
Histone Deacetylases and Their Potential as Targets to Enhance Tumour Radiosensitisation



Abstract
:Simple Summary
Abstract
1. Introduction
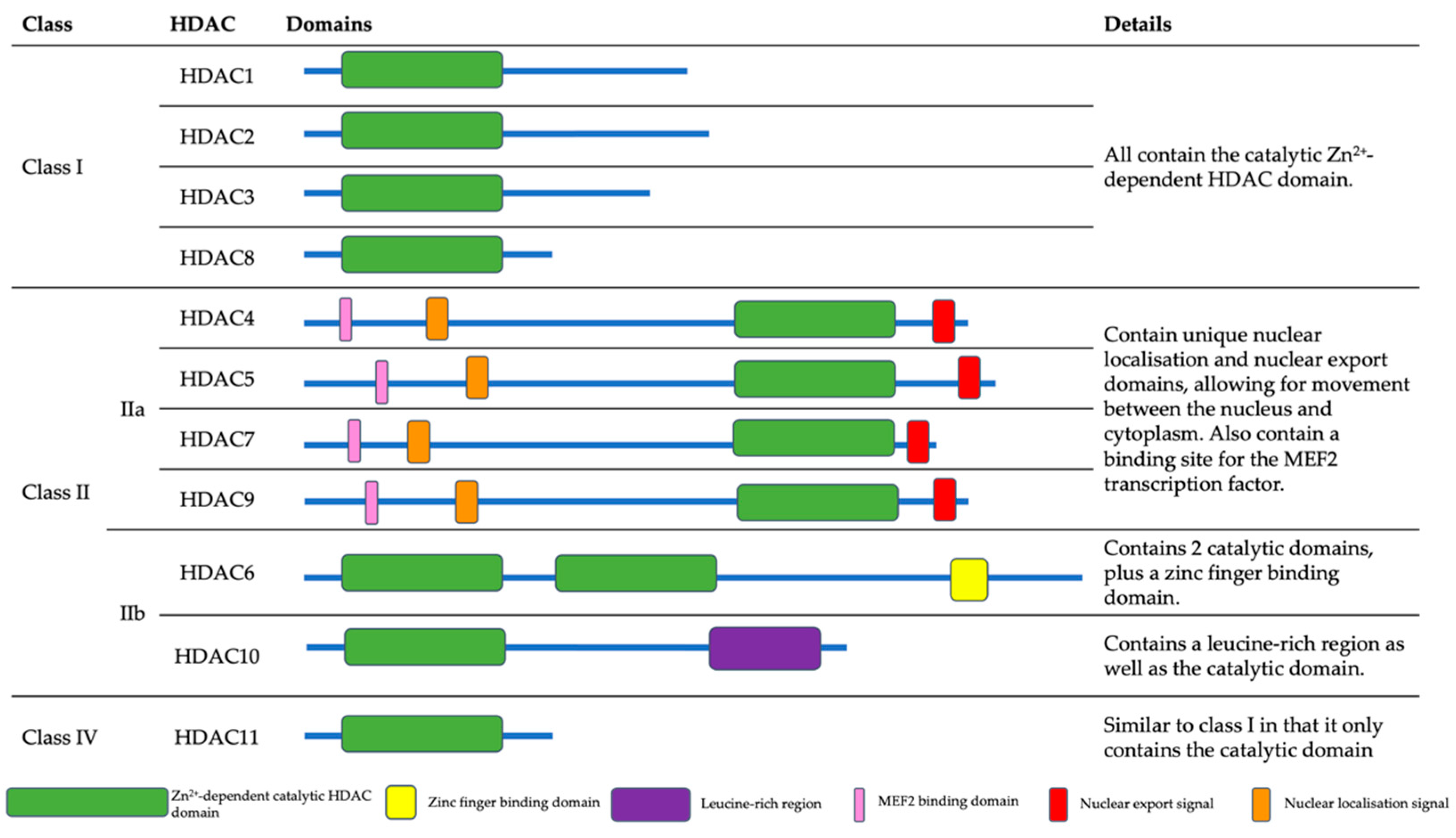
2. Histone Deacetylases (HDACs)
2.1. Class I HDACs
2.1.1. HDAC1
2.1.2. HDAC2
2.1.3. HDAC3
2.1.4. HDAC8

{kind=link}
| HDAC Class | HDAC | Cellular Targets | Tumour Association | References |
|---|---|---|---|---|
| I | HDAC1 | CoREST transcriptional complex, BRG1-RB1-HDAC1 complex, SP1/3 transcription factors, NF-κB, HIF-1α | Gastric, pancreatic, colorectal, prostate, hepatocellular, lung, breast | [26,27,28,29,30,31,32,33,34,35,36,37,43,56] |
| HDAC2 | CoREST transcriptional complex, DNMT/DMAP1, MTA1, p53 | Cervical, gastric, breast, prostate | [26,31,38,39,40,41,42,43] | |
| HDAC3 | BCL6, XBP1, AKT1, N-CoR | Acute promyelocytic leukaemia, hepatocellular, colorectal, breast | [44,45,46,47,48,49,50] | |
| HDAC8 | Telomerase, ERRα | Neuroblastoma, breast | [51,53,54,55] | |
| II | HDAC4 | HIF-1α, CDK | Oesophageal, glioma, gastric, colorectal | [57,58,59,60,61,62] |
| HDAC5 | MEF2C, MTA1, ESR1, Rb, RARA | Hepatocellular, colorectal, breast, lung | [63,64,65,66,67,68,69,70] | |
| HDAC6 | ESR1, HIF-1α, Aggresome | Oral squamous cell carcinoma, ovarian, breast | [57,63,71,72,73,74] | |
| HDAC7 | RARA, FOXP3, STAT3 | Nasopharyngeal, lung, breast, gastric | [64,75,76,77,78,79] | |
| HDAC9 | MEF2, C-Jun, Aldehyde Dehydrogenase 1A3 | Oral squamous cell carcinoma, lung, hepatocellular, breast | [80,81,82,83,84,85] | |
| HDAC10 | PTPN22, AKT, DNA Repair | Lung, colorectal, ovarian | [86,87,88,89,90,91] | |
| IV | HDAC11 | p53, cell cycle progression, glycolysis | Pituitary, neuroblastoma, hepatocellular | [92,93,94] |
2.2. Class II HDACs
2.2.1. HDAC4
2.2.2. HDAC5
2.2.3. HDAC6
2.2.4. HDAC7
2.2.5. HDAC9
2.2.6. HDAC10
2.3. Class IV HDACs
3. HDACs in the Cellular DDR
3.1. Class I HDACs
3.2. Class II HDACs
4. HDAC Inhibitors in Enhancing IR Sensitivity
4.1. Valproic Acid (VPA)
4.2. Vorinostat
4.3. CUDC-101
4.4. Panobinostat
4.5. Romidepsin
4.6. Mocetinostat
4.7. Belinostat
4.8. Abexinostat
4.9. HDACi in Response to PBT
| HDAC Inhibitor | Target | Radiosensitivity Impact | Reference |
|---|---|---|---|
| Valproic Acid | Non-Selective | Effectively radiosensitised colorectal cancer, oesophageal cancer and thyroid cancer cells to photons, and hepatocellular carcinoma cells to PBT | [114,115,116,128] |
| Vorinostat | Non-Selective | Increased sensitivity of melanoma, lung cancer, breast cancer, and colorectal cancer cells to photons | [117,118,119] |
| CUDC-101 | HDAC1–10, EGFR, HER2 | Radiosensitised pancreatic cancer, breast cancer, and glioblastoma cells to photons | [120,121] |
| Panobinostat | Non-Selective | Sensitised bladder cancer, hepatocellular carcinoma, prostate cancer cells to photons, and hepatocellular carcinoma cells to PBT | [122,123,129] |
| Romidepsin | Class I | Increases the sensitivity of bladder cancer cells to photons | [124] |
| Mocetinostat | Class I | Radiosensitised bladder cancer cells to photons | [123] |
| Belinostat | Non-Selective | Sensitises rhabdomyosarcoma, cervical cancer and colorectal cancer cell lines to photons | [125,126] |
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, C.L.; Dimitrov, S. Higher-order structure of chromatin and chromosomes. Curr. Opin. Genet. Dev. 2001, 11, 130–135. [Google Scholar] [CrossRef]
- Finch, J.T.; Klug, A. Solenoidal model for superstructure in chromatin. Proc. Nat. Acad. Sci. USA 1976, 73, 1897–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, N.; Allan, J. Supercoiling in DNA and chromatin. Curr. Opin. Genet. Dev. 2014, 25, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Allfrey, V.G.; Faulkner, R.; Mirskey, A. Acetylation and Methylation of Histones and Their Possible Role in thr Regulation of RNA Synthesis. Proc. Nat. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, V.; Richard-Foy, H. Role of Histone N-Terminal Tails and Their Acetylation in Nucleosome Dynamics. Mol. Cell Biol. 2000, 20, 7230–7237. [Google Scholar] [CrossRef] [Green Version]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef]
- Garcia-Ramirez, M.; Rocchini, C.; Ausio, J. Modulation of chromatin folding by histone acetylation. J. Biol. Chem. 1995, 270, 17923–17928. [Google Scholar] [CrossRef] [Green Version]
- Tamburini, B.A.; Tyler, J.K. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol. Cell Biol. 2005, 25, 4903–4913. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Nagaria, P.K.; Pawar, N.; Adewuyi, A.; Gojo, I.; Meyers, D.J.; Cole, P.A.; Rassool, F.V. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk. Res. 2016, 45, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.G.; So, S.; Gupta, A.; Kumar, R.; Cayrou, C.; Avvakumov, N.; Bhadra, U.; Pandita, R.K.; Porteus, M.H.; Chen, D.J.; et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell Biol. 2010, 30, 3582–3595. [Google Scholar] [CrossRef] [Green Version]
- Vitti, E.T.; Parsons, J.L. The Radiobiological Effects of Proton Beam Therapy: Impact on DNA Damage and Repair. Cancers 2019, 11, 946. [Google Scholar] [CrossRef] [Green Version]
- Carter, R.J.; Parsons, J.L. Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Mol. Cell Biol. 2016, 36, 1426–1437. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008, 7, 2902–2906. [Google Scholar] [CrossRef]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Complex DNA Damage Induced by High Linear Energy Transfer Alpha-Particles and Protons Triggers a Specific Cellular DNA Damage Response. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Characterisation of Deubiquitylating Enzymes in the Cellular Response to High-LET Ionizing Radiation and Complex DNA Damage. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 656–665. [Google Scholar] [CrossRef] [Green Version]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 15, 737–749. [Google Scholar] [CrossRef]
- Muslin, A.J.; Xing, H. 14-3-3 proteins: Regulation of subcellular localization by molecular interference. Cell Signal. 2000, 12, 703–709. [Google Scholar] [CrossRef]
- Jayathilaka, N.; Han, A.; Gaffney, K.J.; Dey, R.; Jarusiewicz, J.A.; Noridomi, K.; Philips, M.A.; Lei, X.; He, J.; Ye, J.; et al. Inhibition of the function of class IIa HDACs by blocking their interaction with MEF2. Nucleic. Acids. Res. 2012, 40, 5378–5388. [Google Scholar] [CrossRef] [Green Version]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Faller, D.V. Transcription Regulation by Class III Histone Deacetylases (HDACs)—Sirtuins. Transl. Oncogenomics 2008, 3, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.J.; Millard, C.J.; Riley, A.M.; Roberston, N.S.; Wright, L.C.; Godage, H.Y.; Cowley, S.M.; Jamieson, A.G.; Potter, B.V.L.; Schwabe, J.W.R. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nature Commun. 2016, 48, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Myers, S.J.; Dingledine, R. Transcriptional repression by REST: Recruitment of Sin3A and histone deacetylase to neuronal genes. Nat. Neurosci. 1999, 2, 867–872. [Google Scholar] [CrossRef]
- Qiu, Z.; Ghosh, A. A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron 2008, 60, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Ammanamanchi, S.; Freeman, J.W.; Brattain, M.G. Acetylated sp3 is a transcriptional activator. J. Biol. Chem. 2003, 278, 35775–35780. [Google Scholar] [CrossRef] [Green Version]
- Hung, J.J.; Wang, Y.T.; Chang, W.C. Sp1 deacetylation induced by phorbol ester recruits p300 to activate 12(S)-lipoxygenase gene transcription. Mol. Cell Biol. 2006, 26, 1770–1785. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, B.P.; Westerheide, S.D.; Baldwin, A.S., Jr. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell Biol. 2001, 21, 7065–7077. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.A.; Pross, M.; Dietel, M.; Denkert, C.; Röcken, C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef]
- Choi, J.-H.; Kwon, H.J.; Yoon, B.-I.; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Yoshizumi, T.; Imura, S.; Sugimoto, K.; Batmunkh, E.; Kanemura, H.; Morine, Y.; Shimada, M. Expression of Hypoxia-Inducible Factor-1α, Histone Deacetylase 1, and Metastasis-Associated Protein 1 in Pancreatic Carcinoma.Correlation With Poor Prognosis With Possible Regulation. Pancreas 2008, 36, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef]
- Rikimaru, T.; Taketomi, A.; Yamashita, Y.; Shirabe, K.; Hamatsu, T.; Shimada, M.; Maehara, Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology 2007, 72, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.L.; Song, X.; Pei, L.; Liu, L.; Wang, H.; Jia, M. Histone deacetylase HDAC1 expression correlates with the progression and prognosis of lung cancer: A meta-analysis. Medicine 2017, 96, e7663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef]
- Rountree, M.R.; Bachman, K.E.; Baylin, S.B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 2000, 25, 269–277. [Google Scholar] [CrossRef]
- Cong, L.; Pakala, S.B.; Ohshiro, K.; Li, D.Q.; Kumar, R. SUMOylation and SUMO-interacting motif (SIM) of metastasis tumor antigen 1 (MTA1) synergistically regulate its transcriptional repressor function. J. Biol. Chem. 2011, 286, 43793–43808. [Google Scholar] [CrossRef] [Green Version]
- Harms, K.L.; Chen, X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007, 67, 3145–3152. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.H.; Laban, M.; Leung, C.H.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death. Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.; Dietel, M.; Weichert, W.; Denkert, C. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer—Overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzi, K.; Jiang, Y.; Huang, C.; Garrett-Bakelman, F.; Gearhart, M.D.; Giannopoulou, E.G.; Zumbo, P.; Kirouac, K.; Bhaskara, S.; Polo, J.M.; et al. A hybrid mechanism of action for BCL6 in B cells defined by formation of functionally distinct complexes at enhancers and promoters. Cell Rep. 2013, 4, 578–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.; Li, Y.; Yang, J.; Wang, G.; Margariti, A.; Jiang, Z.; Yu, H.; Zampetaki, A.; Hu, Y.; Xu, Q.; et al. Unspliced X-box-binding protein 1 (XBP1) protects endothelial cells from oxidative stress through interaction with histone deacetylase 3. J. Biol. Chem. 2014, 289, 30625–30634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsumi, A.; Tomita, A.; Kiyoi, H.; Naoe, T. Histone deacetylase 3 (HDAC3) is recruited to target promoters by PML-RARalpha as a component of the N-CoR co-repressor complex to repress transcription in vivo. Biochem. Biophys. Res. Commun. 2006, 345, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Zhou, Y.; Zhuang, X.; Zhu, Y.; Wu, Z.; Lu, Y.; Li, S.; Zeng, Y.; Lu, Q.R.; Huo, Y.; et al. HDAC3 Deficiency Promotes Liver Cancer through a Defect in H3K9ac/H3K9me3 Transition. Cancer Res. 2019, 79, 3676–3688. [Google Scholar] [CrossRef]
- Spurling, C.C.; Godman, C.A.; Noonan, E.J.; Rasmussen, T.P.; Rosenberg, D.W.; Giardina, C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol. Carcinog. 2008, 47, 137–147. [Google Scholar] [CrossRef]
- Li, J.; Hu, M.; Liu, N.; Li, H.; Yu, Z.; Yan, Q.; Zhou, M.; Wang, Y.; Song, Y.; Pan, G.; et al. HDAC3 deteriorates colorectal cancer progression via microRNA-296-3p/TGIF1/TGFbeta axis. J. Exp. Clin Cancer Res. 2020, 39, 248. [Google Scholar] [CrossRef]
- Cui, Z.; Xie, M.; Wu, Z.; Shi, Y. Relationship Between Histone Deacetylase 3 (HDAC3) and Breast Cancer. Med. Sci. Monit. 2018, 24, 2456–2464. [Google Scholar] [CrossRef]
- Lee, H.; Sengupta, N.; Villagra, A.; Rezai-Zadeh, N.; Seto, E. Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (hEST1B) protein from ubiquitin-mediated degradation. Mol. Cell Biol. 2006, 26, 5259–5269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.J.; Tremblay, A.M.; Deblois, G.; Sylvain-Drolet, G.; Giguere, V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol. Endocrinol. 2010, 24, 1349–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [Green Version]
- An, P.; Chen, F.; Li, Z.; Ling, Y.; Peng, Y.; Zhang, H.; Li, J.; Chen, Z.; Wang, H. HDAC8 promotes the dissemination of breast cancer cells via AKT/GSK-3beta/Snail signals. Oncogene 2020, 39, 4956–4969. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Roske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006, 66, 8814–8821. [Google Scholar] [CrossRef] [Green Version]
- Bryant, D.W.; Haynes, R.H. Endonuclease a from Saccharomyces cerevisiae shows increased activity on ultraviolet irradiated native DNA. Mol. Gen. Genet. 1978, 167, 139–145. [Google Scholar] [CrossRef]
- Zeng, L.-S.; Yang, X.-Z.; Wen, Y.-F.; Mai, S.-J.; Wang, M.-H.; Zhang, M.-Y.; Zheng, X.F.S.; Wang, H.-Y. Overexpressed HDAC4 is associated with poor survival and promotes tumor progression in esophageal carcinoma. Aging 2016, 8, 1236–1248. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.Y.; Xu, T.T.; Wang, Y.; Chang, J.J.; Li, J.; Chen, X.Y.; Chen, X.; Yin, Y.F.; Ni, X.J. Histone deacetylase HDAC4 promotes the proliferation and invasion of glioma cells. Int. J. Oncol. 2018, 53, 2758–2768. [Google Scholar] [CrossRef] [Green Version]
- Kang, Z.H.; Wang, C.Y.; Zhang, W.L.; Zhang, J.T.; Yuan, C.H.; Zhao, P.W.; Lin, Y.Y.; Hong, S.; Li, C.Y.; Wang, L. Histone deacetylase HDAC4 promotes gastric cancer SGC-7901 cells progression via p21 repression. PLoS ONE 2014, 9, e98894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.J.; Byun, D.-S.; Nasser, S.; Murray, L.B.; Auyyanar, K.; Arango, D.; Figueroa, M.; Melnick, A.; Kao, G.D.; Augenlicht, L.H.; et al. HDAC4 Promotes Growth of Colon Cancer Cells via Repression of p21. Mol. Biol. Cell 2008, 19, 4062–4075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Lee, M.H.; Kang, H.L.; Kim, S.H.; Ahn, J.R.; Na, H.; Na, T.Y.; Kim, Y.N.; Seong, J.K.; Lee, M.O. Differential regulation of estrogen receptor alpha expression in breast cancer cells by metastasis-associated protein 1. Cancer Res. 2014, 74, 1484–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.Y.; Lin, T.E.; Lee, C.I.; Zhou, J.; Huang, Y.H.; Lee, P.L.; Shih, Y.T.; Chien, S.; Chiu, J.J. MicroRNA-10a is crucial for endothelial response to different flow patterns via interaction of retinoid acid receptors and histone deacetylases. Proc. Natl. Acad. Sci. USA 2017, 114, 2072–2077. [Google Scholar] [CrossRef] [Green Version]
- Wein, M.N.; Spatz, J.; Nishimori, S.; Doench, J.; Root, D.; Babij, P.; Nagano, K.; Baron, R.; Brooks, D.; Bouxsein, M.; et al. HDAC5 controls MEF2C-driven sclerostin expression in osteocytes. J. Bone. Min. Res. 2015, 30, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Jin, X.; Ma, J.; Ding, D.; Huang, Z.; Sheng, H.; Yan, Y.; Pan, Y.; Wei, T.; Wang, L.; et al. HDAC5 Loss Impairs RB Repression of Pro-Oncogenic Genes and Confers CDK4/6 Inhibitor Resistance in Cancer. Cancer Res. 2021, 81, 1486–1499. [Google Scholar] [CrossRef]
- Feng, G.-W.; Dong, L.-D.; Shang, W.-J.; Pang, X.-L.; Li, J.F.; Liu, L.; Wang, Y. HDAC5 promotes cell proliferation in human hepatocellular carcinoma by up-regulating Six1 expression. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 811–816. [Google Scholar]
- He, P.; Liang, J.; Shau, T.; Guo, Y.; Hou, Y.; Li, Y. HDAC5 promotes colorectal cancer cell proliferation by up-regulating DLL4 expression. Int. J. Clin Exp. Med. 2015, 8, 6510–6516. [Google Scholar]
- Li, A.; Liu, Z.; Li, M.; Zhou, S.; Xu, Y.; Xiao, Y.; Yang, W. HDAC5, a potential therapeutic target and prognostic biomarker, promotes proliferation, invasion and migration in human breast cancer. Oncotarget 2016, 7, 37966–37978. [Google Scholar] [CrossRef]
- Zhong, L.; Sun, S.; Yao, S.; Han, X.; Gu, M.; Shi, J. Histone deacetylase 5 promotes the proliferation and invasion of lung cancer cells. Oncol. Rep. 2018, 40, 2224–2232. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [Green Version]
- Bazzaro, M.; Lin, Z.; Santillan, A.; Lee, M.K.; Wang, M.C.; Chan, K.C.; Bristow, R.E.; Mazitschek, R.; Bradner, J.; Roden, R.B. Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin. Cancer Res. 2008, 14, 7340–7347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.; Iwase, H. HDAC6 expression is correlated with better survival in breast cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, T.; Uzawa, K.; Onda, T.; Shiiba, M.; Yokoe, H.; Shiabahara, T.; Tanzawa, H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int. J. Oncol. 2006, 29, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Liu, L.; Zhang, S.; Guo, S.; Li, X.; Wang, J.; Su, B.; Fang, Y.; Chen, X.; Ke, H.; et al. Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation. Mol. Cancer 2017, 16, 170. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Samanta, A.; Song, X.; Iacono, K.T.; Bembas, K.; Tao, R.; Basu, S.; Riley, J.L.; Hancock, W.W.; Shen, Y.; et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc. Natl. Acad. Sci. USA 2007, 104, 4571–4576. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.G.; Xiao, T.; Zhu, W.; Yu, Z.Z.; Huang, X.P.; Yi, H.; Lu, S.S.; Tang, Y.Y.; Huang, W.; Xiao, Z.Q. HDAC7 promotes the oncogenicity of nasopharyngeal carcinoma cells by miR-4465-EphA2 signaling axis. Cell Death. Dis. 2020, 11, 322. [Google Scholar] [CrossRef]
- Uzelac, B.; Krivokuca, A.; Susnjar, S.; Milovanovic, Z.; Supic, G. Histone Deacetylase 7 Gene Overexpression Is Associated with Poor Prognosis of Triple-Negative Breast Cancer Patients. Genet. Test Mol. Biomark. 2021, 25, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cao, F.; Yu, X.; Zhou, P.; Di, Q.; Lei, J.; Tai, Y.; Wu, H.; Li, X.; Wang, X.; et al. The expression of HDAC7 in cancerous gastric tissues is positively associated with distant metastasis and poor patient prognosis. Clin. Transl. Oncol. 2017, 19, 1045–1054. [Google Scholar] [CrossRef]
- Haberland, M.; Arnold, M.A.; McAnally, J.; Phan, D.; Kim, Y.; Olson, E.N. Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol. Cell Biol. 2007, 27, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Kanki, K.; Watanabe, R.; Nguyen Thai, L.; Zhao, C.H.; Naito, K. HDAC9 Is Preferentially Expressed in Dedifferentiated Hepatocellular Carcinoma Cells and Is Involved in an Anchorage-Independent Growth. Cancers 2020, 12, 2734. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wu, Y.; Tang, X.; Xia, Y.; He, G.; Min, Z.; Li, C.; Xiong, S.; Shi, Z.; Lu, Y.; et al. HDAC inhibitors suppress c-Jun/Fra-1-mediated proliferation through transcriptionally downregulating MKK7 and Raf1 in neuroblastoma cells. Oncotarget 2016, 7, 6727–6747. [Google Scholar] [CrossRef] [PubMed]
- Okudela, K.; Mitsui, H.; Suzuki, T.; Woo, T.; Tateishi, Y.; Umeda, S.; Saito, Y.; Tajiri, M.; Masuda, M.; Ohashi, K. Expression of HDAC9 in lung cancer—Potential role in lung carcinogenesis. Int. J. Clin. Exp. Pathol. 2014, 7, 213–220. [Google Scholar] [PubMed]
- Rastogi, B.; Raut, S.K.; Panda, N.K.; Rattan, V.; Radotra, B.D.; Khullar, M. Overexpression of HDAC9 promotes oral squamous cell carcinoma growth, regulates cell cycle progression, and inhibits apoptosis. Mol. Cell Biochem. 2016, 415, 183–196. [Google Scholar] [CrossRef]
- Salgado, E.; Bian, X.; Feng, A.; Shim, H.; Liang, Z. HDAC9 overexpression confers invasive and angiogenic potential to triple negative breast cancer cells via modulating microRNA-206. Biochem. Biophys. Res. Commun. 2018, 503, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Ridinger, J.; Koeneke, E.; Kolbinger, F.R.; Koerholz, K.; Mahboobi, S.; Hellweg, L.; Gunkel, N.; Miller, A.K.; Peterziel, H.; Schmezer, P.; et al. Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Sci. Rep. 2018, 8, 10039. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Yan, Y.; Lu, L.; Chen, B. HDAC10 expression is associated with DNA mismatch repair gene and is a predictor of good prognosis in colon carcinoma. Oncol. Lett. 2017, 14, 4923–4929. [Google Scholar] [CrossRef] [Green Version]
- Duan, B.; Ye, D.; Zhu, S.; Jia, W.; Lu, C.; Wang, G.; Guo, X.; Yu, Y.; Wu, C.; Kang, J. HDAC10 promotes angiogenesis in endothelial cells through the PTPN22/ERK axis. Oncotarget 2017, 8, 61338–61349. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, Y.; Wang, Z.; Wang, H.-T.; Duan, B.; Ye, D.; Wang, C.; Jing, R.; Leng, Y.; Zi, J.; et al. HDAC10 promotes lung cancer proliferation via AKT phosphorylation. Oncotarget 2016, 7, 59388–59401. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.M.; Banerjee, T.; Packard, C.Z.; Kotian, S.; Selvendiran, K.; Cohn, D.E.; Parvin, J.D. HDAC10 as a potential therapeutic target in ovarian cancer. Gynecol. Oncol. 2017, 144, 613–620. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Zhang, R.; Jin, T.; Qu, L.; Jin, Q.; Zheng, J.; Sun, J.; Wu, Z.; Wang, L.; et al. HDAC10 Is Positively Associated With PD-L1 Expression and Poor Prognosis in Patients With NSCLC. Front. Oncol. 2020, 10, 485. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Ren, Y.; Feng, M.; Meng, P.; Wang, Q.; Chen, W.; Jiao, Q.; Wang, Y.; Du, L.; Zhou, F.; et al. HDAC11 Regulates Glycolysis through the LKB1/AMPK Signaling Pathway to Maintain Hepatocellular Carcinoma Stemness. Cancer Res. 2021, 81, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Thole, T.M.; Lodrini, M.; Fabian, J.; Wuenschel, J.; Pfeil, S.; Hielscher, T.; Kopp-Schneider, A.; Heinicke, U.; Fulda, S.; Witt, O.; et al. Neuroblastoma cells depend on HDAC11 for mitotic cell cycle progression and survival. Cell Death. Dis. 2017, 8, e2635. [Google Scholar] [CrossRef]
- Wang, W.; Fu, L.; Li, S.; Xu, Z.; Li, X. Histone deacetylase 11 suppresses p53 expression in pituitary tumor cells. Cell Biol. Int. 2017, 41, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Lemercier, C.; Verdel, A.; Galloo, B.; Curtet, S.; Brocard, M.P.; Khochbin, S. mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J. Biol. Chem. 2000, 275, 15594–15599. [Google Scholar] [CrossRef] [Green Version]
- Dressel, U.; Bailey, P.J.; Wang, S.C.; Downes, M.; Evans, R.M.; Muscat, G.E. A dynamic role for HDAC7 in MEF2-mediated muscle differentiation. J. Biol. Chem. 2001, 276, 17007–17013. [Google Scholar] [CrossRef] [Green Version]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic Activity Associated with Class IIHDACs Is Dependent on a Multiprotein ComplexContaining HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–47. [Google Scholar] [CrossRef] [Green Version]
- Parra, M. Class IIa HDACs—New insights into their functions in physiology and pathology. FEBS J. 2015, 282, 1736–1744. [Google Scholar] [CrossRef]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1a by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J. Biol. Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottet, D.; Bellahcene, A.; Pirotte, S.; Waltregny, D.; Deroanne, C.; Lamour, V.; Lidereau, R.; Castronovo, V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ. Res. 2007, 101, 1237–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhu, W.G. Targeting histone deacetylases for cancer therapy: From molecular mechanisms to clinical implications. Int. J. Biol. Sci. 2014, 10, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.-W.; Hiebert, S.W. Deletion of Histone Deacetylase 3 reveals critical roles in S-phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Kao, G.D.; McKenna, W.G.; Guenther, M.G.; Muschel, R.J.; Lazar, M.A.; Yen, T.J. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J. Cell Biol. 2003, 160, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Hu, C.; Moses, N.; Haakenson, J.; Xiang, S.; Quan, D.; Fang, B.; Yang, Z.; Bai, W.; Bepler, G.; et al. HDAC6 regulates DNA damage response via deacetylating MLH1. J. Biol. Chem. 2019, 294, 5813–5826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xiang, S.; Joo, H.Y.; Wang, L.; Williams, K.A.; Liu, W.; Hu, C.; Tong, D.; Haakenson, J.; Wang, C.; et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSalpha. Mol. Cell 2014, 55, 31–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotian, S.; Liyanarachchi, S.; Zelent, A.; Parvin, J.D. Histone deacetylases 9 and 10 are required for homologous recombination. J. Biol. Chem. 2011, 286, 7722–7726. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wong, P.; Radany, E.; Wong, J.Y.C. HDAC Inhibitor, Valproic Acid, Induces p53-Dependent Radiosensitization of Colon Cancer Cells. Cancer Biother. 2009, 24, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoji, M.; Ninomiya, I.; Makino, I.; Kinoshita, J.; Nakamura, K.; Oyama, K.; Nakagawara, H.; Fujita, H.; Tajima, H.; Takamura, H.; et al. Valproic acid, a histone deacetylase inhibitor, enhances radiosensitivity in esophageal squamous cell carcinoma. Int. J. Oncol. 2012, 40, 2140–2146. [Google Scholar] [CrossRef] [Green Version]
- Perona, M.; Thomasz, L.; Rossich, L.; Rodriguez, C.; Pisarev, M.A.; Rosemblit, C.; Cremaschi, G.A.; Dagrosa, M.A.; Juvenal, G.J. Radiosensitivity enhancement of human thyroid carcinoma cells by the inhibitors of histone deacetylase sodium butyrate and valproic acid. Mol. Cell Endocrinol. 2018, 478, 141–150. [Google Scholar] [CrossRef]
- Munshi, A.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Richon, V.M.; Meyn, R.E. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol. Cancer Ther. 2006, 5, 1967–1974. [Google Scholar] [CrossRef] [Green Version]
- Baschnagel, A.; Russo, A.; Burgan, W.E.; Carter, D.; Beam, K.; Palmieri, D.; Steeg, P.S.; Tofilon, P.; Camphausen, K. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol. Cancer Ther. 2009, 8, 1589–1595. [Google Scholar] [CrossRef] [Green Version]
- Saelen, M.G.; Ree, A.H.; Kristian, A.; Fleten, K.G.; Furee, T.; Hektoen, H.H.; Flatmark, K. Radiosensitization by the histone deacetylase inhibitor vorinostat under hypoxia and with capecitabine in experimental colorectal carcinoma. Radiat. Oncol. 2012, 7, 165. [Google Scholar] [CrossRef] [Green Version]
- Moertl, S.; Payer, S.; Kell, R.; Winkler, K.; Anastasov, N.; Atkinson, M.J. Comparison of Radiosensitization by HDAC Inhibitors CUDC-101 and SAHA in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3295. [Google Scholar] [CrossRef] [Green Version]
- Schlaff, C.D.; Arscott, W.T.; Gordon, I.; Tandle, A.; Tofilon, P.; Camphausen, K. Radiosensitization Effects of Novel Triple-Inhibitor CUDC-101 in Glioblastoma Multiforme and Breast Cancer Cells In Vitro. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, S650. [Google Scholar] [CrossRef]
- Xiao, W.; Graham, P.H.; Hao, J.; Chang, L.; Ni, J.; Power, C.A.; Dong, Q.; Kearsley, J.H.; Li, Y. Combination therapy with the histone deacetylase inhibitor LBH589 and radiation is an effective regimen for prostate cancer cells. PLoS ONE 2013, 8, e74253. [Google Scholar] [CrossRef] [Green Version]
- Groselj, B.; Ruan, J.L.; Scott, H.; Gorrill, J.; Nicholson, J.; Kelly, J.; Anbalagan, S.; Thompson, J.; Stratford, M.R.L.; Jevons, S.J.; et al. Radiosensitization In Vivo by Histone Deacetylase Inhibition with No Increase in Early Normal Tissue Radiation Toxicity. Mol. Cancer Ther. 2018, 17, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paillas, S.; Then, C.K.; Kilgas, S.; Ruan, J.L.; Thompson, J.; Elliott, A.; Smart, S.; Kiltie, A.E. The Histone Deacetylase Inhibitor Romidepsin Spares Normal Tissues While Acting as an Effective Radiosensitizer in Bladder Tumors In Vivo. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Marampon, F.; Di Nisio, V.; Pietrantoni, I.; Petragnano, F.; Fasciani, I.; Scicchitano, B.M.; Ciccarelli, C.; Gravina, G.L.; Festuccia, C.; Del Fattore, A.; et al. Pro-differentiating and radiosensitizing effects of inhibiting HDACs by PXD-101 (Belinostat) in in vitro and in vivo models of human rhabdomyosarcoma cell lines. Cancer Lett. 2019, 461, 90–101. [Google Scholar] [CrossRef]
- Banuelos, C.A.; Banath, J.P.; MacPhail, S.H.; Zhao, J.; Reitsema, T.; Olive, P.L. Radiosensitization by the histone deacetylase inhibitor PCI-24781. Clin Cancer Res. 2007, 13, 6816–6826. [Google Scholar] [CrossRef] [Green Version]
- Rivera, S.; Leteur, C.; Mégnin, F.; Law, F.; Martins, I.; Kloos, I.; Depil, S.; Modjtahedi, N.; Perfettini, J.L.; Hennequin, C.; et al. Time dependent modulation of tumor radiosensitivity by a pan HDAC inhibitor: Abexinostat. Oncotarget 2017, 8, 56210–56227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.I.; Choi, C.; Shin, S.W.; Son, A.; Lee, G.H.; Kim, S.Y.; Park, H.C. Valproic Acid Sensitizes Hepatocellular Carcinoma Cells to Proton Therapy by Suppressing NRF2 Activation. Sci. Rep. 2017, 7, 14986. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Lee, G.H.; Son, A.; Yoo, G.S.; Yu, J.I.; Park, H.C. Downregulation of Mcl-1 by Panobinostat Potentiates Proton Beam Therapy in Hepatocellular Carcinoma Cells. Cells 2021, 10, 554. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antrobus, J.; Parsons, J.L. Histone Deacetylases and Their Potential as Targets to Enhance Tumour Radiosensitisation. Radiation 2022, 2, 149-167. https://doi.org/10.3390/radiation2010011
Antrobus J, Parsons JL. Histone Deacetylases and Their Potential as Targets to Enhance Tumour Radiosensitisation. Radiation. 2022; 2(1):149-167. https://doi.org/10.3390/radiation2010011
Chicago/Turabian StyleAntrobus, Jennifer, and Jason L. Parsons. 2022. "Histone Deacetylases and Their Potential as Targets to Enhance Tumour Radiosensitisation" Radiation 2, no. 1: 149-167. https://doi.org/10.3390/radiation2010011
APA StyleAntrobus, J., & Parsons, J. L. (2022). Histone Deacetylases and Their Potential as Targets to Enhance Tumour Radiosensitisation. Radiation, 2(1), 149-167. https://doi.org/10.3390/radiation2010011