Visible-Light Photoredox Catalyzed Formation of Triarylethylenes Using a Low-Cost Photosensitizer

 , ,
, ,  and
and
Abstract
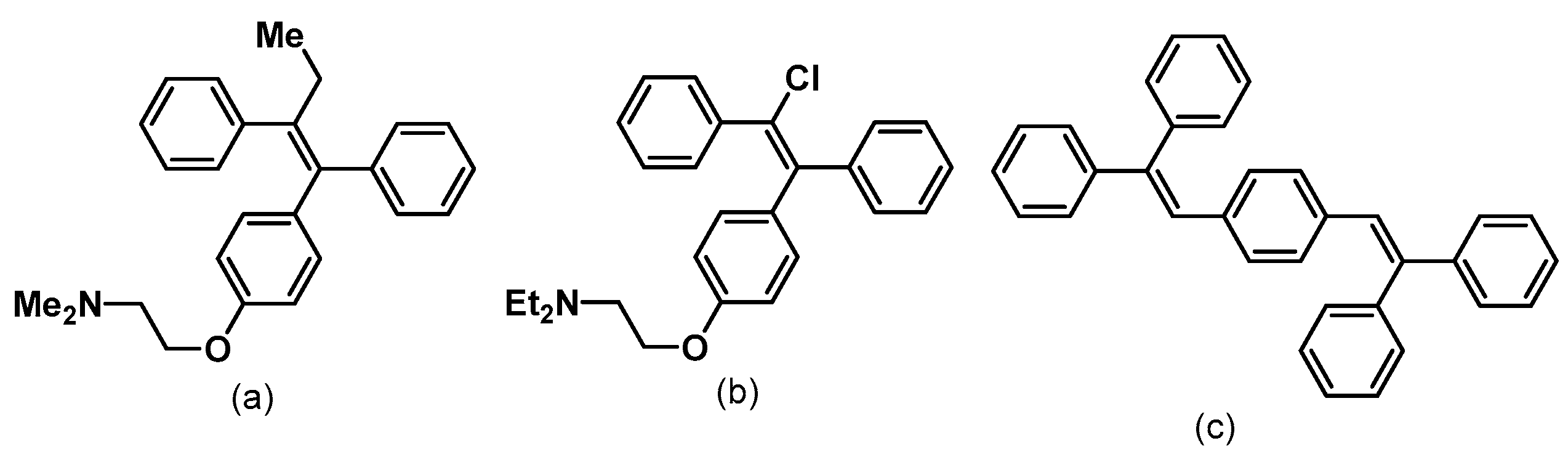
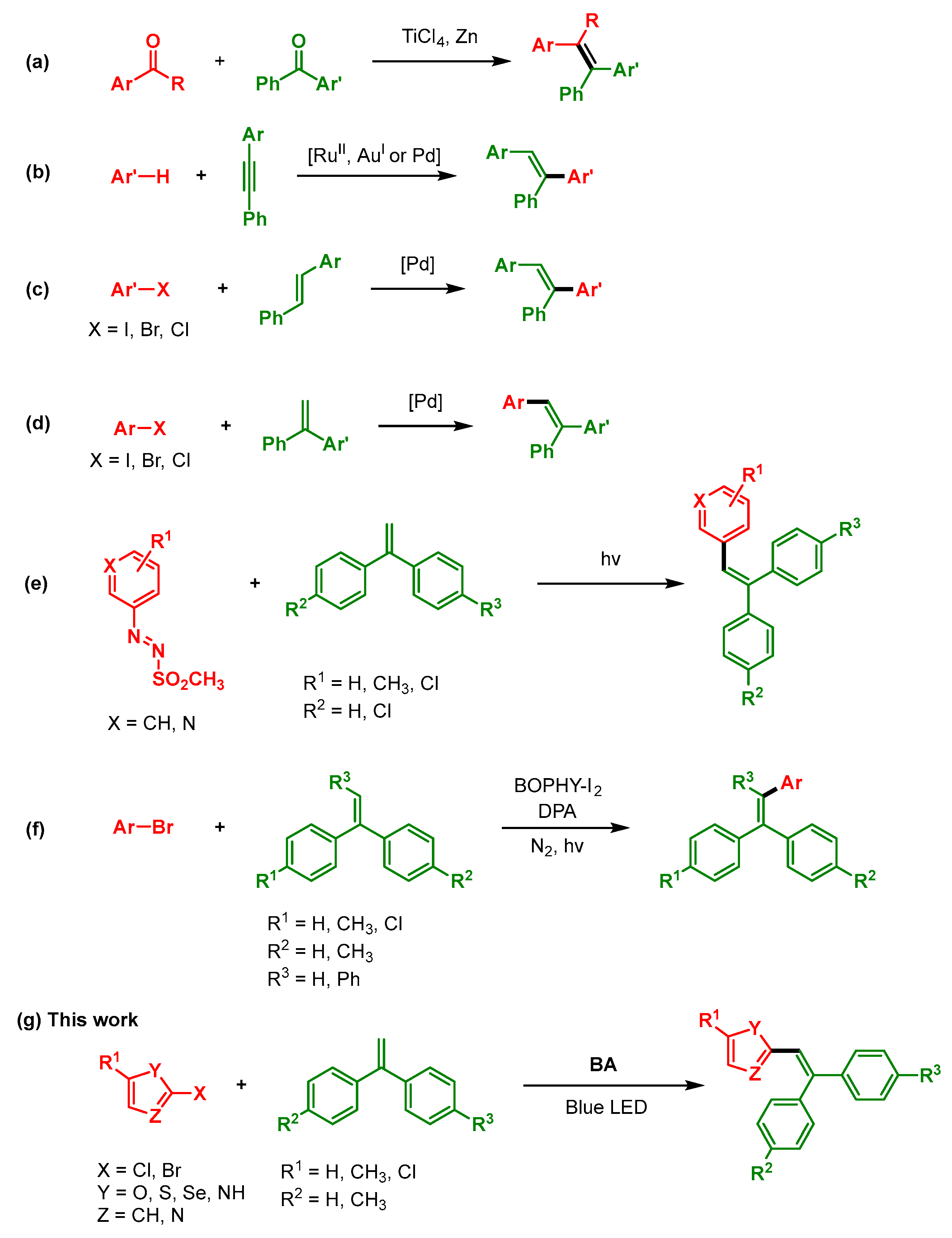
1. Introduction
2. Materials and Methods
2.1. General
2.2. Steady-State Irradiation
2.2.1. Irradiation in Homogeneous Media
2.2.2. Irradiation in Supramolecular Viscoelastic Gel Media
2.3. Characterization of the Supramolecular Viscoelastic Gel Media
2.3.1. Tgel Determination
2.3.2. Oscillatory Rheology
2.3.3. Scanning Electron Microscopy (SEM)
2.3.4. Transmission Electron Microscopy (TEM)
2.3.5. Atomic Force Microscopy (AFM)
2.4. Steady-State and Transient Absorption Spectroscopy
2.5. Reaction Quantum Yield Determination
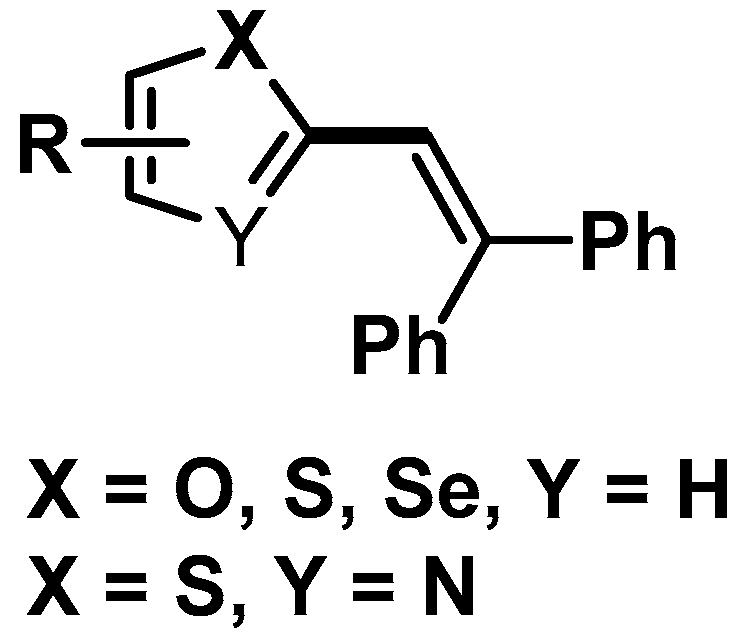
3. Results
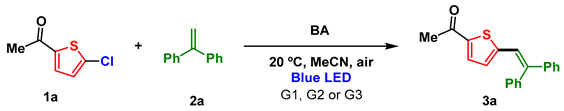
3.1. Optimization of the Reaction Conditions for Photosensitized Irradiation in Organic Solvents
3.2. Optimization of the Reaction Conditions in Gel Media
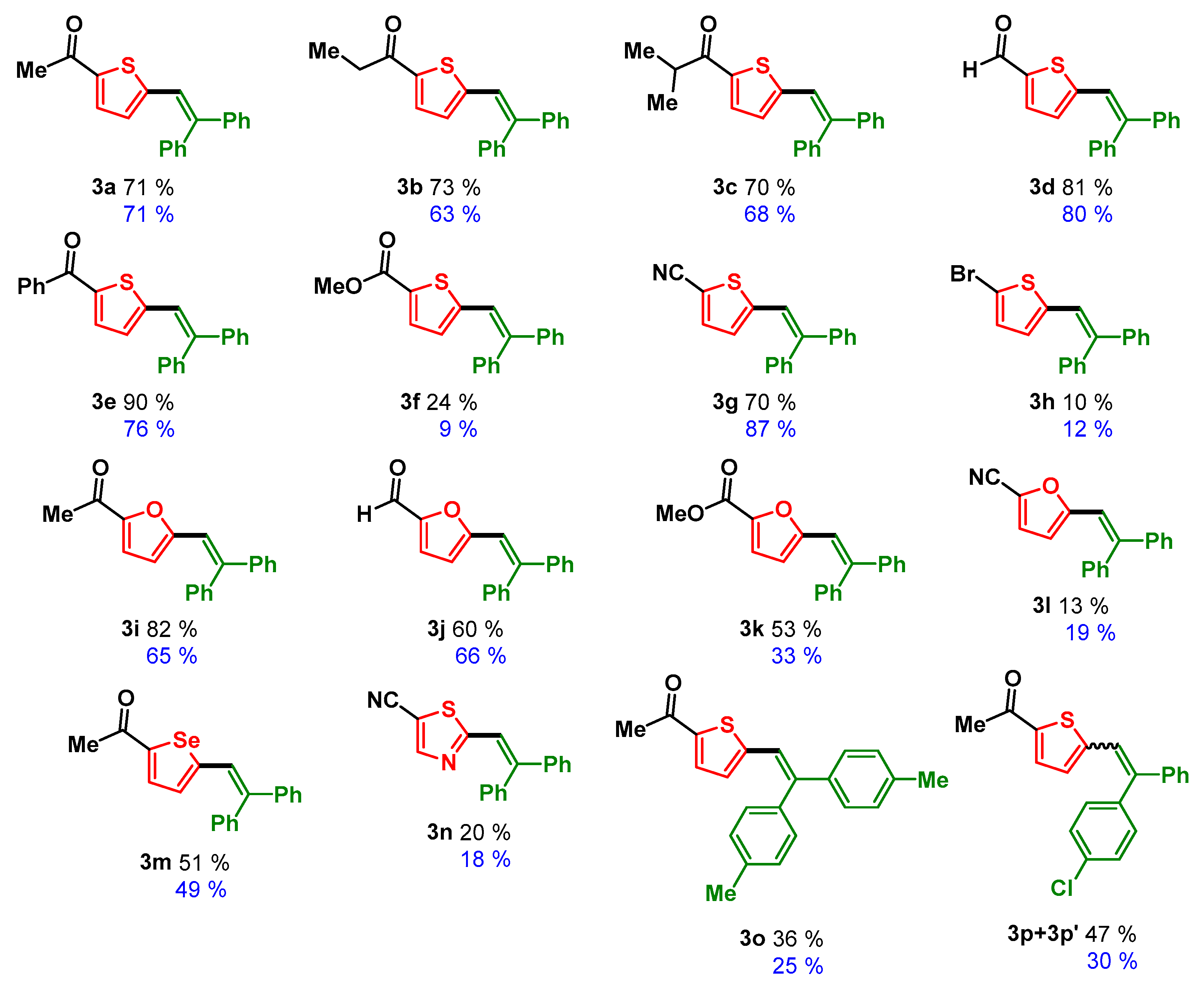
3.3. Scope of the Reaction
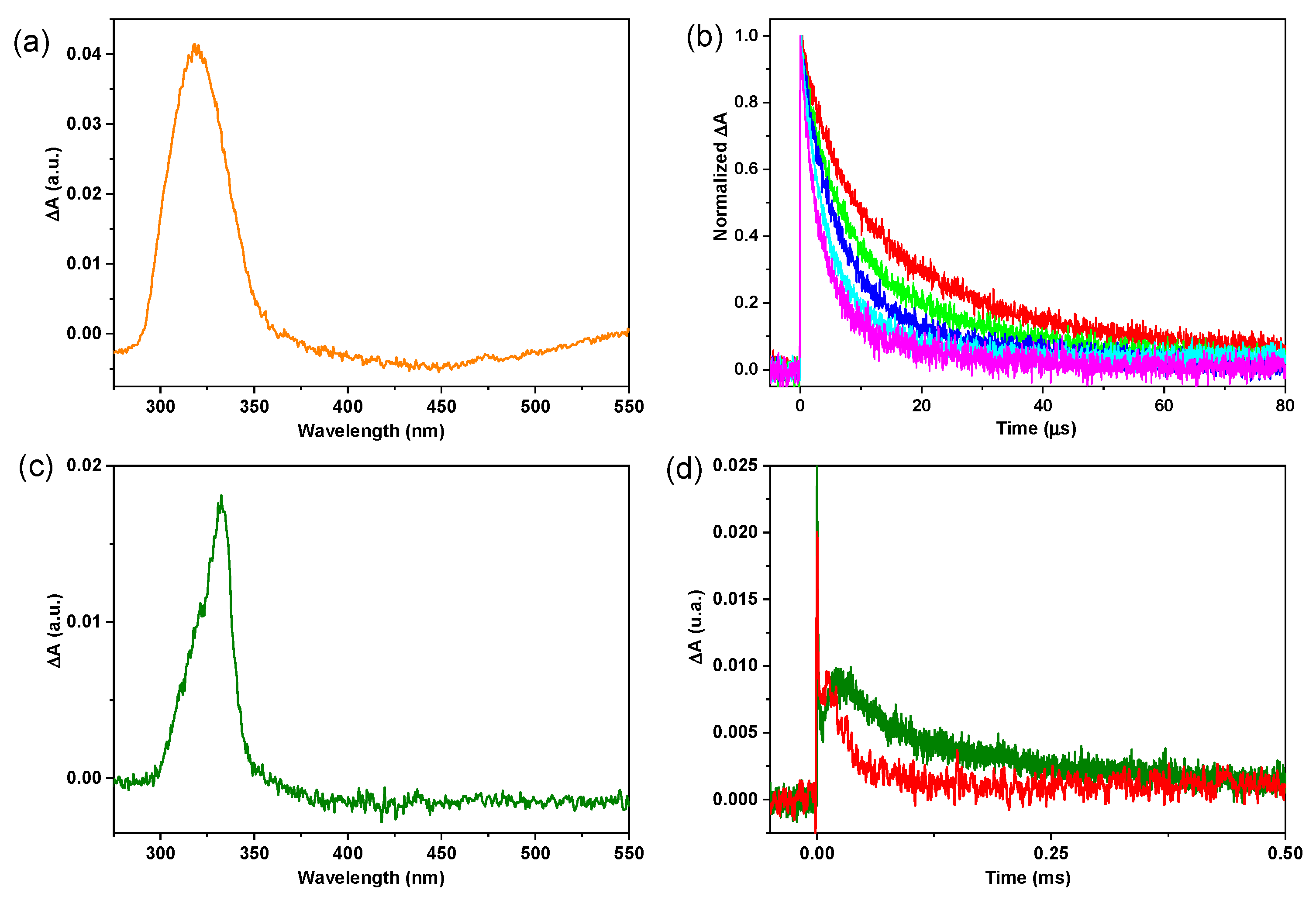
3.4. Laser Flash Photolysis Experiments
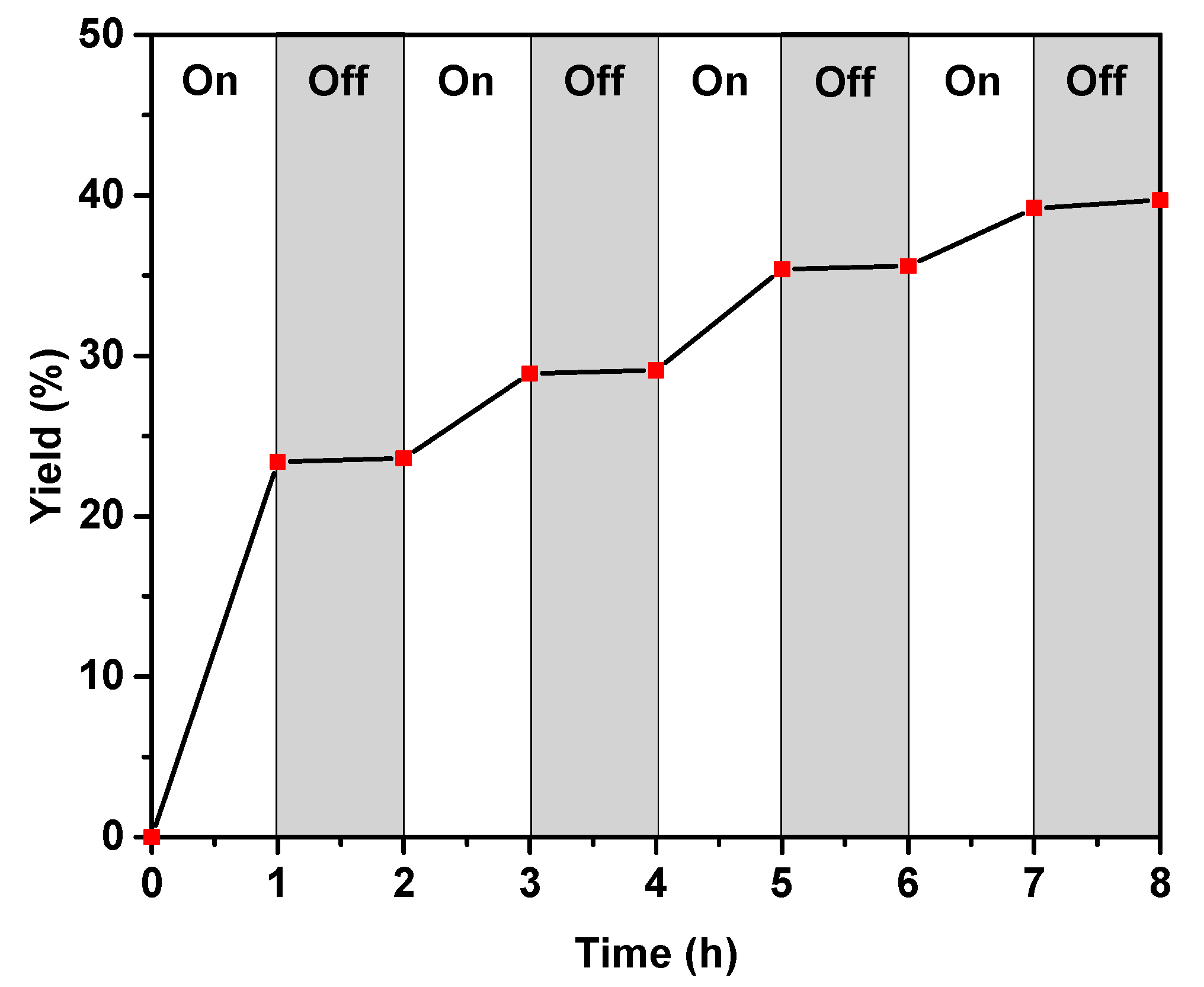
3.5. “Light–Dark” Experiments
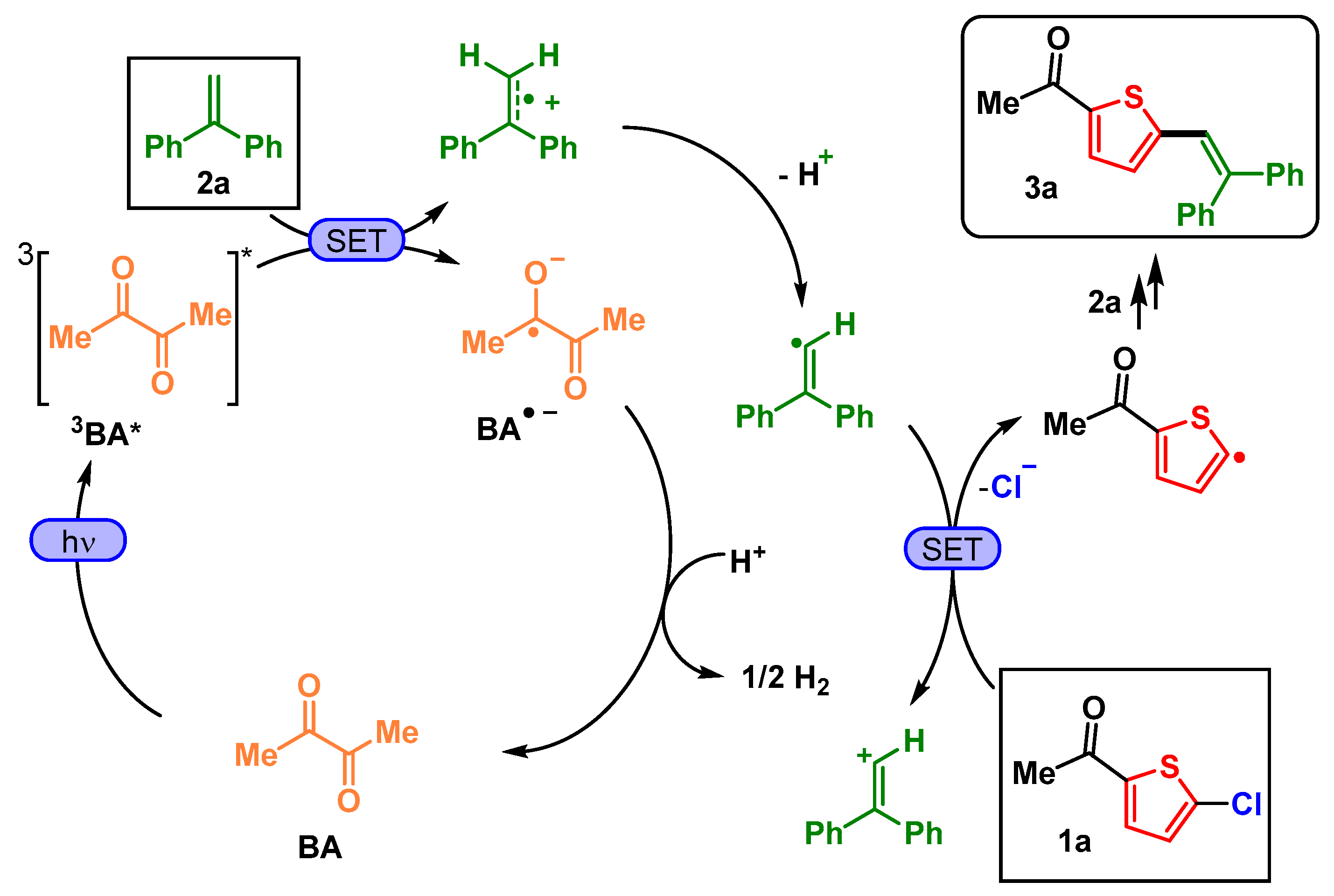
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AcOEt | Ethyl acetate |
| AFM | Atomic Force Microscopy |
| BA | Biacetyl |
| DFS | Dynamic Frequency Sweep |
| DMA | Dimethylacetamide |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DSS | Dynamic Strain Sweep |
| DTS | Dynamic Time Sweep |
| G1 | Tert-butyl-(S)-(1,5-bis(octadecylamino)-1,5-dioxopentan-2-yl)carbamate |
| G2 | N,N′-((1S,2S)-Cyclohexane-1,2-diyl)didodecanamide |
| G3 | (1R)-1-((4R,8aS)-2,6-Diphenyltetrahydro-[1,3]dioxino[5,4-d][1,3]dioxin-4-yl)ethane-1,2-diol) |
| MeCN | Acetonitrile |
| SEM | Scanning electron microscopy |
| SET | Single electron transfer |
| TAE | Triarylethylene |
| Tgel | Gel-to-sol transition temperature |
| TEM | Transmission Electron Microscopy |
| ULL | Universidad de La Laguna |
| UPV | Universitat Politècnica de València |
References
- Wang, L.Y.; Yu, T.; Xie, Z.L.; Chen, X.J.; Yang, Z.; Zhang, Y.; Aldred, M.P.; Chi, Z.G. Design, Synthesis and Photochromism Studies of Thienyl Containing Triarylethylene Derivatives and their Applications in Real-Time Photoresponsive surfaces. J. Mater. Chem. C 2018, 6, 8832–8838. [Google Scholar] [CrossRef]
- Wu, Y.; Hu, F.; Wang, Z. Design and Synthesis of Triarylethylene-Based Organic Semiconductors for Photovoltaic Applications. J. Mater. Chem. C 2020, 8, 4657–4674. [Google Scholar]
- Jiang, X.; Lin, X.; Wang, J. Synthesis and Properties of Triarylamine-Based Liquid Crystals: From “Dumbbells” to Multiarm Star Shapes. Chem. Eur. J. 2019, 25, 9170–9181. [Google Scholar]
- Li, J.; Wang, S.; Chen, K. Recent Advances in the Development of Triarylethylene-Based Anticancer Agent. Eur. J. Med. Chem. 2019, 171, 310–329. [Google Scholar]
- Zhang, X.; Sun, H.; Low, K.-H.; Yu, T.; Au, V.K.-M. Photochromic porous organic crystals constructed by the self-assembly of triarylethylene derivatives. Mater. Chem. Front. 2023, 7, 3332–3339. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, F.; Du, B.; Huang, R.; Zhang, S.; He, Y.; Wang, H.; Cui, J.; Zhang, B.; Yu, T.; et al. Construction of Photoresponsive 3D Structures Based on Triphenylethylene Photochromic Building Blocks. Research 2022, 2022, 9834140. [Google Scholar] [CrossRef] [PubMed]
- Flynn, A.B.; Ogilvie, W.W. Stereocontrolled Synthesis of Tetrasubstituted Olefins. Chem. Rev. 2007, 107, 4698–4745. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Y.; Li, L. Strategies for the Synthesis of Triarylethylenes: A Comprehensive Review. Chem. Rev. 2021, 121, 1120–1157. [Google Scholar]
- Wu, Y.; Hu, F.; Wang, Z. Recent Advances in the Synthesis and Functionalization of Triarylethylenes. Org. Lett. 2021, 23, 3710–3718. [Google Scholar]
- Chatterjee, A.K.; Choi, T.L.; Sanders, D.P. A Concise Synthesis of Triarylethylenes. Angew. Chem. Int. Ed. 2018, 57, 6489–6493. [Google Scholar]
- McMurry, J.E. Titanium-Induced Dicarbonyl-Coupling Reactions. Acc. Chem. Res. 1983, 16, 405–411. [Google Scholar] [CrossRef]
- Ladipo, F.T. Low-Valent Titanium-Mediated Reductive Coupling of Carbonyl Compounds. Curr. Org. Chem. 2006, 10, 965. [Google Scholar] [CrossRef]
- Duan, X.-F.; Zeng, J.; Lü, J.-W.; Zhang, Z.-B. Insights into the General and Efficient Cross McMurry Reactions between Ketones. J. Org. Chem. 2006, 71, 9873–9876. [Google Scholar] [CrossRef] [PubMed]
- Pigeon, P.; Görmen, M.; Kowalski, K.; Müller-Bunz, H.; McGlinchey, M.J.; Top, S.; Jaouen, G. Atypical McMurry Cross-Coupling Reactions Leading to a New Series of Potent Antiproliferative Compounds Bearing the Key [Ferrocenyl-Ene-Phenol] Motif. Molecules 2014, 19, 10350–10369. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Liu, J.; Skaar, T.C.; Flockhart, D.A.; Cushman, M. Design and Synthesis of Norendoxifen Analogues with Dual Aromatase Inhibitory and Estrogen Receptor Modulatory Activities. J. Med. Chem. 2015, 58, 2623–2648. [Google Scholar] [CrossRef]
- Kaur, G.; Mahajan, M.P.; Pandey, M.K.; Singh, P.; Ramisetti, S.R.; Sharma, A.K. Design, Synthesis, and Anti-Breast Cancer Evaluation of New Triarylethylene Analogs Bearing Short Alkyl- and Polar Amino-/Amido-Ethyl Chains. Bioorg. Med. Chem. Lett. 2016, 26, 1963–1969. [Google Scholar] [CrossRef]
- Mostafa, T.; Albeir, M.; Wober, J.; Abadi, A.; Salama, I.; Ahmed, N.S. Design, Synthesis, and in-Silico Study of Novel Triarylethylene Analogs with Dual Anti-Estrogenic and Serotonergic Activity. Drug Dev. Res. 2024, 85, e22127. [Google Scholar] [CrossRef]
- Nevado, C.; Echavarren, A.M. Transition Metal-Catalyzed Hydroarylation of Alkynes. Synthesis 2005, 2005, 167–182. [Google Scholar] [CrossRef]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef]
- Manikandan, R.; Jeganmohan, M. Recent Advances in the Ruthenium-Catalyzed Hydroarylation of Alkynes with Aromatics: Synthesis of Trisubstituted Alkenes. Org. Biomol. Chem. 2015, 13, 10420–10436. [Google Scholar] [CrossRef]
- Lee, W.; Shin, C.; Park, S.E.; Joo, J.M. Regio- and Stereoselective Synthesis of Thiazole-Containing Triarylethylenes by Hydroarylation of Alkynes. J. Org. Chem. 2019, 84, 12913–12924. [Google Scholar] [CrossRef]
- Cartney, D.M.; Guiry, P.J. The Asymmetric Heck and Related Reactions. Chem. Soc. Rev. 2011, 40, 5122–5150. [Google Scholar] [CrossRef]
- Lee, D.-H.; Taher, A.; Hossain, S.; Jin, M.-J. An Efficient and General Method for the Heck and Buchwald–Hartwig Coupling Reactions of Aryl Chlorides. Org. Lett. 2011, 13, 5540–5543. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Huang, Y.; Wei, Z.; Ding, Y.; Su, C.; Xu, Q. Heck Reactions Catalyzed by Ultrasmall and Uniform Pd Nanoparticles Supported on Polyaniline. J. Org. Chem. 2015, 80, 8677–8683. [Google Scholar] [CrossRef]
- Liu, P.; Pan, Y.; Hu, K.; Huang, X.; Liang, Y.; Wang, H. Ligand-Free Indium(III)-Catalyzed Heck Reaction. Tetrahedron 2013, 69, 7925–7930. [Google Scholar] [CrossRef]
- Khanizeman, R.N.; Barde, E.; Bates, R.W.; Guérinot, A.; Cossy, J. Modular Access to Triarylethylene Units from Arylvinyl MIDA Boronates Using a Regioselective Heck Coupling. Org. Lett. 2017, 19, 5046–5049. [Google Scholar] [CrossRef] [PubMed]
- Onuigbo, L.; Raviola, C.; Di Fonzo, A.; Protti, S.; Fagnoni, M. Sunlight-Driven Synthesis of Triarylethylenes (TAEs) via Metal-Free Mizoroki–Heck-Type Coupling. Eur. J. Org. Chem. 2018, 2018, 5297–5303. [Google Scholar] [CrossRef]
- Chawla, R.; Singh, A.K.; Dutta, P.K. Arylazo sulfones: Multifaceted photochemical reagents and beyond. Org. Biomol. Chem. 2024, 22, 869–893. [Google Scholar] [CrossRef]
- Castellanos-Soriano, J.; Garnes-Portolés, F.; Jiménez, M.C.; Leyva-Pérez, A.; Pérez-Ruiz, R. In-Flow Heterogeneous Triplet–Triplet Annihilation Upconversion. ACS Phys. Chem. Au 2024, 4, 242–246. [Google Scholar] [CrossRef]
- Herrera-Luna, J.C.; Díaz, D.D.; Jiménez, M.C.; Pérez-Ruiz, R. Highly Efficient Production of Heteroarene Phosphonates by Dichromatic Photoredox Catalysis. ACS Appl. Mater. Interfaces 2021, 13, 48784–48794. [Google Scholar] [CrossRef]
- Li, Y.; Wang, T.; Liu, M. Gelating-Induced Supramolecular Chirality of Achiral Porphyrins: Chiroptical Switch between Achiral Molecules and Chiral Assemblies. Soft Matter 2007, 3, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- van Esch, J.H.; Schoonbeek, F.; de Loos, M.; Kooijman, H.; Spek, A.L.; Kellogg, R.M.; Feringa, B.L. Cyclic Bis-Urea Compounds as Gelators for Organic Solvents. Chem. Eur. J. 1999, 5, 937–950. [Google Scholar] [CrossRef]
- Cismesia, M.A.; Yoon, T.P. Characterizing chain processes in visible light photoredox catalysis. Chem. Sci. 2015, 6, 5426–5434. [Google Scholar] [CrossRef] [PubMed]
- Hamai, S.; Hirayama, F. Actinometric determination of absolute fluorescence quantum yields. J. Phys. Chem. 1983, 87, 83–89. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; De Sarkar, S.; Das, A. Supramolecular Engineering and Self-Assembly Strategies in Photoredox Catalysis. ACS Catal. 2021, 11, 710–733. [Google Scholar] [CrossRef]
- Häring, M.; Pérez-Ruiz, R.; von Wangelin, A.J.; Díaz, D.D. Intragel Photoreduction of Aryl Halides by Green-to-Blue Upconversion under Aerobic Conditions. Chem. Commun. 2015, 51, 16848–16851. [Google Scholar] [CrossRef]
- Häring, M.; Abramov, A.; Okumura, K.; Ghosh, I.; König, B.; Yanai, N.; Kimizuka, N.; Díaz, D.D. Air-sensitive photoredox catalysis performed under aerobic conditions in gel networks. J. Org. Chem. 2018, 83, 7928–7938. [Google Scholar] [CrossRef]
- Montalti, M.; Credi, A.; Prodi, A.; Gandolfi, M.T. Handbook of Photochemistry, 3rd ed.; CRC Press: Boca Raton, FA, USA, 2005. [Google Scholar]
- Singh-Rachford, T.N.; Castellano, F.N. Low Power Visible-to-UV Upconversion. J. Phys. Chem. A 2009, 113, 5912–5917. [Google Scholar] [CrossRef] [PubMed]
- Görner, H. Triplet States of Phenylethylenes in Solution. Energies, Lifetimes, and AbsorptionSpectra of 1,1-Dlphenyl-, Triphenyl-, and Tetraphenylethylene Triplets. J. Phys. Chem. 1982, 86, 2028–2035. [Google Scholar] [CrossRef]
- Weller, A.Z. Photoinduced electron transfer in solution: Exciplex and radical ion pair formation free enthalpies and their solvent dependence. Z. Phys. Chem. 1982, 133, 93–98. [Google Scholar] [CrossRef]
- Hayashi, K.; Irie, M.; Lindenau, D.; Schnabel, W. Primary Ionic Species of 1,1-Diphenylethylene Produced by High Energy Radiation. Radiat. Phys. Chem. 1978, 11, 139–144. [Google Scholar] [CrossRef]
- Clennan, E.L.; Speth, D.R.; Bartlett, P.D. Structural effects in photoepoxidation sensitized by α-diketones. J. Org. Chem. 1983, 48, 1246–1250. [Google Scholar] [CrossRef]
- Garnes-Portolés, F.; Greco, R.; Oliver-Meseguer, J.; Castellanos-Soriano, J.; Consuelo Jiménez, M.; López-Haro, M.; Hernández-Garrido, J.C.; Boronat, M.; Pérez-Ruiz, R.; Leyva-Pérez, A. Regioirregular and Catalytic Mizoroki–Heck Reactions. Nat. Catal. 2021, 4, 293–303. [Google Scholar] [CrossRef]
- Herrera-Luna, J.C.; Pérez-Aguilar, M.C.; Gerken, L.; García Mancheño, O.; Consuelo Jiménez, M.; Pérez-Ruiz, R. Effective Formation of New C(sp2)−S Bonds via Photoactivation of Alkylamine-Based Electron Donor-Acceptor Complexes. Chem. Eur. J. 2023, 29, e202203353. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Entry | Solvent | Time (h) | 2a (equiv) | BA (equiv) | Yield (%) b |
|---|---|---|---|---|---|
| 1 | MeCN | 24 | 10 | 3 | 74 |
| 2 | MeCN | 24 | 5 | 3 | 71 |
| 3 | MeCN | 24 | 3 | 3 | 71 |
| 4 | MeCN | 24 | 2 | 3 | 64 |
| 5 | MeCN | 24 | 1 | 3 | 54 |
| 6 | MeCN | 24 | 3 | 2 | 65 |
| 7 | MeCN | 24 | 3 | 1 | 58 |
| 8 | MeCN | 24 | 3 | 0.5 | 50 |
| 9 | MeCN | 24 | 3 | 0.3 | 46 |
| 10 | MeCN | 48 | 3 | 0.3 | 42 |
| 11 | MeCN | 72 | 3 | 0.3 | 53 |
| 12 | MeCN | 96 | 3 | 0.3 | 54 |
| 13 c | MeCN | 24 | 3 | 3 | 53 |
| 14 d | MeCN | 24 | 3 | 3 | 64 |
| 15 e | MeCN/H2O | 24 | 3 | 3 | 55 |
| 16 f | MeCN/H2O | 24 | 3 | 3 | 57 |
| 17 g | MeCN/H2O | 24 | 3 | 3 | 46 |
| 18 | DMF | 24 | 3 | 3 | 33 |
| 19 | DMA | 24 | 3 | 3 | 21 |
| 20 | DMSO | 24 | 3 | 3 | 39 |
| 21 | Acetone | 24 | 3 | 3 | 56 |
| 22 | AcOEt | 24 | 3 | 3 | 45 |
| 23 | DCM | 24 | 3 | 3 | 44 |
| 24 h | MeCN | 24 | 3 | 3 | 0 |
| 25 | MeCN | 24 | 3 | 0 | 0 |
| 26 i | MeCN | 24 | 3 | 3 | 2 |
| 27 j | MeCN | 24 | 3 | 3 | <1 |
 | |||||
| Entry | Solvent | Time (h) | 2 (equiv) | BA (equiv) | Yield (%) b |
|---|---|---|---|---|---|
| 1 | MeCN | 4 | 50 | 8 | 48 |
| 2 | DMF | 4 | 50 | 8 | 35 |
| 3 | DMA | 4 | 50 | 8 | 26 |
| 4 | AcOEt | 4 | 50 | 8 | 38 |
| 5 | Acetone | 4 | 50 | 8 | 43 |
| 6 | DMSO | 4 | 50 | 8 | 14 |
| 7 | MeCN | 8 | 50 | 8 | 65 |
| 8 | MeCN | 24 | 50 | 8 | 87 |
| 9 | MeCN | 24 | 25 | 8 | 83 |
| 10 | MeCN | 24 | 20 | 8 | 83 |
| 11 | MeCN | 24 | 15 | 8 | 82 |
| 12 | MeCN | 24 | 15 | 2 | 70 |
| 13 | MeCN | 24 | 15 | 3 | 74 |
| 14 | MeCN | 24 | 15 | 4 | 74 |
| 15 | MeCN | 24 | 10 | 3 | 71 |
| 16 | MeCN | 24 | 5 | 3 | 24 |
| 17 | MeCN | 24 | 1 | 3 | n.d. |
| 18 | MeCN | 24 | 10 | 3 | 66 |
| 19 | MeCN | 24 | 10 | 3 | 61 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Gutiérrez, D.; Domínguez, P.D.; Pérez-Ruiz, R.; Díaz, D.D.; Jiménez, M.C. Visible-Light Photoredox Catalyzed Formation of Triarylethylenes Using a Low-Cost Photosensitizer. Photochem 2025, 5, 13. https://doi.org/10.3390/photochem5020013
Álvarez-Gutiérrez D, Domínguez PD, Pérez-Ruiz R, Díaz DD, Jiménez MC. Visible-Light Photoredox Catalyzed Formation of Triarylethylenes Using a Low-Cost Photosensitizer. Photochem. 2025; 5(2):13. https://doi.org/10.3390/photochem5020013
Chicago/Turabian StyleÁlvarez-Gutiérrez, Daniel, Paola Domínguez Domínguez, Raúl Pérez-Ruiz, David Díaz Díaz, and M. Consuelo Jiménez. 2025. "Visible-Light Photoredox Catalyzed Formation of Triarylethylenes Using a Low-Cost Photosensitizer" Photochem 5, no. 2: 13. https://doi.org/10.3390/photochem5020013
APA StyleÁlvarez-Gutiérrez, D., Domínguez, P. D., Pérez-Ruiz, R., Díaz, D. D., & Jiménez, M. C. (2025). Visible-Light Photoredox Catalyzed Formation of Triarylethylenes Using a Low-Cost Photosensitizer. Photochem, 5(2), 13. https://doi.org/10.3390/photochem5020013