
Abraham Model Solute Descriptors for Favipiravir: Case of Tautomeric Equilibrium and Intramolecular Hydrogen-Bond Formation



Abstract
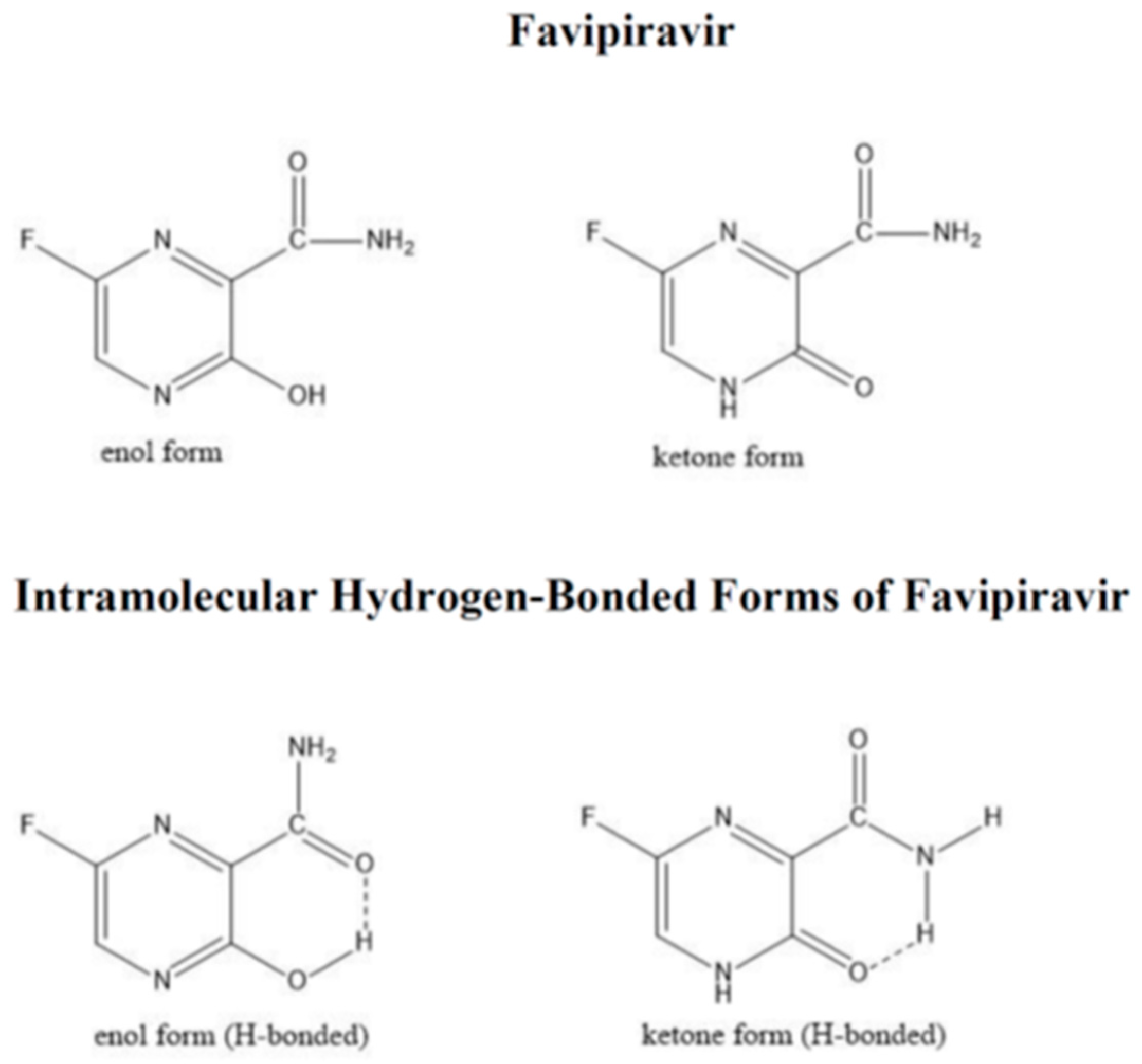
:1. Introduction
2. Solute Descriptor Calculations
3. Results
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yalamanchi, K.K.; van Oudenhoven, V.C.O.; Tutino, F.; Monge-Palacios, M.; Alshehri, A.; Gao, X.; Sarathy, S.M. Machine learning to predict standard enthalpy of formation of hydrocarbons. J. Phys. Chem. A 2019, 123, 8305–8313. [Google Scholar] [CrossRef]
- Trinh, C.; Meimaroglou, D.; Lasala, S.; Herbinet, O. Machine learning for the prediction of the thermo-chemical properties (enthalpy and entropy of formation) of a molecule from its molecular descriptors. Comp. Aided Chem. Eng. 2022, 51, 1471–1476. [Google Scholar] [CrossRef]
- Alshehri, A.S.; Tula, A.K.; You, F.; Gani, R. Next generation pure component property estimation models: With and without machine learning techniques. AIChE J. 2022, 68, e17469. [Google Scholar] [CrossRef]
- Grambow, C.A.; Li, Y.-P.; Green, W.H. Accurate thermochemistry with small data sets: A bond additivity correction and transfer learning approach. J. Phys. Chem. A 2019, 123, 5826–5835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, D.H.; Paffenroth, R.C.; Timko, M.T.; Teixeira, A.R. Dimensionally reduced machine learning model for predicting single component octanol-water partition coefficients. J. Cheminform. 2023, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Hu, B.; Perdana, M.C.; Zhao, Y.; Chen, Z. Extreme learning machine models for predicting the n-octanol/water partition coefficient (Kow) data of organic compounds. J. Environ. Chem. Eng. 2022, 10, 108552. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, M.; Xie, L.; Gao, Q.; Xu, X.; Xu, L. In silico prediction of boiling point, octanol-water partition coefficient, and retention time index of polycyclic aromatic hydrocarbons through machine learning. Chem. Biol. Drug Des. 2023, 101, 52–68. [Google Scholar] [CrossRef]
- Pozzan, A. QM Calculations in ADMET Prediction. Methods Mol. Biol. 2020, 2114, 285–305. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Ketkar, R.; Tao, P. ADMETboost: A web server for accurate ADMET prediction. J. Mol. Model. 2022, 28, 408. [Google Scholar] [CrossRef]
- Matyushin, D.D.; Sholokhova, A.Y.; Buryak, A.K. Deep learning-based prediction of gas chromatographic retention indices for a wide variety of polar and mid-polar liquid stationary phases. Int. J. Mol. Sci. 2021, 22, 9194. [Google Scholar] [CrossRef]
- Wu, J.; Kang, Y.; Pan, P.; Hou, T. Machine learning methods for pKa prediction of small molecules: Advances and challenges. Drug Discov. Today 2022, 27, 103372. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Li, Y.; Yang, J.D.; Liu, L.; Zhang, L.; Luo, S.; Cheng, J.-P. Holistic prediction of the pKa in diverse solvents based on a machine-learning approach. Solvation free energy and enthalpy. Angew. Chem. Int. Ed. 2020, 59, 19282–19291. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Vermeire, F.H.; Wu, H.; Walker, P.J.; Abraham, M.H.; Green, W.H. Group contribution and machine learning approaches to predict Abraham solute parameters, solvation free energy, and solvation enthalpy. J. Chem. Inf. Model. 2022, 62, 433–446. [Google Scholar] [CrossRef]
- Jaquis, B.J.; Li, A.; Monnier, N.D.; Sisk, R.S.; Acree, W.E., Jr.; Lang, A.S.I.D. Using machine learning to predict enthalpy of solvation. J. Solut. Chem. 2019, 48, 564–573. [Google Scholar] [CrossRef]
- Fowles, D.J.; Palmer, D.S. Solvation entropy, enthalpy and free energy prediction using a multi-task deep learning functional in 1D-RISM. Phys. Chem. Chem. Phys. 2023, 25, 6944–6954. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, N.; Endo, S.; Brown, T.N.; Watanabe, N.; Bronner, G.; Abraham, M.H.; Goss, K.-U. UFZ-LSER Database v 3.2.1 [Internet], Leipzig, Germany, Helmholtz Centre for Environmental Research-UFZ. 2017. Available online: http://www.ufz.de/lserd (accessed on 27 May 2023).
- Sinha, S.; Yang, C.; Wu, E.; Acree, W.E., Jr. Abraham solvation parameter model: Examination of possible intramolecular hydrogen-bonding using calculated solute descriptors. Liquids 2022, 2, 131–146. [Google Scholar] [CrossRef]
- Acree, W.E., Jr.; Smart, K.; Abraham, M.H. Abraham model solute descriptors reveal strong intramolecular hydrogen bonding in 1,4-dihydroxyanthraquinone and 1,8-dihydroxyanthraquinone. Phys. Chem. Liq. 2018, 56, 416–420. [Google Scholar] [CrossRef]
- Cui, X.; Yan, Y.; Zhang, X. Solubility determination, model correlation, thermodynamic analysis, molecular dynamic simulation of Favipiravir in twelve pure solvents. J. Mol. Liq. 2023, 348, 122208. [Google Scholar] [CrossRef]
- Joshi, S.; Parkar, J.; Ansari, A.; Vora, A.; Talwar, D.; Tiwaskar, M.; Patil, S.; Barkate, H. Barkat, Role of favipiravir in the treatment of COVID-19. Int. J. Infect. Dis. 2021, 102, 501–508. [Google Scholar] [CrossRef]
- Deneva, V.; Slavova, S.; Kumanova, A.; Vassilev, N.; Nedeltcheva-Antonova, D.; Antonov, L. Favipiravir-tautomeric and complexation properties in solution. Pharmaceuticals 2023, 16, 45. [Google Scholar] [CrossRef]
- Antonov, L. Favipiravir tautomerism: A theoretical insight. Theor. Chem. Acc. 2020, 139, 145. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Li, Z.; Kong, L.; Xie, Y.; Zhang, T.; Xu, W. Synthesis and crystal structure of 6-fluoro-3-hydroxypyrazine-2-carboxamide. Drug Discov. Ther. 2014, 8, 117–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.; Park, J.; Kim, S.M. Self-assembly driven by an aromatic primary amide motif. Org. Biomol. Chem. 2012, 10, 5332–5342. [Google Scholar] [CrossRef]
- Solvation Tools. Available online: https://rmg.mit.edu/database/solvation/search/ (accessed on 15 July 2023).
- Megahed, S.M.; Habib, A.A.; Hammad, S.F.; Kamal, A.H. Experimental design approach for development of spectrofluorimetric method for determination of favipiravir; a potential therapeutic agent against COVID-19 virus: Application to spiked human plasma. Spectrochim. Acta, Part A 2021, 249, 119241. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Yao, C.; Xie, G.; Song, S.; Li, H.; Qu, Y.; Tao, X. Novel formulations of the antiviral drug favipiravir: Improving permeability and tabletability. Cryst. Growth Des. 2021, 21, 3807–3817. [Google Scholar] [CrossRef]
- Deyab, M.A.; El-Shamy, O.A.A.; Thabet, H.K.; Ashmawy, A.M. Electrochemical and theoretical investigations of favipiravir drug performance as ecologically benign corrosion inhibitor for aluminum alloy in acid solution. Sci. Rep. 2023, 13, 8680. [Google Scholar] [CrossRef]
- Pari, A.A.; Yousefi, M. Interactions between favipiravir and a BNC cage towards drug delivery applications. Struct. Chem. 2022, 33, 159–167. [Google Scholar] [CrossRef]
- Mishra, G.P.; Panigrahi, D. Computational studies of drugs for possible action against Covid-19 infections. J. Drug Deliv. Ther. 2020, 10, 99–105. [Google Scholar] [CrossRef]
- SciFinder; Chemical Abstracts Service: Columbus, OH. Available online: https://scifinder.cas.org (accessed on 28 July 2023).
- Green, C.E.; Abraham, M.H.; Acree, W.E., Jr.; De Fina, K.M.; Sharp, T.L. Solvation descriptors for pesticides from the solubility of solids: Diuron as an example. Pest Manag. Sci. 2000, 56, 1043–1053. [Google Scholar] [CrossRef]
- Abraham, M.H.; Green, C.E.; Acree, W.E., Jr.; Hernandez, C.E.; Roy, L.E. Descriptors for solutes from the solubility of solids: Trans-stilbene as an example. J. Chem. Soc. Perkin Trans. 2 1998, 2677–2682. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.H.; Benjelloun-Dakhama, N.; Gola, J.M.R.; Acree, W.E., Jr.; Cain, W.S.; Cometto-Muniz, J.E. Solvation descriptors for ferrocene, and the estimation of some physicochemical and biochemical properties. New J. Chem. 2000, 24, 825–829. [Google Scholar] [CrossRef]
- Jalan, A.; Ashcraft, R.W.; West, R.H.; Green, W.H. Predicting solvation energies for kinetic modeling. Annu. Rep. Prog. Chem. Sec. C Phys. Chem. 2010, 106, 211–258. [Google Scholar] [CrossRef]
- Clarke, E.D.; Mallon, L. The determination of Abraham descriptors and their application to crop protection research. In Modern Methods in Crop Protection Research; Jeschke, P., Krämer, W., Schirmer, U., Witschel, M., Eds.; Wiley: New York, NY, USA, 2012. [Google Scholar]
- Clarke, E.D. Beyond physical properties—Application of Abraham descriptors and LFER analysis in agrochemical research. Bioorg. Med. Chem. 2009, 17, 4153–4159. [Google Scholar] [CrossRef]
- Poole, C.F.; Ariyasena, T.C.; Lenca, N. Estimation of the environmental properties of compounds from chromatographic properties and the solvation parameter method. J. Chromatogr. A 2013, 1317, 85–104. [Google Scholar] [CrossRef]
- Poole, C.F.; Atapattu, S.N. Recent advances for estimating environmental properties for small molecules from chromatographic measurements and the solvation parameter model. J. Chromatogr. A 2023, 1687, 463682. [Google Scholar] [CrossRef]
- Endo, S.; Goss, K.-U. Applications of polyparameter linear free energy relationships in environmental chemistry. Environ. Sci. Technol. 2014, 48, 12477–12491. [Google Scholar] [CrossRef]
- Moshikur, R.M.; Ali, K.; Wakabayashi, R.; Moniruzzaman, M.; Goto, M. Favipiravir-based ionic liquids as potent antiviral drugs for oral delivery: Synthesis, solubility, and pharmacokinetic evaluation. Mol. Pharm. 2021, 18, 3108–3115. [Google Scholar] [CrossRef]
- Abraham, M.H.; McGowan, J.C. The use of characteristic volumes to measure cavity terms in reversed phase liquid chromatography. Chromatographia 1987, 23, 243–246. [Google Scholar] [CrossRef]
- Martin, Y.C. Experimental and pKa prediction aspects of tautomerism of drug-like molecules. Drug Discov. Today Technol. 2018, 27, 59–64. [Google Scholar] [CrossRef]

{kind=link}
| Solvent | log CS,organic | Log P |
|---|---|---|
| 1-Octanol (wet) | 0.72 | |
| Methanol | −1.428 | |
| Ethanol | −1.373 | |
| 1-Propanol | −1.581 | |
| 2-Propanol | −1.594 | |
| 1-Butanol | −1.493 | |
| Acetone | −1.203 | |
| Acetonitrile | −1.450 | |
| Dichloromethane | −1.377 | |
| N,N-Dimethylformamide | −0.827 | |
| Methyl acetate | −1.292 | |
| Ethyl acetate | −1.396 | |
| Butyl acetate | −1.412 |
| Solvent | c | e | s | a | b | l | v |
| Equation (1) Coefficients | |||||||
| 1-Octanol (wet) | 0.088 | 0.562 | −1.054 | 0.034 | −3.460 | 0.000 | 3.814 |
| Methanol (dry) | 0.276 | 0.334 | −0.714 | 0.243 | −3.320 | 0.000 | 3.549 |
| Ethanol (dry) | 0.222 | 0.471 | −1.035 | 0.326 | −3.596 | 0.000 | 3.857 |
| 1-Propanol (dry) | 0.139 | 0.405 | −1.029 | 0.247 | −3.767 | 0.000 | 3.986 |
| 1-Butanol (dry) | 0.165 | 0.401 | −1.011 | 0.056 | −3.958 | 0.000 | 4.044 |
| 2-Propanol (dry) | 0.099 | 0.344 | −1.049 | 0.406 | −3.827 | 0.000 | 4.033 |
| Methyl acetate (dry) | 0.351 | 0.223 | −0.150 | −1.035 | −4.527 | 0.000 | 3.972 |
| Ethyl acetate (dry) | 0.328 | 0.314 | −0.348 | −0.847 | −4.899 | 0.000 | 4.142 |
| Butyl acetate (dry) | 0.289 | 0.336 | −0.501 | −0.913 | −4.964 | 0.000 | 4.262 |
| Acetone (dry) | 0.313 | 0.312 | −0.121 | −0.608 | −4.753 | 0.000 | 3.942 |
| Acetonitrile (dry) | 0.413 | 0.077 | 0.326 | −1.566 | −4.391 | 0.000 | 3.364 |
| N,N-Dimethylformamide (dry) | −0.305 | −0.058 | 0.343 | 0.358 | −4.865 | 0.000 | 4.486 |
| Dichloromethane (dry) | 0.319 | 0.102 | −0.187 | −3.058 | −4.090 | 0.000 | 4.324 |
| Gas-to-water | −0.994 | 0.577 | 2.549 | 3.813 | 4.841 | 0.000 | −0.869 |
| Equation (2) Coefficients | |||||||
| 1-Octanol (wet) | −0.198 | 0.002 | 0.709 | 3.519 | 1.429 | 0.858 | 0.000 |
| Methanol (dry) | −0.039 | −0.338 | 1.317 | 3.826 | 1.396 | 0.773 | 0.000 |
| Ethanol (dry) | 0.017 | −0.232 | 0.867 | 3.894 | 1.192 | 0.846 | 0.000 |
| 1-Propanol (dry) | −0.042 | −0.246 | 0.749 | 3.888 | 1.076 | 0.874 | 0.000 |
| 1-Butanol (dry) | −0.004 | −0.285 | 0.768 | 3.705 | 0.879 | 0.890 | 0.000 |
| 2-Propanol (dry) | −0.048 | −0.324 | 0.713 | 4.036 | 1.055 | 0.884 | 0.000 |
| Methyl acetate (dry) | 0.134 | −0.477 | 1.749 | 2.678 | 0.000 | 0.876 | 0.000 |
| Ethyl acetate (dry) | 0.171 | −0.403 | 1.428 | 2.726 | 0.000 | 0.914 | 0.000 |
| Butyl acetate (dry) | 0.154 | −0.439 | 1.223 | 2.586 | 0.000 | 0.953 | 0.000 |
| Acetone (dry) | 0.127 | −0.387 | 1.733 | 3.060 | 0.000 | 0.866 | 0.000 |
| Acetonitrile (dry) | 0.192 | −0.572 | 1.492 | 0.460 | 0.847 | 0.965 | 0.000 |
| N,N-Dimethylformamide (dry) | −0.391 | −0.869 | 2.107 | 3.774 | 0.000 | 1.011 | 0.000 |
| Dichloromethane (dry) | −0.007 | −0.595 | 2.461 | 2.085 | 0.418 | 0.738 | 0.000 |
| Gas-to-water | −1.271 | 0.822 | 2.743 | 3.904 | 4.814 | −0.213 | 0.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, E.; Acree, W.E., Jr. Abraham Model Solute Descriptors for Favipiravir: Case of Tautomeric Equilibrium and Intramolecular Hydrogen-Bond Formation. Thermo 2023, 3, 443-451. https://doi.org/10.3390/thermo3030027
Yao E, Acree WE Jr. Abraham Model Solute Descriptors for Favipiravir: Case of Tautomeric Equilibrium and Intramolecular Hydrogen-Bond Formation. Thermo. 2023; 3(3):443-451. https://doi.org/10.3390/thermo3030027
Chicago/Turabian StyleYao, Emily, and William E. Acree, Jr. 2023. "Abraham Model Solute Descriptors for Favipiravir: Case of Tautomeric Equilibrium and Intramolecular Hydrogen-Bond Formation" Thermo 3, no. 3: 443-451. https://doi.org/10.3390/thermo3030027
APA StyleYao, E., & Acree, W. E., Jr. (2023). Abraham Model Solute Descriptors for Favipiravir: Case of Tautomeric Equilibrium and Intramolecular Hydrogen-Bond Formation. Thermo, 3(3), 443-451. https://doi.org/10.3390/thermo3030027