
Pathophysiology of Myelodysplastic Syndromes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Natural Course of MDS Disease
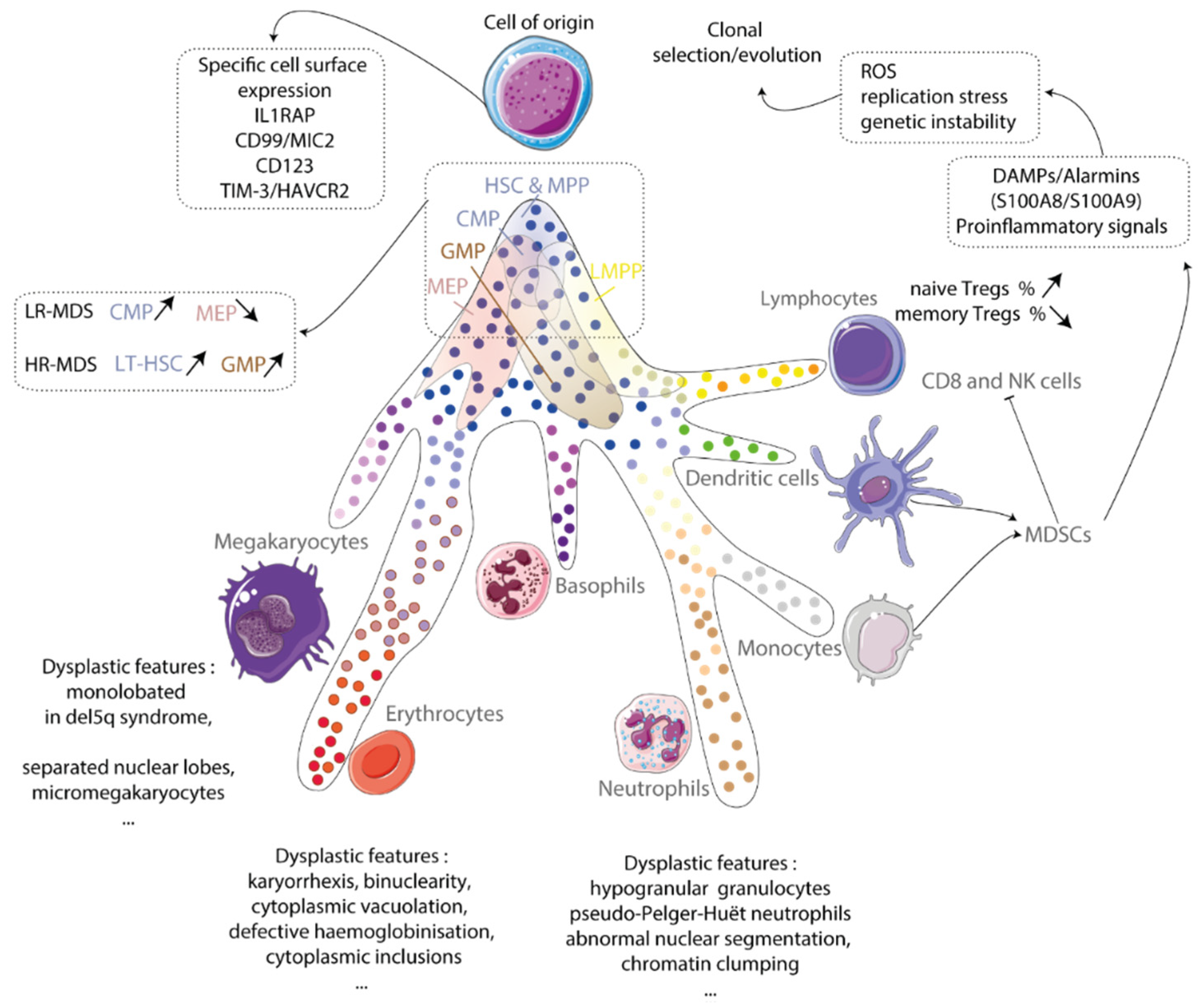
2.1. Cell of Origin
2.2. Clonal Hematopoiesis
2.3. Early MDS
2.4. Clonal Evolution to AML
2.5. Epistatic Interactions of Genes Involved in Co-/Post-Transcriptional Regulation of Transcription
3. Mechanisms of Cytopenias in Early MDS
3.1. Hematopoietic Stem Cell Aging
3.1.1. Aged HSC Functional Defects
3.1.2. Aged HSC Changes in Gene Expression Profiles
3.1.3. Epigenetic Changes with Aging in HSC
3.1.4. Replication Stress in Aged HSC
3.2. Ineffective Erythropoiesis
3.2.1. Excessive Cell Death
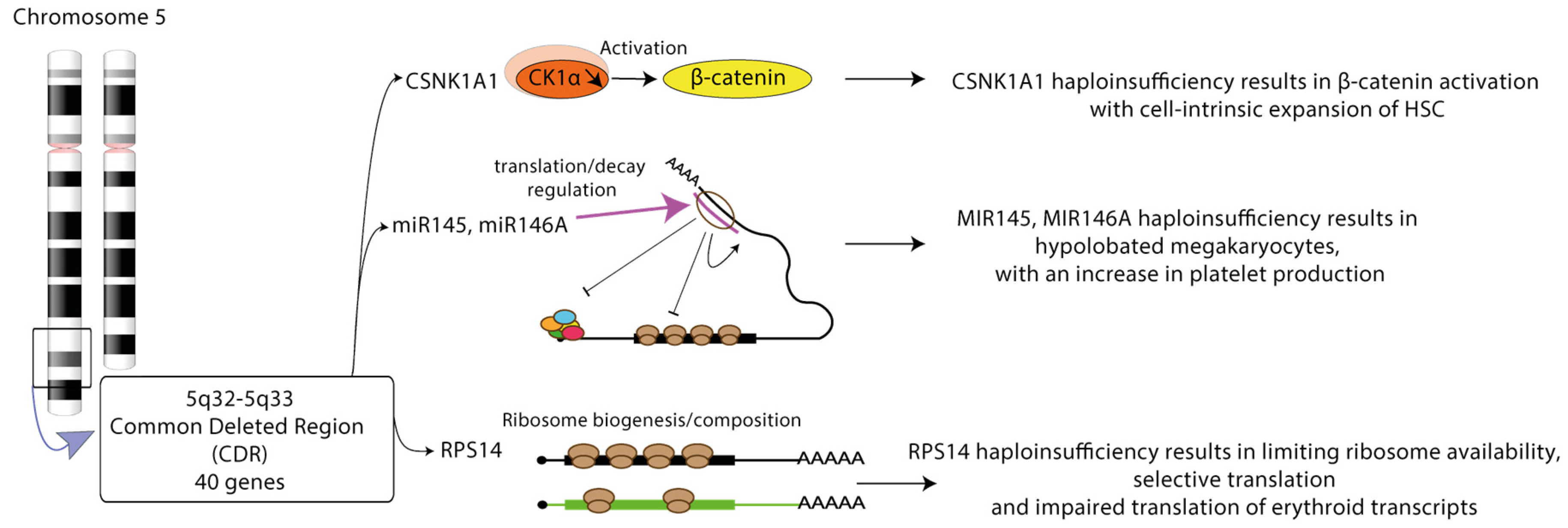
3.2.2. Post-Transcriptional Deregulation of Gene Expression in Del(5q) MDS
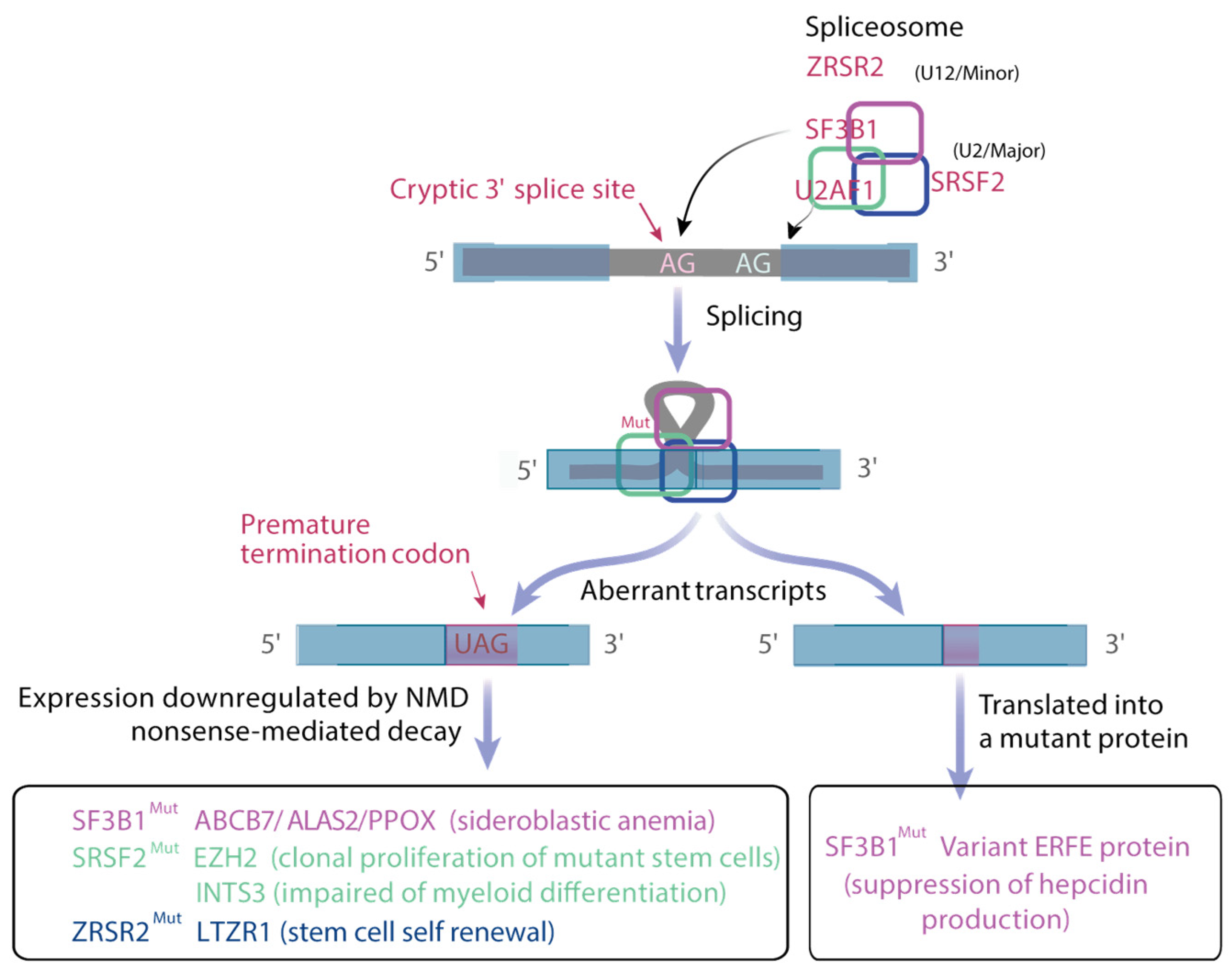
3.2.3. Co-Transcriptional and Post-Transcriptional Deregulation of Gene Expression by Mutant Splicing Factors
DNA Replication Stress in Splicing Factor Mutated MDS
Deregulation of the Splicing Machinery
Defective Metabolic Reprogramming
4. Immune Dysregulation in MDS
Immunity in Aging
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fenaux, P.; Platzbecker, U.; Ades, L. How we manage adults with myelodysplastic syndrome. Br. J. Haematol. 2020, 189, 1016–1027. [Google Scholar] [CrossRef]
- Santini, V. How I treat MDS after hypomethylating agent failure. Blood 2019, 133, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Will, B.; Zhou, L.; Vogler, T.O.; Ben-Neriah, S.; Schinke, C.; Tamari, R.; Yu, Y.; Bhagat, T.D.; Bhattacharyya, S.; Barreyro, L.; et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood 2012, 120, 2076–2086. [Google Scholar] [CrossRef]
- Pang, W.W.; Pluvinage, J.V.; Price, E.A.; Sridhar, K.; Arber, D.A.; Greenberg, P.L.; Schrier, S.L.; Park, C.Y.; Weissman, I.L. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc. Natl. Acad. Sci. USA 2013, 110, 3011–3016. [Google Scholar] [CrossRef]
- Woll, P.S.; Kjällquist, U.; Chowdhury, O.; Doolittle, H.; Wedge, D.; Thongjuea, S.; Erlandsson, R.; Ngara, M.; Anderson, K.; Deng, Q.; et al. Myelodysplastic Syndromes Are Propagated by Rare and Distinct Human Cancer Stem Cells In Vivo. Cancer Cell 2014, 25, 794–808. [Google Scholar] [CrossRef]
- Chesnais, V.; Arcangeli, M.-L.; Delette, C.; Rousseau, A.; Guermouche, H.; Lefevre, C.; Bondu, S.; Diop, M.; Cheok, M.; Chapuis, N.; et al. Architectural and functional heterogeneity of hematopoietic stem/progenitor cells in non-del(5q) myelodysplastic syndromes. Blood 2017, 129, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Mian, S.A.; Rouault-Pierre, K.; Smith, A.E.; Seidl, T.; Pizzitola, I.; Kizilors, A.; Kulasekararaj, A.G.; Bonnet, D.; Mufti, G.J. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat. Commun. 2015, 6, 10004. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, L.; Will, B.; Bartholdy, B.; Zhou, L.; Todorova, T.I.; Stanley, R.F.; Ben-Neriah, S.; Montagna, C.; Parekh, S.; Pellagatti, A.; et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood 2012, 120, 1290–1298. [Google Scholar] [CrossRef]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.S.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like Leukemia Stem Cells in Acute Myeloid Leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef]
- Shastri, A.; Will, B.; Steidl, U.; Verma, A. Stem and progenitor cell alterations in myelodysplastic syndromes. Blood 2017, 129, 1586–1594. [Google Scholar] [CrossRef]
- Chung, S.S.; Eng, W.S.; Hu, W.; Khalaj, M.; Garrett-Bakelman, F.E.; Tavakkoli, M.; Levine, R.L.; Carroll, M.; Klimek, V.M.; Melnick, A.M.; et al. CD99 is a therapeutic target on disease stem cells in myeloid malignancies. Sci. Transl. Med. 2017, 9, eaaj2025. [Google Scholar] [CrossRef]
- Li, L.J.; Tao, J.L.; Fu, R.; Wang, H.Q.; Jiang, H.J.; Yue, L.; Zhang, W.; Liu, H.; Shao, Z.H. Increased CD34+CD38−CD123+ cells in myelodysplastic syndrome displaying malignant features similar to those in AML. Int. J. Hematol. 2014, 100, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Asayama, T.; Tamura, H.; Ishibashi, M.; Kuribayashi-Hamada, Y.; Onodera-Kondo, A.; Okuyama, N.; Yamada, A.; Shimizu, M.; Moriya, K.; Takahashi, H.; et al. Functional expression of Tim-3 on blasts and clinical impact of its ligand galectin-9 in myelodysplastic syndromes. Oncotarget 2017, 8, 88904–88917. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Shima, T.; Takayanagi, S.I.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010, 7, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haemato-poietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.; Lindberg, J.; Rose, S.; Bakhoum, S.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Van Zeventer, I.A.; de Graaf, A.O.; Wouters, H.J.C.M.; van der Reijden, B.A.; van der Klauw, M.M.; de Witte, T.; Jonker, M.A.; Malcovati, L.; Jansen, J.H.; Huls, G. Mutational spectrum and dynamics of clonal hematopoiesis in anemia of older individuals. Blood 2020, 135, 1161–1170. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Athero-sclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-mutant MDS as a distinct disease subtype: A proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2016, 49, 204–212. [Google Scholar] [CrossRef]
- Da Silva-Coelho, P.; Kroeze, L.I.; Yoshida, K.; Koorenhof-Scheele, T.N.; Knops, R.; van de Locht, L.T.; de Graaf, A.O.; Massop, M.; Sandmann, S.; Dugas, M.; et al. Clonal evolution in myel-odysplastic syndromes. Nat. Commun. 2017, 8, 15099. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2013, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kao, Y.-R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.-R.; Montagna, C.; Will, B.; Verma, A.; Steidl, A.U. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med. 2018, 25, 103–110. [Google Scholar] [CrossRef]
- Jaedersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Goehring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 Mutations in Low-Risk Myelodysplastic Syndromes With del(5q) Predict Disease Progression. J. Clin. Oncol. 2011, 29, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 33, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, A.; Lin, K.-T.; Wiseman, D.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.; Micol, J.-B.; Zhang, X.J.; De Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nat. Cell Biol. 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the Elongation Phase of Transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef]
- Ji, X.; Zhou, Y.; Pandit, S.; Huang, J.; Li, H.; Lin, C.Y.; Xiao, R.; Burge, C.B.; Fu, X.D. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 2013, 153, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Elrod, N.D.; Henriques, T.; Huang, K.-L.; Tatomer, D.C.; Wilusz, J.; Wagner, E.J.; Adelman, K. The Integrator Complex Attenuates Promoter-Proximal Transcription at Protein-Coding Genes. Mol. Cell 2019, 76, 738–752.e7. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J.Y.; Huang, Y.J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The augmented R-loop is a unifying mechanism for myelodys-plastic syndromes induced by high-risk splicing factor mutations. Mol. Cell 2018, 69, 412–425. [Google Scholar] [CrossRef]
- Ochi, Y.; Kon, A.; Sakata, T.; Nakagawa, M.M.; Nakazawa, N.; Kakuta, M.; Kataoka, K.; Koseki, H.; Nakayama, M.; Morishita, D.; et al. Combined Cohesin-RUNX1 Deficiency Syner-gistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes. Cancer Discov. 2020, 10, 836–853. [Google Scholar] [CrossRef]
- Morrison, S.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef]
- Nakamura-Ishizu, A.; Suda, T. Aging of the hematopoietic stem cells niche. Int. J. Hematol. 2014, 100, 317–325. [Google Scholar] [CrossRef]
- Chambers, S.M.; Shaw, C.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; A Goodell, M. Aging Hematopoietic Stem Cells Decline in Function and Exhibit Epigenetic Dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef]
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef] [PubMed]
- Bernitz, J.; Kim, H.S.; MacArthur, B.; Sieburg, H.; Moore, K. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 2016, 167, 1296–1309.e10. [Google Scholar] [CrossRef] [PubMed]
- Säwen, P.; Eldeeb, M.; Erlandsson, E.; Kristiansen, T.A.; Laterza, C.; Kokaia, Z.; Karlsson, G.; Yuan, J.; Soneji, S.; Mandal, P.K.; et al. Murine HSCs contribute actively to native hematopoiesis but with reduced differentiation capacity upon aging. Elife 2018, 7, e41258. [Google Scholar] [CrossRef]
- Kollman, C.; Howe, C.W.S.; Anasetti, C.; Antin, J.H.; Davies, S.M.; Filipovich, A.H.; Hegland, J.; Kamani, N.; Kernan, N.; King, R.; et al. Donor characteristics as risk factors in recipients after transplantation of bone marrow from unrelated donors: The effect of donor age. Blood 2001, 98, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.W.; Price, E.A.; Sahoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and mye-loid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef]
- Kuranda, K.; Vargaftig, J.; De La Rochère, P.; Dosquet, C.; Charron, D.; Bardin, F.; Tonnelle, C.; Bonnet, D.; Goodhardt, M. Age-related changes in human hematopoietic stem/progenitor cells. Aging Cell 2011, 10, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.A.; Balaian, L.; Santos, N.D.; Leu, H.S.; Court, A.C.; Lazzari, E.; Sadarangani, A.; Zipeto, M.A.; La Clair, J.J.; Villa, R.; et al. RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML. Cell Stem Cell 2016, 19, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Bocker, M.T.; Hellwig, I.; Breiling, A.; Eckstein, V.; Ho, A.D.; Lyko, F. Genome-wide promoter DNA methylation dynamics of human hematopoietic pro-genitor cells during differentiation and aging. Blood 2011, 117, e182–e189. [Google Scholar] [CrossRef]
- Bacalini, M.G.; Boattini, A.; Gentilini, D.; Giampieri, E.; Pirazzini, C.; Giuliani, C.; Fontanesi, E.; Remondini, D.; Capri, M.; Del Rio, A.; et al. A meta-analysis on age-associated changes in blood DNA methylation: Results from an original analysis pipeline for Infinium 450k data. Aging 2015, 7, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Vallania, F.; Warsinske, H.C.; Donato, M.; Schaffert, S.; Chang, S.E.; Dvorak, M.; Dekker, C.L.; Davis, M.M.; Utz, P.J.; et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 2018, 173, 1385–1397.e14. [Google Scholar] [CrossRef]
- Adelman, E.R.; Huang, H.T.; Roisman, A.; Olsson, A.; Colaprico, A.; Qin, T.; Lindsley, R.C.; Bejar, R.; Salomonis, N.; Grimes, H.L.; et al. Aging human hematopoietic stem cells manifest profound epigenetic repro-gramming of enhancers that may predispose to leukemia. Cancer Discov. 2019, 9, 1080–1101. [Google Scholar] [CrossRef]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nat. Cell Biol. 2015, 520, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of func-tional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef]
- Flach, J.; Jann, J.-C.; Knaflic, A.; Riabov, V.; Streuer, A.; Altrock, E.; Xu, Q.; Schmitt, N.; Obländer, J.; Nowak, V.; et al. Replication stress signaling is a therapeutic target in myelodysplastic syndromes with splicing factor mutations. Haematology 2020. [Google Scholar] [CrossRef]
- Raval, A.; Sridhar, K.J.; Patel, S.; Turnbull, B.B.; Greenberg, P.L.; Mitchell, B.S. Reduced rRNA expression and increased rDNA promoter methylation in CD34+ cells of patients with myelodysplastic syndromes. Blood 2012, 120, 4812–4818. [Google Scholar] [CrossRef]
- Tehranchi, R.; Invernizzi, R.; Grandien, A.; Zhivotovsky, B.; Fadeel, B.; Forsblom, A.-M.; Travaglino, E.; Samuelsson, J.; Hast, R.; Nilsson, L.; et al. Aberrant mitochondrial iron distribution and maturation arrest characterize early erythroid precursors in low-risk myelodysplastic syndromes. Blood 2005, 106, 247–253. [Google Scholar] [CrossRef]
- Gyan, E.; Frisan, E.; Beyne-Rauzy, O.; Deschemin, J.-C.; Pierre-Eugene, C.; Randriamampita, C.; Dubart-Kupperschmitt, A.; Garrido, C.; Dreyfus, F.; Mayeux, P.; et al. Spontaneous and Fas-induced apoptosis of low-grade MDS erythroid precursors involves the endoplasmic reticulum. Leukemia 2008, 22, 1864–1873. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; Class, C.A.; Ganan-Gomez, I.; Kanagal-Shamanna, R.; Sasaki, K.; Richard-Carpentier, G.; Naqvi, K.; Wei, Y.; Yang, H.; Soltysiak, K.A.; et al. Tran-scriptomic analysis implicates necroptosis in disease progression and prognosis in myelodysplastic syndromes. Leukemia 2020, 34, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.N.; Shi, Q.; Salisbury-Ruf, C.T.; Zou, J.; Savona, M.R.; Fedoriw, Y.; Zinkel, S.S. Increased Ripk1-mediated bone marrow necroptosis leads to myelodysplasia and bone marrow failure in mice. Blood 2019, 133, 107–120. [Google Scholar] [CrossRef]
- Sallman, D.A.; Cluzeau, T.; Basiorka, A.A.; List, A. Unraveling the Pathogenesis of MDS: The NLRP3 Inflammasome and Pyroptosis Drive the MDS Phenotype. Front. Oncol. 2016, 6, 151. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- A Basiorka, A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; McLemore, A.; Vincelette, N.D.; A Ward, G.; Eksioglu, E.A.; Sallman, D.A.; Al Ali, N.; Padron, E.; et al. Assessment of ASC specks as a putative biomarker of pyroptosis in myelodysplastic syndromes: An observational cohort study. Lancet Haematol. 2018, 5, e393–e402. [Google Scholar] [CrossRef]
- Mei, Y.; Zhao, B.; Basiorka, A.A.; Yang, J.; Cao, L.; Zhang, J.; List, A.; Ji, P. Age-related inflammatory bone marrow microen-vironment induces ineffective erythropoiesis mimicking del(5q) MDS. Leukemia 2018, 32, 1023–1033. [Google Scholar] [CrossRef]
- Cluzeau, T.; McGraw, K.L.; Irvine, B.; Masala, E.; Ades, L.; Basiorka, A.A.; Maciejewski, J.; Auberger, P.; Wei, S.; Fenaux, P.; et al. Pro-inflammatory proteins S100A9 and tumor necrosis factor-α suppress erythropoietin elaboration in myelodysplastic syndromes. Haematology 2017, 102, 2015–2020. [Google Scholar] [CrossRef]
- Schneider, R.K.; Schenone, M.; Ferreira, M.S.V.; Kramann, R.; Joyce, C.E.; Hartigan, C.; Beier, F.; Brümmendorf, T.H.; Gehrming, U.; Platzbecker, U.; et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A. Nat. Med. 2016, 22, 288–297. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.; Adisty, M.N.; Van Strien, P.M.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients repro-gram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef]
- Bulycheva, E.; Rauner, M.; Medyouf, H.; Theurl, I.; Bornhauser, M.; Hofbauer, L.C.; Platzbecker, U. Myelodysplasia is in the niche: Novel concepts and emerging therapies. Leukemia 2014, 29, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Pronk, E.; Raaijmakers, M.H.G.P. The mesenchymal niche in MDS. Blood 2019, 133, 1031–1038. [Google Scholar] [CrossRef]
- Schneider, R.K.; Ademà, V.; Heckl, D.; Järås, M.; Mallo, M.; Lord, A.M.; Chu, L.P.; McConkey, M.E.; Kramann, R.; Mullally, A.; et al. Role of Casein Kinase 1A1 in the Biology and Targeted Therapy of del(5q) MDS. Cancer Cell 2014, 26, 509–520. [Google Scholar] [CrossRef]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and deg-radation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; Wells, R.A.; et al. Identification of miR-145 and miR-146a as mediators of the 5q–syndrome phenotype. Nat. Med. 2009, 16, 49–58. [Google Scholar] [CrossRef]
- Ribezzo, F.; Snoeren, I.A.; Ziegler, S.; Stoelben, J.; Olofsen, P.A.; Henic, A.; Ferreira, M.V.; Chen, S.; Stalmann, U.S.; Buesche, G.; et al. Rps14, Csnk1a1 and miRNA145/miRNA146a defi-ciency cooperate in the clinical phenotype and activation of the innate immune system in the 5q- syndrome. Leukemia 2019, 33, 1759–1772. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nat. Cell Biol. 2008, 451, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, S.; Boussaid, I.; Floquet, C.; Raimbault, A.; Hatin, I.; Andrieu-Soler, C.; Salma, M.; LeDuc, M.; Gautier, E.-F.; Guyot, B.; et al. p53 activation during ribosome biogenesis regulates normal erythroid differentiation. Blood 2021, 137, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef]
- Moniz, H.; Gastou, M.; Leblanc, T.; Hurtaud, C.; Cretien, A.; Lecluse, Y.; Raslova, H.; Larghero, J.; Croisille, L.; Faubladier, M.; et al. Group of Société d’Hématologie et d’Immunologie Pédiatrique-SHIP. Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 2012, 3, e356. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.L.; Drynan, L.F.; Hewett, D.R.; Holmes, L.R.; Lorenzo-Abalde, S.; Lane, A.L.; Jolin, H.E.; Pannell, R.; Middleton, A.J.; Wong, S.H.; et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat. Med. 2010, 16, 59–66. [Google Scholar] [CrossRef]
- Frisan, E.; Vandekerckhove, J.; de Thonel, A.; Pierre-Eugène, C.; Sternberg, A.; Arlet, J.B.; Floquet, C.; Gyan, E.; Kosmider, O.; Dreyfus, F.; et al. Defective nuclear localization of Hsp70 is associated with dyserythropoiesis and GATA-1 cleavage in myelodysplastic syndromes. Blood 2012, 119, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Gastou, M.; Rio, S.; Dussiot, M.; Karboul, N.; Moniz, H.; Leblanc, T.; Sevin, M.; Gonin, P.; Larghéro, J.; Garrido, C.; et al. The severe phenotype of Diamond-Blackfan anemia is modulated by heat shock protein 70. Blood Adv. 2017, 1, 1959–1976. [Google Scholar] [CrossRef]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Boussaid, I.; Le Goff, S.; Floquet, C.; Gautier, E.F.; Raimbault, A.; Viailly, P.J.; Al Dulaimi, D.; Burroni, B.; Dusanter-Fourt, I.; Hatin, I.; et al. Integrated analyses of translatome and proteome identify the rules of translation selectivity in RPS14-deficient cells. Haematologica 2021, 106, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Keel, S.B.; Shimamura, A.; Liu, L.; Gerds, A.T.; Li, H.Y.; Wood, B.L.; Scott, B.L.; Abkowitz, J.L. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci. Transl. Med. 2016, 8, 338ra67. [Google Scholar] [CrossRef]
- Singh, S.; Ahmed, D.; Dolatshad, H.; Tatwavedi, D.; Schulze, U.; Sanchi, A.; Ryley, S.; Dhir, A.; Carpenter, L.; Watt, S.M.; et al. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 2020, 34, 2525–2530. [Google Scholar] [CrossRef]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Malcovati, L.; Galli, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. Faculty Opinions recommendation of SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2018, 27, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Lin, K.T.; Bradley, R.K.; Abdel-Wahab, O.; Krainer, A.R. Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev. 2020, 34, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Polaski, J.T.; Taylor, J.; Castel, P.; Chen, S.; Kobayashi, S.; Hogg, S.J.; Hayashi, Y.; Pineda, J.M.B.; El Marabti, E.; et al. Faculty Opinions recommendation of Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat. Genet. 2021, 53, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Damm, F.; Kosmider, O.; Gelsi-Boyer, V.; Renneville, A.; Carbuccia, N.; Hidalgo-Curtis, C.; Della Valle, V.; Couronné, L.; Scourzic, L.; Chesnais, V.; et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood 2012, 119, 3211–3218. [Google Scholar] [CrossRef]
- Pellagatti, A.; Armstrong, R.N.; Steeples, V.; Sharma, E.; Repapi, E.; Singh, S.; Sanchi, A.; Radujkovic, A.; Horn, P.; Dolatshad, H.; et al. Impact of spliceosome mutations on RNA splicing in myelodysplasia: Dysregulated genes/pathways and clinical associations. Blood 2018, 132, 1225–1240. [Google Scholar] [CrossRef]
- Dolatshad, H.; Pellagatti, A.; Liberante, F.G.; Llorian, M.; Repapi, E.; Steeples, V.; Roy, S.; Scifo, L.; Armstrong, R.N.; Shaw, J.; et al. Cryptic splicing events in the iron trans-porter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia 2016, 30, 2322–2331. [Google Scholar] [CrossRef]
- Bondu, S.; Alary, A.-S.; Lefèvre, C.; Houy, A.; Jung, G.; Lefebvre, T.; Rombaut, D.; Boussaid, I.; Bousta, A.; Guillonneau, F.; et al. A variant erythroferrone disrupts iron homeostasis inSF3B1-mutated myelodysplastic syndrome. Sci. Transl. Med. 2019, 11, eaav5467. [Google Scholar] [CrossRef]
- Kordasti, S.Y.; Ingram, W.; Hayden, J.; Darling, D.; Barber, L.; Afzali, B.; Lombardi, G.; Wlodarski, M.; Maciejewski, J.P.; Farzaneh, F.; et al. CD4+CD25high Foxp3+ regulatory T cells in myelodysplastic syndrome (MDS). Blood 2007, 110, 847–850. [Google Scholar] [CrossRef]
- Kotsianidis, I.; Bouchliou, I.; Nakou, E.; Spanoudakis, E.; Margaritis, D.; Christophoridou, A.V.; Anastasiades, A.; Tsigalou, C.; Bourikas, G.; Karadimitris, A.; et al. Kinetics, function and bone marrow trafficking of CD4+CD25+FOXP3+ regulatory T cells in myelodysplastic syndromes (MDS). Leukemia 2008, 23, 510–518. [Google Scholar] [CrossRef]
- Kittang, A.O.; Kordasti, S.; Sand, K.E.; Costantini, B.; Kramer, A.M.; Perezabellan, P.; Seidl, T.; Rye, K.P.; Hagen, K.M.; Kulasekararaj, A.; et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. OncoImmunology 2015, 5, e1062208. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen-Kerkhoff, N.; Westers, T.M.; Poddighe, P.J.; Povoleri, G.A.; Timms, J.A.; Kordasti, S.; De Gruijl, T.D.; Van de Loosdrecht, A.A. Reduced frequencies and functional impairment of dendritic cell subsets and non-classical monocytes in myelodysplastic syndromes. Haematologica 2021. [Google Scholar] [CrossRef]
- Mailloux, A.W.; Sugimori, C.; Komrokji, R.S.; Yang, L.; Maciejewski, J.P.; Sekeres, M.A.; Paquette, R.; Loughran, T.P.; List, A.F.; Epling-Burnette, P.K. Expansion of effector memory regulatory T cells represents a novel prognostic factor in lower risk myelodysplastic syndrome. J. Immunol. 2012, 189, 3198–3208. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.J.; Bourgeois, E.; Lobe, I. Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia 2006, 20, 463–470. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Bai, F.; Painter, J.S.; Rollison, D.E.; Salih, H.R.; Krusch, M.; Zou, J.; Ku, E.; Zhong, B.; Boulware, D.; et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood 2007, 109, 4816–4824. [Google Scholar] [CrossRef]
- Hejazi, M.; Manser, A.R.; Fröbel, J.; Kündgen, A.; Zhao, X.; Schönberg, K.; Germing, U.; Haas, R.; Gattermann, N.; Uhrberg, M. Impaired cytotoxicity associated with defective natural killer cell differentiation in myelodysplastic syndromes. Haematologica 2015, 100, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Mekinian, A.; Dervin, G.; Lapidus, N.; Kahn, J.E.; Terriou, L.; Liozon, E.; Grignano, E.; Piette, J.C.; Rauzy, O.B.; Grobost, V.; et al. Biologics in myelodysplastic syndrome-related systemic inflammatory and autoimmune diseases: French multicenter retrospective study of 29 patients. Autoimmun. Rev. 2017, 16, 903–910. [Google Scholar] [CrossRef][Green Version]
- Roupie, A.L.; Guedon, A.; Terrier, B.; Lahuna, C.; Jachiet, V.; Regent, A.; de Boysson, H.; Carrat, F.; Seguier, J.; Terriou, L.; et al. Vasculitis associated with myelodysplastic syndrome and chronic mye-lomonocytic leukemia: French multicenter case-control study. Semin. Arthritis Rheum. 2020, 50, 879–884. [Google Scholar] [CrossRef]
- Zhao, L.-P.; Boy, M.; Azoulay, C.; Clappier, E.; Sébert, M.; Amable, L.; Klibi, J.; Benlagha, K.; Espéli, M.; Balabanian, K.; et al. Genomic landscape of MDS/CMML associated with systemic inflammatory and autoimmune disease. Leukemia 2021, 1–5. [Google Scholar] [CrossRef]
- Moskorz, W.; Cosmovici, C.; Jäger, P.S.; Cadeddu, R.P.; Timm, J.; Haas, R. Myelodysplastic syndrome patients display alterations in their immune status reflected by increased PD-L1-expressing stem cells and highly dynamic exhausted T-cell frequencies. Br. J. Haematol. 2021, 193, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Eksioglu, E.A.; Chen, X.; Kandell, W.; Le Trinh, T.; Cen, L.; Qi, J.; Sallman, D.A.; Zhang, Y.; Tu, N.; et al. S100A9-induced overexpression of PD-1/PD-L1 contributes to ineffective hematopoiesis in myelodysplastic syndromes. Leukemia 2019, 33, 2034–2046. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontenay, M.; Farhat, B.; Boussaid, I. Pathophysiology of Myelodysplastic Syndromes. Hemato 2021, 2, 477-495. https://doi.org/10.3390/hemato2030030
Fontenay M, Farhat B, Boussaid I. Pathophysiology of Myelodysplastic Syndromes. Hemato. 2021; 2(3):477-495. https://doi.org/10.3390/hemato2030030
Chicago/Turabian StyleFontenay, Michaela, Batoul Farhat, and Ismael Boussaid. 2021. "Pathophysiology of Myelodysplastic Syndromes" Hemato 2, no. 3: 477-495. https://doi.org/10.3390/hemato2030030
APA StyleFontenay, M., Farhat, B., & Boussaid, I. (2021). Pathophysiology of Myelodysplastic Syndromes. Hemato, 2(3), 477-495. https://doi.org/10.3390/hemato2030030