
Genetic Diversity and Phylogeography of Plasmodium vivax Transmission-Blocking Vaccine Candidate Genes pvs47 and pvs48/45 in Honduras

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
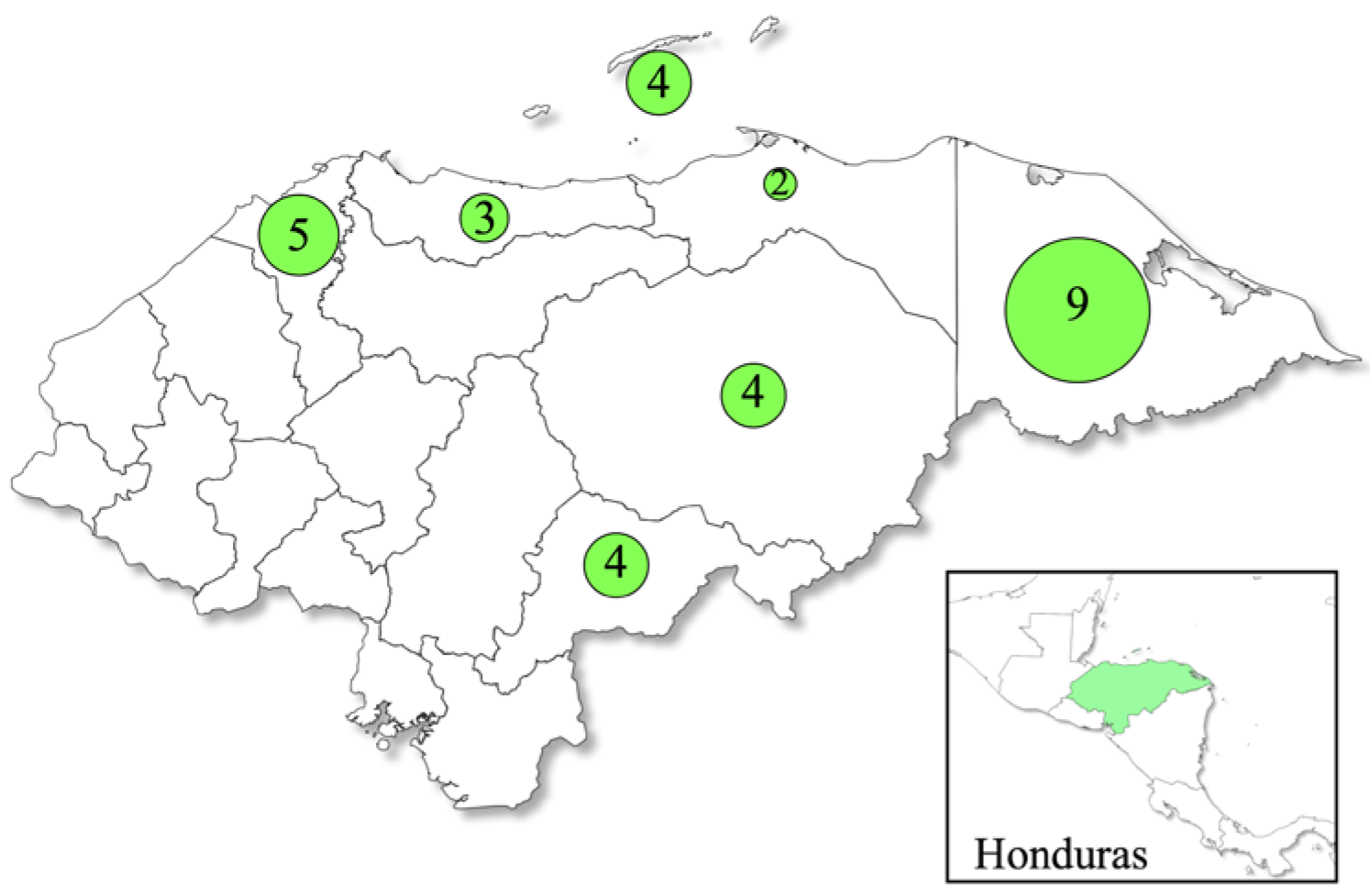
2.1. Sample Collection and Ethical Approval
2.2. DNA Extraction and Molecular Detection of Plasmodium vivax
2.3. PCR Amplification and Sequencing of pvs47 and pvs48/45 Genes
2.3.1. Primary and Secondary PCR Amplifications
2.3.2. Molecular Characterization and Population Genetic Analyses
2.4. Polymorphism Identification, Phylogenetic Tree Construction, and Haplotype Network Analysis
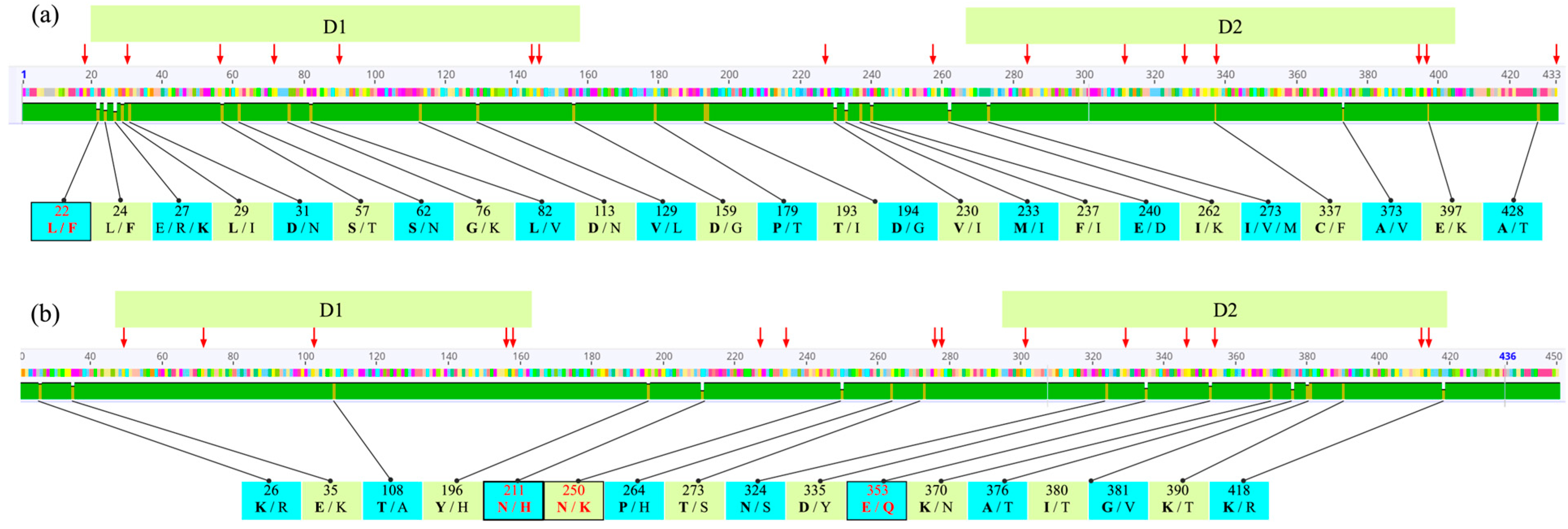
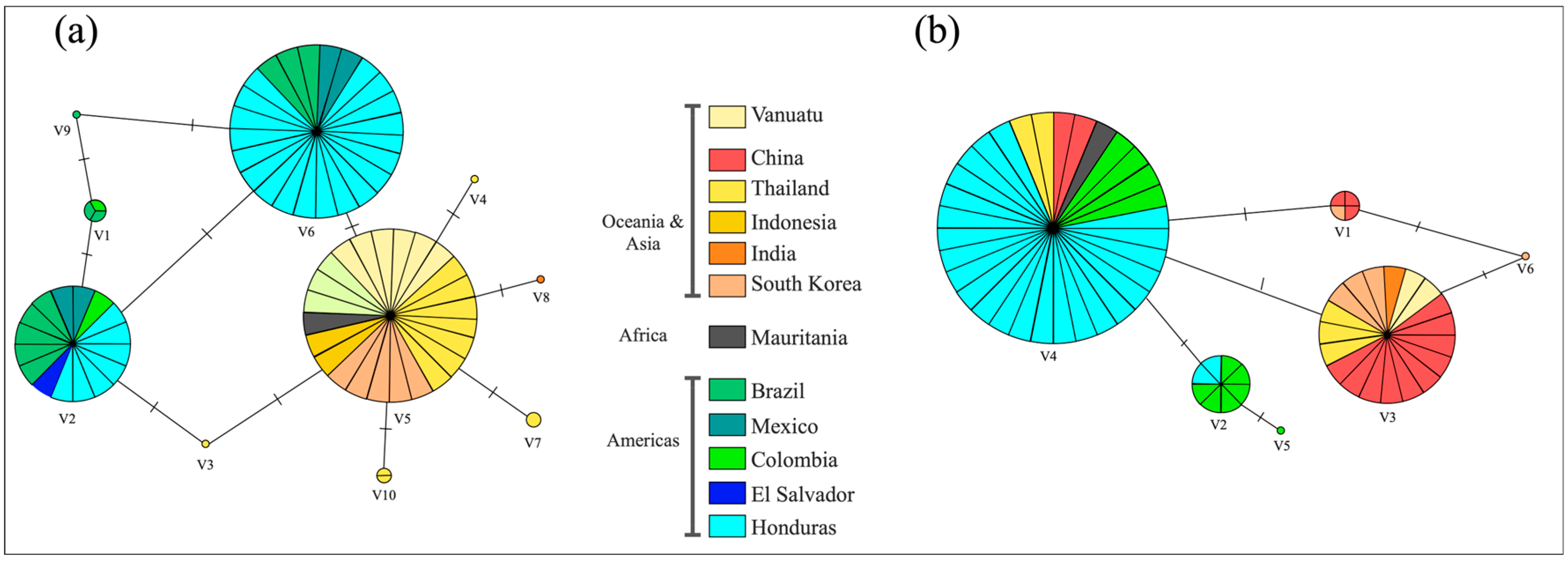
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Pvs47 | Plasmodium vivax surface protein 47 |
| Pvs48/45 | Plasmodium vivax surface protein 48/45 |
| TBVs | Transmission-blocking vaccines |
| DBS | Dried blood spot |
| RDTs | Rapid diagnostic tests |
| PET-PCR | Photo-induced electron transfer polymerase chain reaction |
References
- World Health Organization. World Malaria Report 2024; Global Malaria Programme (GMP), WHO: Geneva, Switzerland, 2024; p. 316. [Google Scholar]
- Jennison, C.; Arnott, A.; Tessier, N.; Tavul, L.; Koepfli, C.; Felger, I.; Siba, P.M.; Reeder, J.C.; Bahlo, M.; Mueller, I.; et al. Plasmodium vivax populations are more genetically diverse and less structured than sympatric Plasmodium falciparum populations. PLoS Negl. Trop. Dis. 2015, 9, e0003634. [Google Scholar] [CrossRef] [PubMed]
- Price, R.N.; Commons, R.J.; Battle, K.E.; Thriemer, K.; Mendis, K. Plasmodium vivax in the Era of the Shrinking P. falciparum Map. Trends Parasitol. 2020, 36, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.; Pinto, A.; Escobar, D.; Valdivia, H.O.; Chaver, L.; Ardon, G.; Carranza, E.; Fontecha, G. Genetic diversity of Plasmodium vivax and Plasmodium falciparum field isolates from Honduras in the malaria elimination phase. Curr. Res. Parasitol. Vector Borne Dis. 2025, 7, 100230. [Google Scholar] [CrossRef] [PubMed]
- de Jong, R.M.; Tebeje, S.K.; Meerstein-Kessel, L.; Tadesse, F.G.; Jore, M.M.; Stone, W.; Bousema, T. Immunity against sexual stage Plasmodium falciparum and Plasmodium vivax parasites. Immunol. Rev. 2020, 293, 190–215. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cruz, A.; Barillas-Mury, C. Pfs47 as a Malaria Transmission-Blocking Vaccine Target. Am. J. Trop. Med. Hyg. 2022. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Suwanabun, N.; Kaneko, O.; Iriko, H.; Otsuki, H.; Sattabongkot, J.; Kaneko, A.; Herrera, S.; Torii, M.; Tsuboi, T. Plasmodium vivax gametocyte proteins, Pvs48/45 and Pvs47, induce transmission-reducing antibodies by DNA immunization. Vaccine 2015, 33, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Takashima, E.; Morita, M.; Sattabongkot, J.; Ishino, T.; Culleton, R.; Torii, M.; Tsuboi, T. Plasmodium vivax transmission-blocking vaccines: Progress, challenges and innovation. Parasitol. Int. 2022, 87, 102525. [Google Scholar] [CrossRef] [PubMed]
- Anthony, T.G.; Polley, S.D.; Vogler, A.P.; Conway, D.J. Evidence of non-neutral polymorphism in Plasmodium falciparum gamete surface protein genes Pfs47 and Pfs48/45. Mol. Biochem. Parasitol. 2007, 156, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Canepa, G.E.; Molina-Cruz, A.; Barillas-Mury, C. Molecular Analysis of Pfs47-Mediated Plasmodium Evasion of Mosquito Immunity. PLoS ONE 2016, 11, e0168279. [Google Scholar] [CrossRef] [PubMed]
- Canepa, G.E.; Molina-Cruz, A.; Yenkoidiok-Douti, L.; Calvo, E.; Williams, A.E.; Burkhardt, M.; Peng, F.; Narum, D.; Boulanger, M.J.; Valenzuela, J.G.; et al. Antibody targeting of a specific region of Pfs47 blocks Plasmodium falciparum malaria transmission. NPJ Vaccines 2018, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cruz, A.; Garver, L.S.; Alabaster, A.; Bangiolo, L.; Haile, A.; Winikor, J.; Ortega, C.; van Schaijk, B.C.; Sauerwein, R.W.; Taylor-Salmon, E.; et al. The human malaria parasite Pfs47 gene mediates evasion of the mosquito immune system. Science 2013, 340, 984–987. [Google Scholar] [CrossRef] [PubMed]
- Conway, D.J.; Machado, R.L.; Singh, B.; Dessert, P.; Mikes, Z.S.; Povoa, M.M.; Oduola, A.M.; Roper, C. Extreme geographical fixation of variation in the Plasmodium falciparum gamete surface protein gene Pfs48/45 compared with microsatellite loci. Mol. Biochem. Parasitol. 2001, 115, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Escalante, A.A.; Grebert, H.M.; Chaiyaroj, S.C.; Riggione, F.; Biswas, S.; Nahlen, B.L.; Lal, A.A. Polymorphism in the gene encoding the Pfs48/45 antigen of Plasmodium falciparum. XI. Asembo Bay Cohort Project. Mol. Biochem. Parasitol. 2002, 119, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Ciubotariu, I.I.; Broyles, B.K.; Xie, S.; Thimmapuram, J.; Mwenda, M.C.; Mambwe, B.; Mulube, C.; Matoba, J.; Schue, J.L.; Moss, W.J.; et al. Diversity and selection analyses identify transmission-blocking antigens as the optimal vaccine candidates in Plasmodium falciparum. eBioMedicine 2024, 106, 105227. [CrossRef]
- Sauerwein, R.W.; Plieskatt, J.; Theisen, M. 40 Years of Pfs48/45 Research as a Transmission-Blocking Vaccine Target of Plasmodium falciparum Malaria. Am. J. Trop. Med. Hyg. 2022, 107, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, A.F.; Martinez, N.L.; Tobon, A.; Alger, J.; Lacerda, M.V.; Kajava, A.V.; Arevalo-Herrera, M.; Herrera, S. Global genetic diversity of the Plasmodium vivax transmission-blocking vaccine candidate Pvs48/45. Malar. J. 2016, 15, 202. [Google Scholar] [CrossRef] [PubMed]
- Kuesap, J.; Suphakhonchuwong, N.; Eksonthi, B.; Huaihongthong, S. Genetic polymorphisms of Plasmodium vivax transmission-blocking vaccine candidates Pvs48/45 and Pvs47 in Thailand. Malar. J. 2025, 24, 63. [Google Scholar] [CrossRef] [PubMed]
- Asali, S.; Raz, A.; Turki, H.; Mafakher, L.; Razmjou, E.; Solaymani-Mohammadi, S. Restricted genetic heterogeneity of the Plasmodium vivax transmission-blocking vaccine (TBV) candidate Pvs48/45 in a low transmission setting: Implications for the Plasmodium vivax malaria vaccine development. Infect. Genet. Evol. 2021, 89, 104710. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Hart, R.J.; Bansal, G.P.; Kumar, N. Functional Conservation of P48/45 Proteins in the Transmission Stages of Plasmodium vivax (Human Malaria Parasite) and P. berghei (Murine Malaria Parasite). mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Gupta, B.; Wang, M.; Zheng, W.; Zheng, L.; Zhu, X.; Yang, Y.; Fang, Q.; Luo, E.; Fan, Q.; et al. Genetic diversity of transmission-blocking vaccine candidate Pvs48/45 in Plasmodium vivax populations in China. Parasit. Vectors 2015, 8, 615. [Google Scholar] [CrossRef] [PubMed]
- Barillas-Mury, C.; Molina-Cruz, A.; Torres, T.Z.; Raytselis, N.; Young, M.; Kamath, N.; McNich, C.; Xin-zhuan, S.; Ankit, D.; Joana, S.; et al. Worldwide genetic diversity and population structure of Plasmodium vivax Pv47 is consistent with natural selection by anopheline mosquitoes. Res. Sq. 2024. [Google Scholar] [CrossRef]
- Lucchi, N.W.; Narayanan, J.; Karell, M.A.; Xayavong, M.; Kariuki, S.; DaSilva, A.J.; Hill, V.; Udhayakumar, V. Molecular diagnosis of malaria by photo-induced electron transfer fluorogenic primers: PET-PCR. PLoS ONE 2013, 8, e56677. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, G.; Escobar, D.; Pinto, A.; Serrano, D.; Ksandrova, E.; Grimaldi, N.; Juarez-Fontecha, G.; Moncada, M.; Valdivia, H.O.; Fontecha, G. PET-PCR reveals low parasitaemia and submicroscopic malarial infections in Honduran Moskitia. Malar. J. 2023, 22, 110. [Google Scholar] [CrossRef] [PubMed]
- Kudyba, H.M.; Louzada, J.; Ljolje, D.; Kudyba, K.A.; Muralidharan, V.; Oliveira-Ferreira, J.; Lucchi, N.W. Field evaluation of malaria malachite green loop-mediated isothermal amplification in health posts in Roraima state, Brazil. Malar. J. 2019, 18, 98. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.K.; Kim, K.A.; Kim, J.; Oh, J.S.; Han, E.T.; An, S.S.; Lim, C.S. Sequence polymorphisms in Pvs48/45 and Pvs47 gametocyte and gamete surface proteins in Plasmodium vivax isolated in Korea. Mem. Inst. Oswaldo Cruz 2013, 108, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef] [PubMed]
- JukesTH, C.C. Evolution of Protein Molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press: New York, NY, USA, 1969. [Google Scholar]
- Molina-Cruz, A.; Raytselis, N.; Withers, R.; Dwivedi, A.; Crompton, P.D.; Traore, B.; Carpi, G.; Silva, J.C.; Barillas-Mury, C. A genotyping assay to determine geographic origin and transmission potential of Plasmodium falciparum malaria cases. Commun. Biol. 2021, 4, 1145. [Google Scholar] [CrossRef] [PubMed]
- Onyango, S.A.; Ochwedo, K.O.; Machani, M.G.; Omondi, C.J.; Debrah, I.; Ogolla, S.O.; Lee, M.C.; Zhou, G.; Kokwaro, E.; Kazura, J.W.; et al. Genetic diversity and population structure of the human malaria parasite Plasmodium falciparum surface protein Pfs47 in isolates from the lowlands in Western Kenya. PLoS ONE 2021, 16, e0260434. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Morazan, A.S.; Moncada, M.M.; Escobar, D.; Cabrera-Moreno, L.A.; Fontecha, G. Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach. Bioinform. Biol. Insights 2024, 18, 11779322241284223. [Google Scholar] [CrossRef] [PubMed]
- Onyango, S.A.; Machani, M.G.; Ochwedo, K.O.; Oriango, R.M.; Lee, M.C.; Kokwaro, E.; Afrane, Y.A.; Githeko, A.K.; Zhong, D.; Yan, G. Plasmodium falciparum Pfs47 haplotype compatibility to Anopheles gambiae in Kisumu, a malaria-endemic region of Kenya. Sci. Rep. 2025, 15, 6550. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.C.; Ortiz, A.; Coello, J.; Sosa-Ochoa, W.; Torres, R.E.; Banegas, E.I.; Jovel, I.; Fontecha, G.A. Genetic diversity of Plasmodium vivax and Plasmodium falciparum in Honduras. Malar. J. 2012, 11, 391. [Google Scholar] [CrossRef] [PubMed]
- Larranaga, N.; Mejia, R.E.; Hormaza, J.I.; Montoya, A.; Soto, A.; Fontecha, G.A. Genetic structure of Plasmodium falciparum populations across the Honduras-Nicaragua border. Malar. J. 2013, 12, 354. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.; Archaga, O.; Mejia, A.; Escober, L.; Henriquez, J.; Montoya, A.; Valdivia, H.O.; Fontecha, G. Evidence of a Recent Bottleneck in Plasmodium falciparum Populations on the Honduran-Nicaraguan Border. Pathogens 2021, 10, 1432. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, M.J.M.; Degrugillier, F.; Arnathau, C.; Fontecha, G.A.; Noya, O.; Houze, S.; Severini, C.; Pradines, B.; Berry, A.; Trape, J.F.; et al. Genomic exploration of the journey of Plasmodium vivax in Latin America. PLoS Pathog. 2025, 21, e1012811. [Google Scholar] [CrossRef] [PubMed]
- Joy, D.A.; Gonzalez-Ceron, L.; Carlton, J.M.; Gueye, A.; Fay, M.; McCutchan, T.F.; Su, X.Z. Local adaptation and vector-mediated population structure in Plasmodium vivax malaria. Mol. Biol. Evol. 2008, 25, 1245–1252. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Reaction | Primer Name | Primer Sequence (5′-3′) | References |
|---|---|---|---|---|
| 18Sr RNA gene | PET-PCR for genus Plasmodium | Genus forward | GGC CTA ACA TGG CTA TGA CG | [23,24] |
| Genus reverse | 6FAM-agg cgc ata gcg cct gg CTG CCT TCC TTA GAT GTG GTA GCT | |||
| 18Sr RNA gene | PET-PCR for P. vivax | Vivax forward | ACT GAC ACT GAT GAT TTA GAA CCC ATT T | [25] |
| Vivax reverse | HEX- agg cgc ata gcg cct ggT GGA GAG ATC TTT CCA TCC TAA ACC T | |||
| Pvs47 | 1st round | CM 000453 Fwd | CAC ACC ACC GCA AAC AGG | [26] |
| CM 000453 Rev | GTG CAC ATT CCG CGG TTG | |||
| 2nd round | nst 1440 Fwd | GCG GTC CAC CCT AAC TGT AA | * | |
| nst 1440 Rev | TGC TGC AAA CCA CAC ATG T | [7] | ||
| Sequencing primers | pPvs47-F | ATA TTT CCA ACG AAG CAT TTA TGC | ||
| pPvs47-R | TTT TCC ATT ATG CTC ACA AAC GC | |||
| Pvs47F2 | GAA GAA AGG GGA GGA CCA AG | [26] | ||
| Pvs48/45 | 1st round | Pvs4845F | GGA ATA ATT TCG ACC ACT C | [17] |
| 887 | TCA GAA GTA CAA CAG GAG GAG CAC | [20] | ||
| 2nd round | 866 | ATG TTG AAG CGC CAG CTC GCC AA | ||
| 887 | TCA GAA GTA CAA CAG GAG GAG CAC | |||
| Sequencing primers | pPvs4845F | ATG GCC AAA GGA GAG GTC AAG TAC | [7] | |
| pPvs48/45-6C-F | TCG GCA GAT GCA AGT GAA GGA AGT C | |||
| Pvs4845FF | TGT AAA ATC TGC GGA CGT GA | [26] | ||
| Pvs4845RR | CGGGTGCTTTAAAAATGGAA | |||
| 1001F | GAATGAGTTGCCCTGGGGAA | This study | ||
| 1025F | TGCCCGAGTGCTTCTTTCAA | |||
| 249R | TCCTGGGATCTTCTTCGGGA | |||
| 494R | AAGGCCACTCTTCCCTTCAC |
| Molecular Diversity Indices and Neutrality Tests | pvs47 | pvs48/45 |
|---|---|---|
| Nucleotide diversity (π) | 0.00374 | 0.00258 |
| Segregating sites (S) | 33 | 21 |
| Average number of nucleotide differences (k) | 4.87560 | 3.49423 |
| Number of haplotypes (h) | 45 | 32 |
| Haplotype diversity (Hd) | 0.918 | 0.900 |
| Tajima’s D | −1.02707 | −0.65000 |
| Statistical significance | Not significant, p > 0.10 | Not significant, p > 0.10 |
| Fu and Li’s D* test statistic | −2.08212 | −2.12009 |
| Statistical significance | Not significant, 0.10 > p > 0.05 | Not significant, p > 0.10 |
| Fu and Li’s F* test statistic | −2.01002 | −1.89449 |
| Statistical significance | Not significant, 0.10 > p > 0.05 | Not significant, p > 0.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Euceda, K.; Matamoros, G.; Araujo, M.E.; Chaver, L.; Ardón, G.; Fontecha, G. Genetic Diversity and Phylogeography of Plasmodium vivax Transmission-Blocking Vaccine Candidate Genes pvs47 and pvs48/45 in Honduras. Parasitologia 2025, 5, 36. https://doi.org/10.3390/parasitologia5030036
Euceda K, Matamoros G, Araujo ME, Chaver L, Ardón G, Fontecha G. Genetic Diversity and Phylogeography of Plasmodium vivax Transmission-Blocking Vaccine Candidate Genes pvs47 and pvs48/45 in Honduras. Parasitologia. 2025; 5(3):36. https://doi.org/10.3390/parasitologia5030036
Chicago/Turabian StyleEuceda, Kevin, Gabriela Matamoros, María Esther Araujo, Lesly Chaver, Gloria Ardón, and Gustavo Fontecha. 2025. "Genetic Diversity and Phylogeography of Plasmodium vivax Transmission-Blocking Vaccine Candidate Genes pvs47 and pvs48/45 in Honduras" Parasitologia 5, no. 3: 36. https://doi.org/10.3390/parasitologia5030036
APA StyleEuceda, K., Matamoros, G., Araujo, M. E., Chaver, L., Ardón, G., & Fontecha, G. (2025). Genetic Diversity and Phylogeography of Plasmodium vivax Transmission-Blocking Vaccine Candidate Genes pvs47 and pvs48/45 in Honduras. Parasitologia, 5(3), 36. https://doi.org/10.3390/parasitologia5030036