1. Introduction
Tissue engineering is a multidisciplinary field that aims to restore and improve tissue function by preparing porous three-dimensional scaffolds and seeding them with cells and growth factors [
1,
2,
3]. Bone scaffolds are porous structures inserted into bone defects to help regenerate the surrounding tissues and mechanically support the tissues for stable positioning [
4]. They are often made of hydroxyapatite (HAp), which is the main inorganic component of bone [
5]. Recently, a sodium silicate solution (water glass, WG) has been used in combination with HAp to prepare promising scaffolds by sinterization [
6,
7,
8] also starting from processes at room temperature. This new consolidation process preserves the low crystallinity, non-stoichiometry, and size of the precursor nano-HAp, which are essential properties to mimic bone tissue [
9]. Recent applications also seem promising, as reported by Yang [
10], who utilized microwaves to take advantage of the foaming nature of WG, resulting in a rapid fabrication route, yielding pure WG foams with high, tunable porosity (up to 94%) and sizes of up to tens of centimeters within a few minutes.
Domestic microwave ovens are typically less costly than laboratory microwave ovens and can be found in most homes and kitchens. This makes them a potentially attractive option for scaffold fabrication, especially for small-scale production.
The use of a domestic microwave oven to fabricate scaffolds presents several advantages over the use of a laboratory setup. First, no vacuum is needed, which simplifies the process and reduces costs. Second, the cost of a commercial microwave oven is much lower than the cost of a laboratory setup. Third, the process is faster and takes only 45 min compared to the hours or days required for standard/classical laboratory methods using resistive or gas-fired ovens.
Although domestic microwave ovens offer several advantages for scaffold fabrication, there are also some challenges associated with their use, especially when compared to laboratory ones. One challenge is the limited temperature control of domestic microwave ovens. Without precise temperature control, the reaction mixture may overheat. Another challenge is heterogeneous heating. Microwaves tend to heat materials unevenly, creating hot spots and cold spots in the reaction mixture. This can affect the reaction rate and result in uneven mixing of the reactants. Moreover, microwave-assisted reactions can generate significant heat, potentially raising the pressure in the reaction vessel when vapor or gases are developed. It is thus crucial to use reaction vessels that can withstand pressure and to periodically vent the vessel to release excess pressure. Furthermore, the lack of precise temperature control and the potential for uneven heating can make it difficult to reproduce microwave-assisted reactions from one batch to the next. This can be frustrating for researchers, who need consistent results for their experiments. Finally, there is less published literature on microwave techniques using commercial microwave ovens, making it more difficult to find optimized protocols and troubleshooting guidance. Despite these challenges, commercial microwave ovens have the potential to be a valuable tool for scaffold fabrication. Their lower cost and ease of use can make them more accessible to researchers, especially those working on a limited budget or in resource-constrained settings.
In this study, we aimed to address the challenges described above by developing a novel method for fabricating Na
2SiO
3-HAp-SiO
2 scaffolds using a household microwave oven. This present work builds upon a previous seminal study in which a laboratory microwave oven was employed to prepare water glass foams with controlled porosity, as reported in the literature [
10].
2. Materials and Methods
Ethanol 96%, CaO (anhydrous, powder), H3PO4 (85%), NaHCO3, sodium trisilicate solution (water glass, WG), and fumed silica (SiO2) were purchased from Merck KGaA (Darmstadt, Germany). All reagents and chemicals were of reagent grade. Deionized water was used for preparing both the used solutions and the bath to rinse samples.
Hydroxyapatite (HAp) was synthesized via the wet chemical technique using a precipitation reaction, in which CaO and H
3PO
4 were used as precursors, as described elsewhere in the literature by Dorozhkin in 2010 [
11].
The composite samples were prepared by mixing HAp, WG, and fumed silica in a 50 mL Falcon test tube according to
Table 1; for sample WHSiNa (311), NaHCO
3 was also added to promote the geopolymerization, as suggested in the literature [
12]. To obtain homogenization of the samples, a vortex mixer was employed. The prepared mixture was then placed into a Falcon test tube, where it was sealed. A hole was created in the cap, and a cylinder made of filter paper (Whatman
® qualitative filter paper, Grade 4) was inserted into it to allow space for the water vapors released during the reaction, acting like a valve. The Falcon tube was placed inside a beaker, supported by crumbled filter paper balls placed inside to keep it in a vertical position, as illustrated in
Figure 1. The assembly was subsequently introduced into the microwave oven (SAMSUNG M/O 20LT GE71A of 20 L, Samsung, Suwon, South Korea) for further heat treatments.
Samples were prepared in triplicate.
Thanks to the continuous monitoring and the parameters adopted for the microwave-assisted thermal treatments during the MicroWave Manufacturing Process (MWMP), the temperature never exceeded 180 °C, as verified with an infrared camera (model SEEK Thermal ShotPRO, SEEK thermal INC., Santa Barbara, CA, USA). During the MWMP process, the temperature lies in the range of 110–180 °C.
Figure 1 shows the experimental setup for the heat treatment used for the preparation of the samples.
The following conventions were used to name the samples: W (water glass), H (HAp), Si (silica fume), and Na (sodium bicarbonate). The minimum ratio between the quantities used to prepare the mixture of each ingredient is then indicated in parentheses (i.e., (111)). For example, the name of the sample WHSi (1:1:1) indicates a sample prepared with a ratio of 1 mL of water glass to 1 g of HAp and 1 g of SiO2.
Sample WHC was prepared in order to reproduce the mixture used by Lakrat et al., 2021 [
13]. Samples WHSiNa (311) and WH (31) were prepared to study the foaming effect of the water glass (since in these samples, we have 3 times the content in comparison with WHC (11)). SiO
2 was added to try to decrease the solubility in water (and simulated fluid).
Note that the samples WSi (11), WSi (12), WHSi (111), and W (1) were prepared and investigated to test the manufacturing process and to study the densities and mechanical properties of the materials used. They were excluded from the biological characterization because their mechanical properties were not adequate for this application.
2.1. Physicochemical Characterization
Water stability tests were performed to test the dissolution of the samples. For this purpose, samples were immersed in 1 L of distilled water (27 °C laboratory temperature) for ten days. Every three days, the samples were weighted after drying to reach a constant weight.
The apparent density of the samples was determined with a Mettler Toledo (Mettler AE 240) precision balance (ASTM International, 2013 [
14]), and the data are reported in
Table 1.
Fourier transform infrared spectroscopy in attenuated total reflectance mode (ATR-FTIR) was performed using a Perkin Elmer Spectrum 2 in the range of 4000 to 400 cm−1 with a resolution of 0.5 cm−1 and 16 scans.
The morphological analysis of the HAp particles and developed composite samples were evaluated by a scanning electron microscope (SEM) with a Hitachi TM3000 benchtop SEM (operating at 15 kV) (Hitachi High Tech, Naka, Japan) implemented with a probe for energy dispersive X-ray analysis (EDX).
The mechanical properties of the materials were investigated by means of uniaxial compression tests performed with a Shimadzu (Kyoto, Japan) ASG-X universal testing machine operating at R.T. with a load cell of 10 kN. Cylindrical specimens (20 mm in height and 25 mm in diameter) blade-cut from longer cylinders were compressed up to 30% of their initial height with a compression speed of 10 mm/min. The mechanical properties were measured on three samples for each sample, and the results are reported as their average.
2.2. In Vitro Biological Tests
To perform the conditioning of the samples for the in vitro biological test, the scaffolds were placed in a 24-well plate for conditioning for 4 weeks with 1 mL of phosphate-buffered saline (PBS). The PBS was changed two times each week. Specifically, scaffolds were incubated in PBS, and the media were replaced to promote the release of ions from the materials and the stabilization of the pH in the media close to the physiological one (i.e., 7.4) to be suitable for cell colonization and proliferation.
Human fetal osteoblasts (hFOB 1.19 ATCC CRL-11372TM) were used for cytotoxicity tests. Cells were grown using DMEM/F12 no phenol red with L-glutamine (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) and 0.3 mg/mL G418 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C in an atmosphere with 5% CO2. Cells were detached by using trypsin-EDTA (0.25% w/v trypsin and 0.03% w/v EDTA) and centrifuged, and the cell number and cell viability were assessed with a trypan blue dye exclusion test. The composite scaffolds (width = 8 mm and height = 5 mm) were washed in deionized water in a sonicating bath for 30 min before sterilization. All scaffold types (WHC (11), WHSiNa (311), and WH (31)) were then sterilized with ethanol 70% for 30 min, and then the samples were left to dry under the sterile laminar flow hood for 1 h. Samples were pre-soaked in culture medium for 24 h at 37 °C and then seeded by carefully dropping 20 µL of the cell suspension (5.0 × 104 cells) onto the scaffold’s upper surface and allowing cell attachment for 30 min in the incubator before the addition of 980 µL of cell culture medium. The medium was changed every 3 days. All cell handling procedures were performed in a sterile laminar flow hood. All cell culture incubation steps were performed at 37 °C with 5% CO2.
Qualitative cell viability and cytotoxicity analyses were performed for each group after 1 day by using a Live/Dead™ Assay Kit (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Briefly, cells were rinsed in PBS before incubation with acetoxymethyl calcein (AM-calcein) 2 µM and ethidium homodimer-1 (EthD-1) 4 µM for 15 min at 37 °C in dark conditions. Samples were rinsed in PBS, and images were acquired by an inverted fluorescent microscope Eclipse TS 100 (Nikon, Tokyo, Japan).
The quantitative cell viability and proliferation analyses of cells seeded on scaffolds were performed by the PrestoBlue™ cell viability reagent procedure (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Briefly, 10% (v/v) PrestoBlue Reagent was added for each well containing the scaffold and incubated for 2 h at 37 °C with 5% CO2. For each timeframe, three samples were used. Additionally, a scaffold without cells was used according to the same procedure and then considered as a blank in the data elaboration. After the incubation, 100 µL of supernatant was removed from each well and transferred to a 96-well plate, each of them in triplicate. The resulting 96-well plate was then analyzed using the Fluoroskan™ Microplate Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), setting the excitation wavelength equal to 544 nm, whereas the emission wavelength was set to 590 nm. The same set of analyses were performed at different timeframes equal to 1 and 3 days after cell seeding.
One sample per group was used for actin staining 3 days after cell seeding. In order to visualize actin filaments, seeded materials were washed with PBS, fixed with paraformaldehyde 4% (w/v) for 15 min, and then incubated in Triton X-100 0.1% (v/v, PBS as solvent) for 10 min to promote cell permeabilization. Indeed, the incubation with the latter surfactant, Triton X-100, can promote the formation of pores within the cell and nuclear membrane without modifying the overall cell morphology, and this entails the possibility for different molecules to potentially be used to label specific subcellular structures (e.g., cytoskeleton and nucleus) to permeate within the cells. Cells were incubated with ActinRed™ 555 ReadyProbes™ Reagent (rhodamine phalloidin) (Thermo Fisher Scientific, USA) for 30 min. Cells were then incubated with DAPI 300 nM in PBS for 10 min. Finally, scaffolds were transferred on a coverslip with a drop of PBS and then visualized by an inverted fluorescent microscope (Eclipse TS 100, Nikon, Tokyo, Japan).
To perform the conditioning of the samples for the in vitro biological test, the scaffolds were placed in a 24-well plate for conditioning.
2.3. Statistical Analyses
Statistical analysis and graph elaboration were performed using GraphPad Prism 9.0.0 (GraphPad Software, San Diego, CA, USA). One-way ANOVA (analysis of variance) was performed, followed by Dunnett’s multiple comparisons test to evaluate differences. Differences were considered significant for p-values less than 0.05 (* p-value < 0.05; ** p-value < 0.01; *** p-value < 0.001; **** p-value < 0.0001).
3. Results and Discussion
3.1. Solubility and Density Evaluation
Considering that scaffold solubility, a crucial aspect of bone regeneration, is a complex property, we conducted experiments to ensure that the scaffolds did not completely dissolve in water. The results on samples show they maintained their structural integrity without releasing ions that could potentially alter the pH (i.e., OH− from NaOH).
For this purpose, we weighed the samples and immersed them in deionized water in a 10 L container, ensuring that the water was changed every 24 h. After 10 days, we retrieved the samples and reweighed them, confirming that there were no significant weight variations. To ensure the complete process of polymerization, we monitored the pH, which initially registered a value of 12 before stabilizing at neutral levels.
The density value of the foamed WG (W (1)) sample corresponds to 0.2 g/cm3; that value increases up to 0.5 g/cm3 by adding SiO2 and HAp (WSi and WHSi). The maximum value, as expected/predicted for its 1:1 formulation in WG and HAp, is obtained for the WHC (11) sample, with a value of 0.6 g/cm3. For reference, WG has a density of 1.39 g/cm3 (technical data sheet), silica fume has a density of 3.61 g/cm3 (technical data sheet), and HAp has a theoretical density of 3.16 g/cm3.
The density of the other samples decreases in the following order WHC (11), WHSiNa (311), WHSi (111), and WSi (12). The density value series is summarized in
Table 1.
3.2. Morphological Analysis
The total average percentage porosity was determined to be around 60% for all samples, calculated considering the volumes and weights of three specimens for each sample (for comparison, cancellous bone porosity is in the interval between 30% and 80%). The percentage porosity, varying between 35% and 80%, presents a great ability to induce osteointegration and enhance implant adhesion to the host tissue [
15].
The morphology of the samples was investigated by SEM analysis.
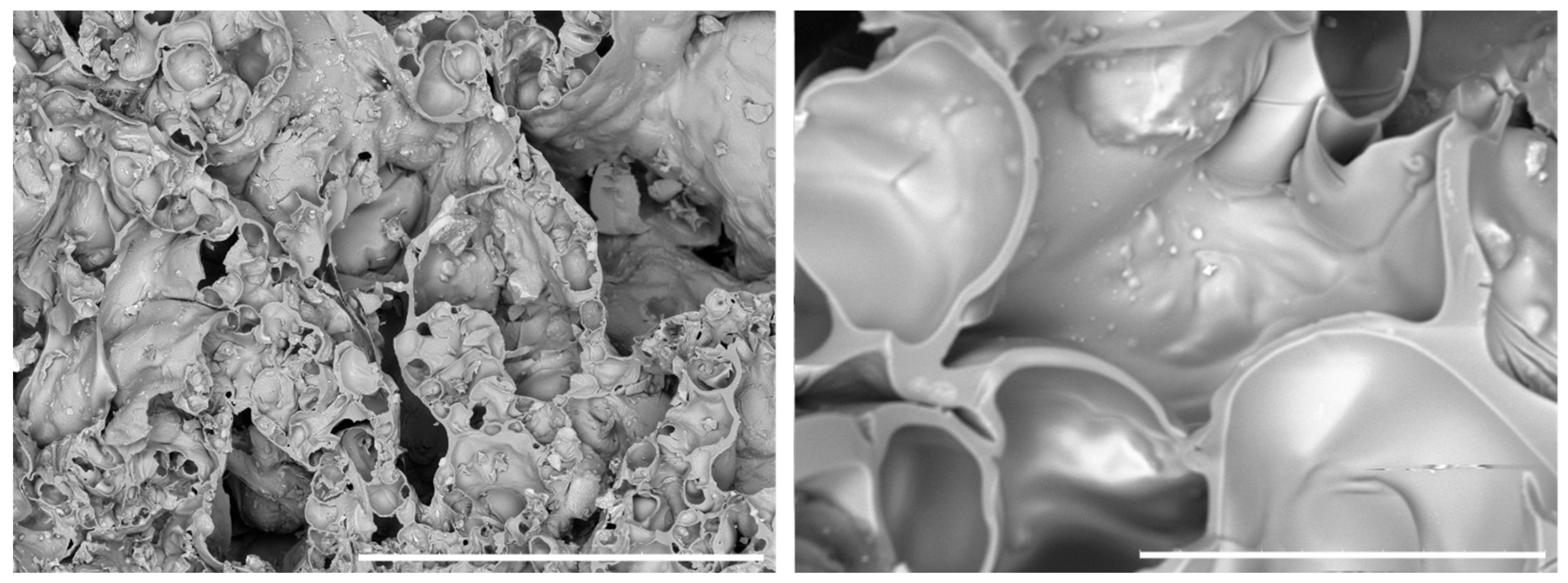
The target was to obtain a porous structure with interconnected pores, useful for promoting cell proliferation without the presence of HAp crystals bigger than a few micrometers (the smaller the better in order to increase the viability of the cells). The particles shown in
Figure 2 have submicrometric features; larger particles (top left left) are the result of the aggregation phenomenon of smaller HAp grains (bottom right). Energy-dispersive X-ray spectroscopy (EDX) (not reported here) analyses showed the presence of calcium (Ca), phosphorus (P), oxygen (O), and carbon (C) in a percentage amount compatible with the theoretical HAp composition. Due to its nonstoichiometric chemical composition, and low crystallinity, it is an optimal candidate to be used in scaffold design and preparation.
SEM images of the prepared 3D composite scaffolds (
Figure 3,
Figure 4 and
Figure 5) point out a densified and cohesive material, where HAp particles, as well as SiO
2 when present, are included within the WG.
WHC (11) prepared with the addition of an equal quantity of WG and HAp shows the formation of a smooth continuous surface with pores that range from a few microns to 20–30 microns. Also, the pores do not look interconnected.
We had anticipated that the morphology of the sample would be different. When we mixed water glass with hydroxyapatite, we observed that instead of forming a liquid mixture, the compound yielded a dense paste. This suggests that the foaming action of water glass would be limited in this case.
For samples WHSiNa (311) and WH (31), where the ratio between the water glass and HAp was 3:1, we can notice the presence of fractures of bubble-like shapes. The radius of the bubbles is in the range of 50 microns to 500 microns for both samples. In these cases, the bubbles are interconnected.
The addition of NaHCO3 in sample WHSiNa (311), used here to increase the foaming and geopolymerization of the water glass, did not cause a significant difference in the morphology of the samples.
3.3. Compressive Tests
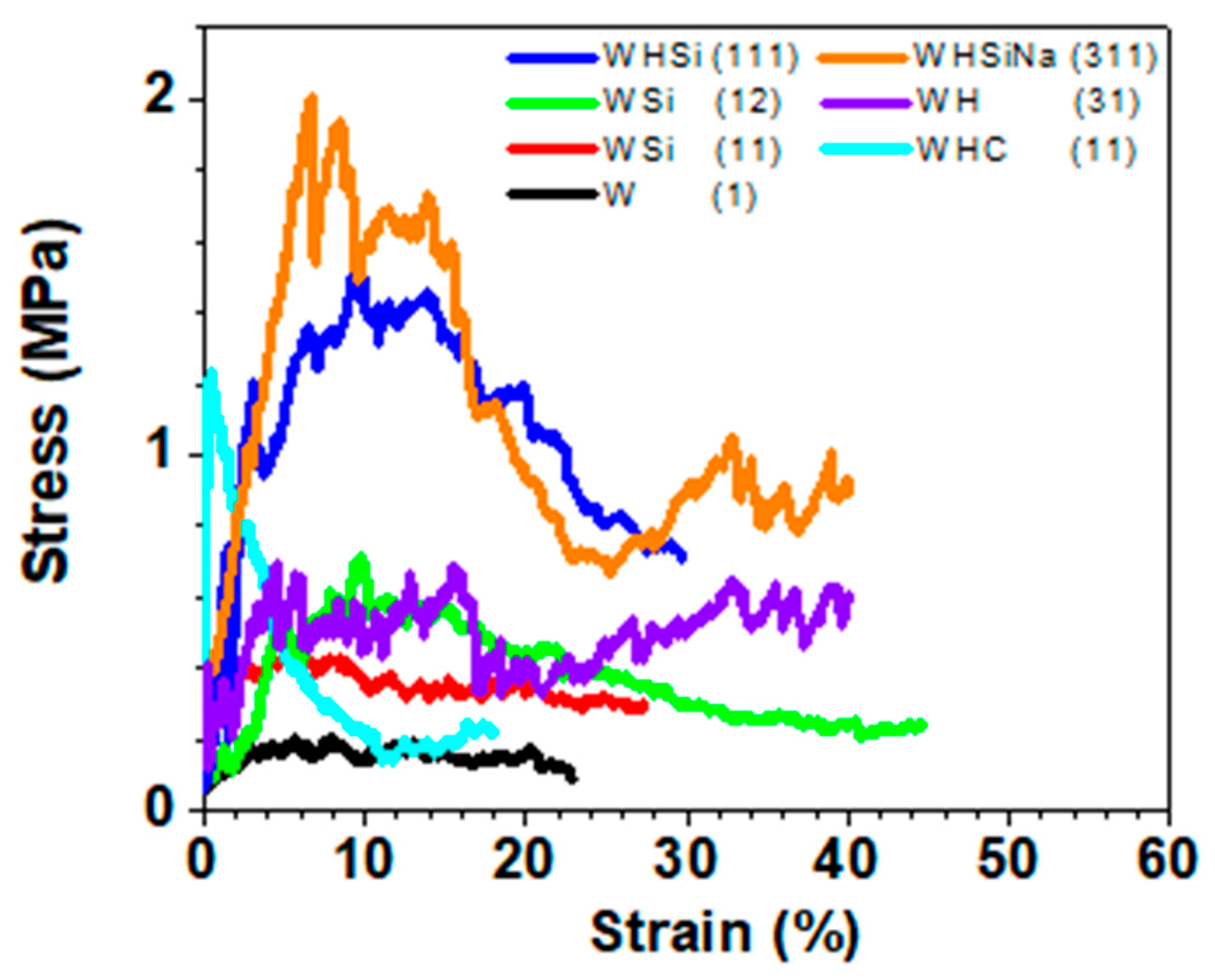
In
Figure 6 and
Table 2, starting with the sample containing only water glass (W (1)), we can see that the addition of SiO
2 increases the mechanical properties, resulting in an increase in the values of maximum stress (σ
max), maximum strain (ε
max), and elastic modulus (E
y). For the WSi (11) sample, the value of 0.2 ± 0.1 MPa increased to a value of 0.4 ± 0.1 MPa for σ
max, while it increased from 22.8 ± 1.0% to 27.9 ± 0.4% for the maximum strain. Finally, the elastic modulus, although presenting a considerable deviation, changed from a range of 0.0–0.5 GPa to a range of 0.0–0.1 GPa. An additional increase in the SiO
2 content leads to a further increase in σ
max and the maximum strain and a decrease in the elastic modulus.
Similar results were found when hydroxyapatite was introduced. In this case as well, a significant increase in the mechanical properties was highlighted. In the case of the WHC (11) sample, we reach 1.2 ± 0.1 MPa, 18.1 ± 0.5%, and 8–10 GPa for maximum stress, maximum strain, and elastic modulus (Ey). In the case of the WH (31) sample, we reach 0.7 ± 0.2 MPa, 40.1 ± 0.6%, and 4–10 GPa.
The elastic modulus is also very high compared to the sample with more water glass. This is also understandable in light of the previous observations regarding morphology, with a lower presence of pores due to a smaller contribution from the foaming effect of the water glass.
The simultaneous contribution of HAp and SiO2 consequently has an increase in the maximum stress, elastic strain, and elastic modulus (values of 2.0 ± 0.1 MPa, 40.2 ± 0.5%, and 6–8.5 GPa for the WHSiNa (311) sample and 1.5 ± 0.1 MPa, 29.5 ± 0.7%, and 1–5 GPa for WHSi (111)). The deviations of the elastic modulus are very high, but they still give us a trend in agreement with both the other mechanical properties and the morphological observations.
In addition, it is worth noting that the shape of the curves for the WHSiNa (311) and WH (31) samples are similar to the load and deformation curves for specimens of cancellous bones, where the yield and ultimate loads are present in the early portion of the curves, followed by a reduction and plateauing of the load due to the collapsing void spaces.
All the mechanical properties are on par with our assigned target of analogous materials, as reported by [
10].
3.4. ATR-FTIR Results
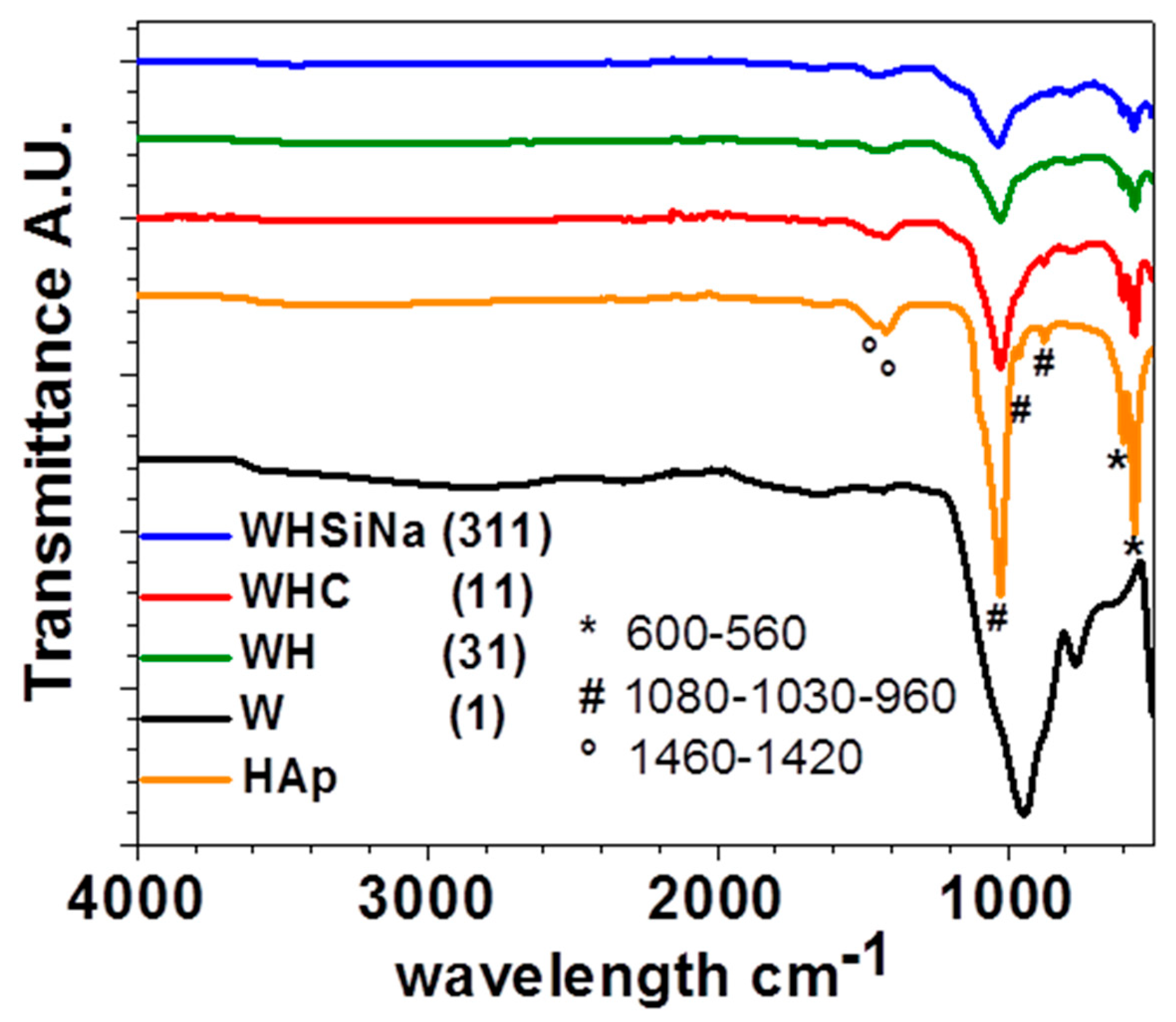
The ATR-FTIR spectra of the composite scaffolds WHSiNa (311), WHC (11), and WH (31) reported in
Figure 7 show the typical vibrational bands of HAp particles before their consolidation by MWMP. The widening of the peak near 1000 cm
−1 can be attributed to the peaks of WG relating to the Si–O band around 850 cm
−1. Peaks in the range of 950 cm
−1 to 1100 cm
−1, assigned to Si–O–Si, overlap with phosphate groups in the HAp structure. An increase in the intensity of the carbonate bands, located between 1400 and 1500 cm
−1, is also evident. Always observing
Figure 7, the phosphate group (PO
43−) shows the characteristic band near 600 cm
−1 (in our case 593–561 cm
−1) (ν4). Furthermore, their characteristic band for HAp at 960 cm
−1 (ν1) and 1030 and 1081 cm
−1 (ν3) are present. Small vibrational bands at ~860 cm
−1, 1420, and 1460 cm
−1 (ν3), which correspond to carbonate ions, are also present. According to literature data [
15], bands suggest that the as-prepared phase is a B-type carbonated apatite. The ATR-FTIR spectrum of microwaved dried water glass (
Figure 7) presents a large band around 950 cm
−1 and a smaller one at 880 cm
−1. They can be attributed to the asymmetric stretching of the Si–O–Si band, which can be assigned to the Q2 and Q1 silica units, respectively [
16]. No water seems to be present (stretching between 3600 cm
−1 and 2700 cm
−1 with a small vibrational band at 1650 cm
−1 is absent from our spectra). No carbonates are present because in that case, an asymmetric stretching band at 1435 cm
−1 would have been present.
3.5. Effects of Scaffolds on Osteoblasts Viability
Considering the potential use of hydroxyapatite-base scaffolds for bone regenerative medicine, and that the interaction of materials and devices with cells determines the successful outcome of the implant [
17], we next sought to investigate their interaction with hFOB cells. The latter is a validated and widely used cell culture model to study the potential of materials intended for regenerative approaches. Osteoblasts are the main component of bone tissue and are the main cells involved in the interaction with the implanted material [
18,
19]. To this aim, cells were seeded on scaffolds to determine the impact of the scaffolds on osteoblast viability, adhesion, and morphology. We thus investigated the cell viability by adopting a qualitative and quantitative assay.
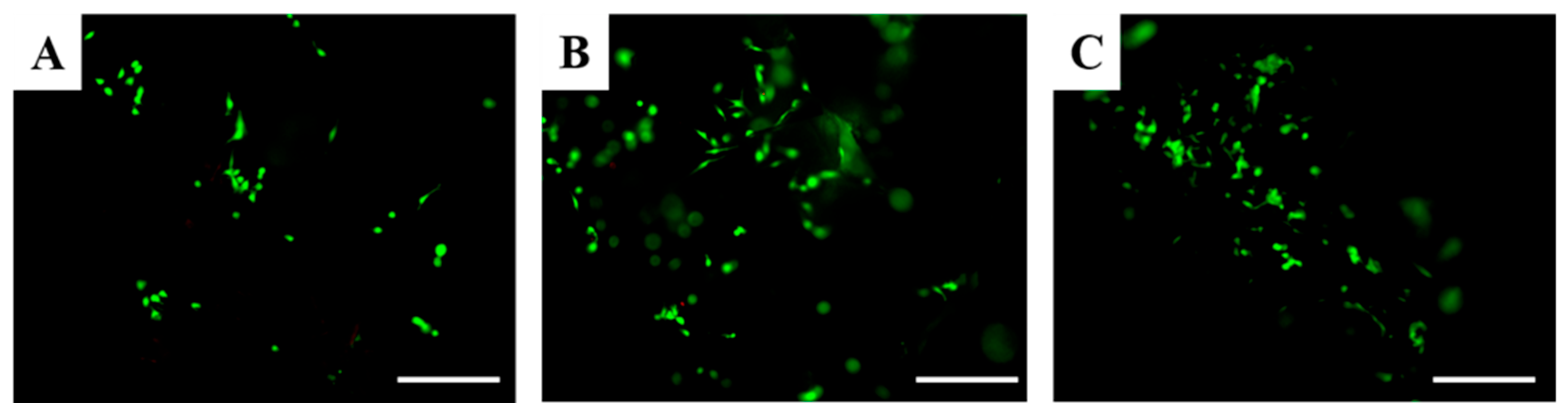
The Live/Dead assay allows the discrimination of live cells from dead cells by simultaneously staining their esterase activity and their loss of plasma membrane integrity, respectively. According to these analyses, viable cells are stained green, whereas dead cells are stained red. Considering the Live/Dead assay, it is possible to assess that only live osteoblasts were detected after 1 day in all scaffolds (
Figure 8). These results suggest that scaffolds based on hydroxyapatite and water glass are not cytotoxic.
The viability and proliferation of human osteoblasts were then quantitatively investigated by the PrestoBlue assay, according to which the fluorescence intensity is proportional to the cell metabolic activity and correlates with cell proliferation [
20]. After 1 day, comparable cell viability in all scaffolds was detected (
Figure 9), similar to the qualitative trend detected by the Live/Dead assay (
Figure 8). After 3 days, a significant increase in cell viability was detected (for all scaffold types, day 1 vs. day 3
p-value < 0.0001), suggesting that scaffolds significantly support osteoblast proliferation. On the other hand, lower cell viability was detected for the WH (31) scaffolds, suggesting that a partial release of their components partially limited cell proliferation. This is not surprising given that this sample has the highest amount of WG, which is known to contain sodium oxide. Sodium oxide can release sodium hydroxide in water or body fluids, making it toxic to cells.
3.6. Effects of Scaffolds on Cell Morphology
The appropriate cell interaction with the implanted materials is crucial to determine the successful cell colonization and integration with the native tissues, and therefore, it affects the outcome of the regeneration approach [
17]. Specifically, cell adhesion to the biomaterial affects the consequent cell proliferation and involves complex processes, especially the reorganization of cytoskeleton proteins like actin. Considering the potential use of scaffolds for bone regenerative medicine, we investigated the influence of the scaffolds on the organization of F-actin filaments of osteoblasts (
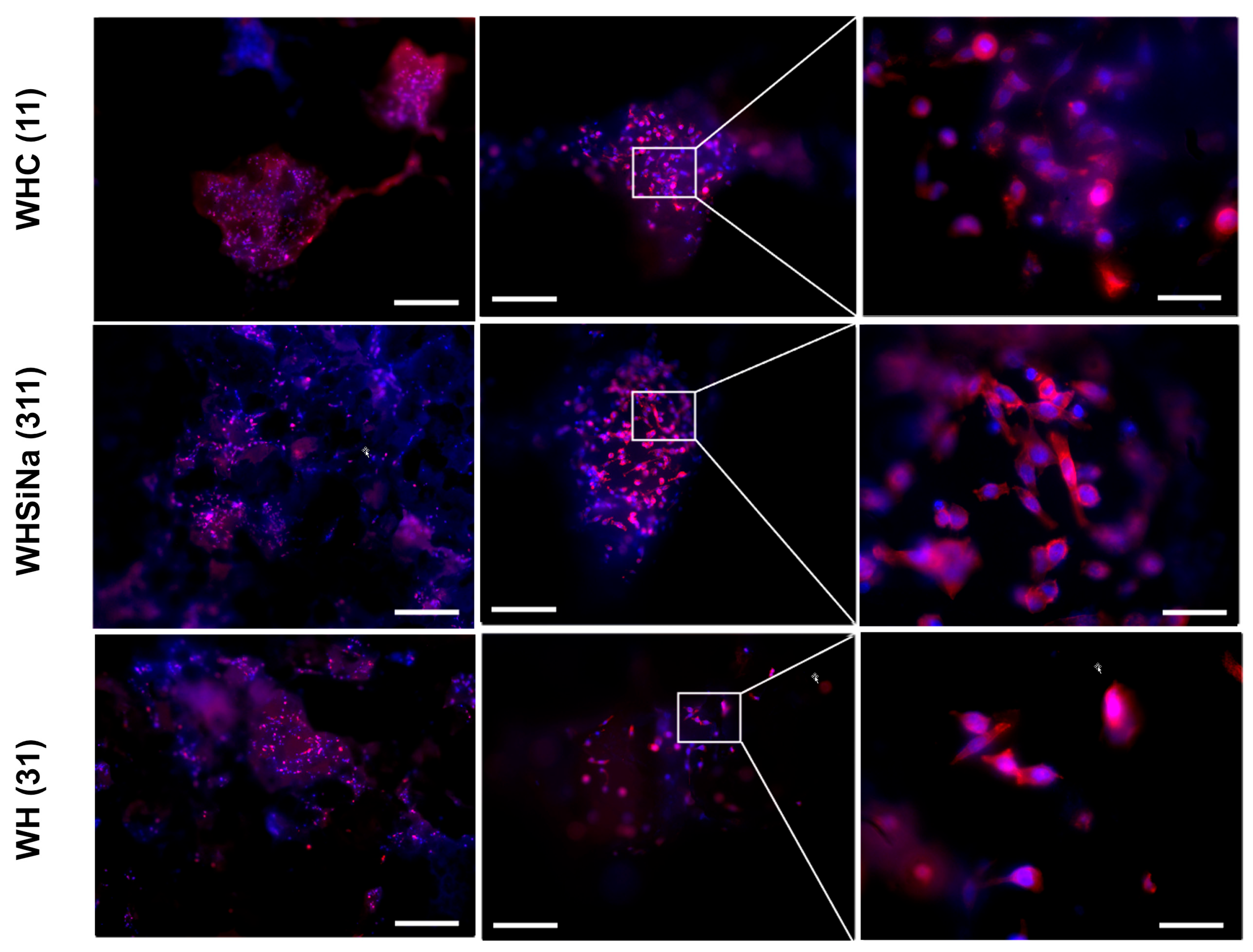
Figure 10).
Three days after cell seeding, the typical osteoblast phenotype well-adherent to the biomaterial was detected, suggesting a good cell adhesion to the different types of scaffolds. In addition, the typical round-shaped nuclear morphology confirmed the absence of any evident cytotoxic effect (e.g., condensed chromatin and apoptotic bodies) (
Figure 10). Furthermore, the number of cells present on the scaffolds correlates with cell viability and proliferation data detected by the PrestoBlue assay after the same timeframe, and it confirms a lower number of cells seeded on sample C, probably due to the release of the components from these scaffolds that partially limit the cell adhesion and spreading to these scaffolds.
Similar findings were previously reported by Lakrat and collaborators for MG63 cells incubated with nano-hydroxyapatite sodium silicate glass composite [
13]. Specifically, they incubated these osteosarcoma cells plated on plastic with different concentrations of the composite, and no cytotoxicity was detected. Additionally, cells displayed the appropriate shape. Nevertheless, they did not investigate the ability of cells to adhere to the scaffolds, and they used a cell model—osteosarcoma cells—which is not appropriate to investigate the potential for bone regeneration.
Different findings were previously reported by Velusamy and co-workers for a lung cancer cell line exposed to HAp nanoparticles doped with different ions (iron and zinc) and produced with a microwave-assisted reaction [
21]. In detail, a cell death coupled to a lower spreading ability proportional to the concentrations of nanoparticle cells was reported. The different results can be correlated to the presence of zinc ions, which are considered cytotoxic [
22,
23].
Considering these reports, the scaffolds reported in the present paper were found to be stable, without releasing any toxic compound, and they can be considered a safe material intended for bone regenerative medicine applications.
4. Conclusions
We successfully fabricated Na2SiO3-SiO2-HAp scaffolds using a commercial and domestic microwave oven, adopting a heat treatment of 45 min, without using vacuum or modified atmosphere. The scaffolds, prepared by MWMP heat treatment, have a morphology comparable to scaffolds obtained via a laboratory microwave oven or via a process involving a laboratory heater, as reported in the references. The scaffolds have a porosity comparable to cancellous bones. The mechanical properties are also on par with the references, and in vitro tests have shown that the scaffolds are biocompatible. In particular, the in vitro growth and adhesion tests showed substantial biocompatibility between the scaffolds and osteoblast cells without significant differences depending on the formulation adopted. Still, considering the three best formulations suggested by the compression tests, it can be seen that after three days, the proliferation decreased from WHC (11) to WH (31) passing through the WHSiNa (311) sample, for which the SEM images show the presence of optimal porosity useful for vascularization and thus the sustenance and proliferation of cells. As stated above, the WHSiNa (311) sample represents an excellent compromise between the other two compositions with regard to mechanical resistance, biocompatibility, and bioregeneration properties.
Last but not least, we avoided pitfalls due to:
- -
Reproducibility: at least three batches of samples were produced, and all had the same characteristics;
- -
Heating control: to avoid overheating of the samples, three cycles of 10 min at a medium power of 450 W, with a waiting time of 5 min between cycles;
- -
Overpressure: to avoid overpressure inside the Falcon test tube, a hole was made in its cap in order to easily release the vapor formed during the MWMP heat treatment. The material losses were avoided by using a cylindrical filter paper, which permits the venting of the water vapor.
The scaffolds we obtained exhibited comparable properties to those fabricated using a laboratory microwave oven, including the ability to induce the formation of bone-like tissue in vitro.
The setup still needs to be improved in order to obtain samples with a more uniform morphology. According to the results obtained, we believe that it is worthwhile to continue research in this area and to consider integrating these methods into clinical practice in the future, as 3D printing technology has been integrated into laboratories when costs have become acceptable.



 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}