comparing nucleotide or protein sequences against databases to find similar sequences. | BLAST can be used to identify homologous sequences of genes or proteins relevant to CFPS experiments. Researchers can search for known protein sequences to compare with sequences of interest for CFPS template design. |
| EMBOSS (European Molecular Biology Open Software Suite) | Collection of bioinformatics tools for sequence analysis, alignment, and manipulation. | EMBOSS provides a suite of tools for analyzing DNA and protein sequences relevant to CFPS. Researchers can use EMBOSS tools for sequence alignment, motif search, and statistical analysis to characterize genes and regulatory elements for CFPS template design. |
| UCSC Genome Browser | A powerful tool for visualizing and analyzing genome sequences and annotations. | The UCSC Genome Browser allows researchers to explore genomic regions containing genes of interest for CFPS. It provides access to genome-wide data, including gene annotations, regulatory elements, and conservation tracks, to inform the design of DNA templates for CFPS reactions. |
| NCBI Entrez | Provides access to a wide range of biomedical databases, including nucleotide and protein sequences, PubMed, and more. | NCBI Entrez enables researchers to search for genetic sequences, literature, and resources relevant to CFPS experiments. It provides access to nucleotide databases for retrieving DNA sequences of interest and PubMed for accessing research articles on CFPS methodologies and applications. |
| Ensembl | Genome browser and bioinformatics platform offering comprehensive genomic data and analysis tools for a wide range of organisms. | Ensembl provides genomic data and analysis tools for various organisms, facilitating the identification of genes and regulatory elements relevant to CFPS. Researchers can explore gene annotations, sequence variations, and functional annotations to inform the design of DNA templates for CFPS experiments. |
| Aspects | Actual Experiment | Virtual Experiment |
|---|---|---|
| Experimental set up | CFPS reactions conducted in the lab using biological components and controlled conditions (e.g., SDS-PAGE, Western blotting). | Computational simulations using mathematical models to predict system behavior. |
| Validation strategy | Comparison with experimental data; sensitivity analysis; iterative optimization. | Cross-validation with experimental datasets; wet lab validation. |
| Examples | Protein synthesis yields measured experimentally (e.g., SDS-PAGE). | Computational prediction. |
| Comparison | Small difference of 2 μg/mL between predicted and experimental yields. | Computational model accurately predicts protein synthesis outcomes. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kathirvel, I.; Gayathri Ganesan, N. Computational Strategies to Enhance Cell-Free Protein Synthesis Efficiency. BioMedInformatics 2024, 4, 2022-2042. https://doi.org/10.3390/biomedinformatics4030110
Kathirvel I, Gayathri Ganesan N. Computational Strategies to Enhance Cell-Free Protein Synthesis Efficiency. BioMedInformatics. 2024; 4(3):2022-2042. https://doi.org/10.3390/biomedinformatics4030110
Chicago/Turabian StyleKathirvel, Iyappan, and Neela Gayathri Ganesan. 2024. "Computational Strategies to Enhance Cell-Free Protein Synthesis Efficiency" BioMedInformatics 4, no. 3: 2022-2042. https://doi.org/10.3390/biomedinformatics4030110
APA StyleKathirvel, I., & Gayathri Ganesan, N. (2024). Computational Strategies to Enhance Cell-Free Protein Synthesis Efficiency. BioMedInformatics, 4(3), 2022-2042. https://doi.org/10.3390/biomedinformatics4030110