



A Long Journey into the Investigation of the Structure–Dynamics–Function Paradigm in Proteins through the Activities of the Palermo Biophysics Group


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Protein Dynamics: Equilibrium Fluctuations, Dynamics in Confinement, and Structural Relaxation
1.1. Protein Equilibrium Fluctuations
1.2. Dynamics of Proteins in Confinement
1.3. Structural Relaxations
1.4. Myoglobin Structure–Dynamics–Function with Simulations
2. Protein Aggregation Processes
2.1. Kinetics of Protein Aggregation

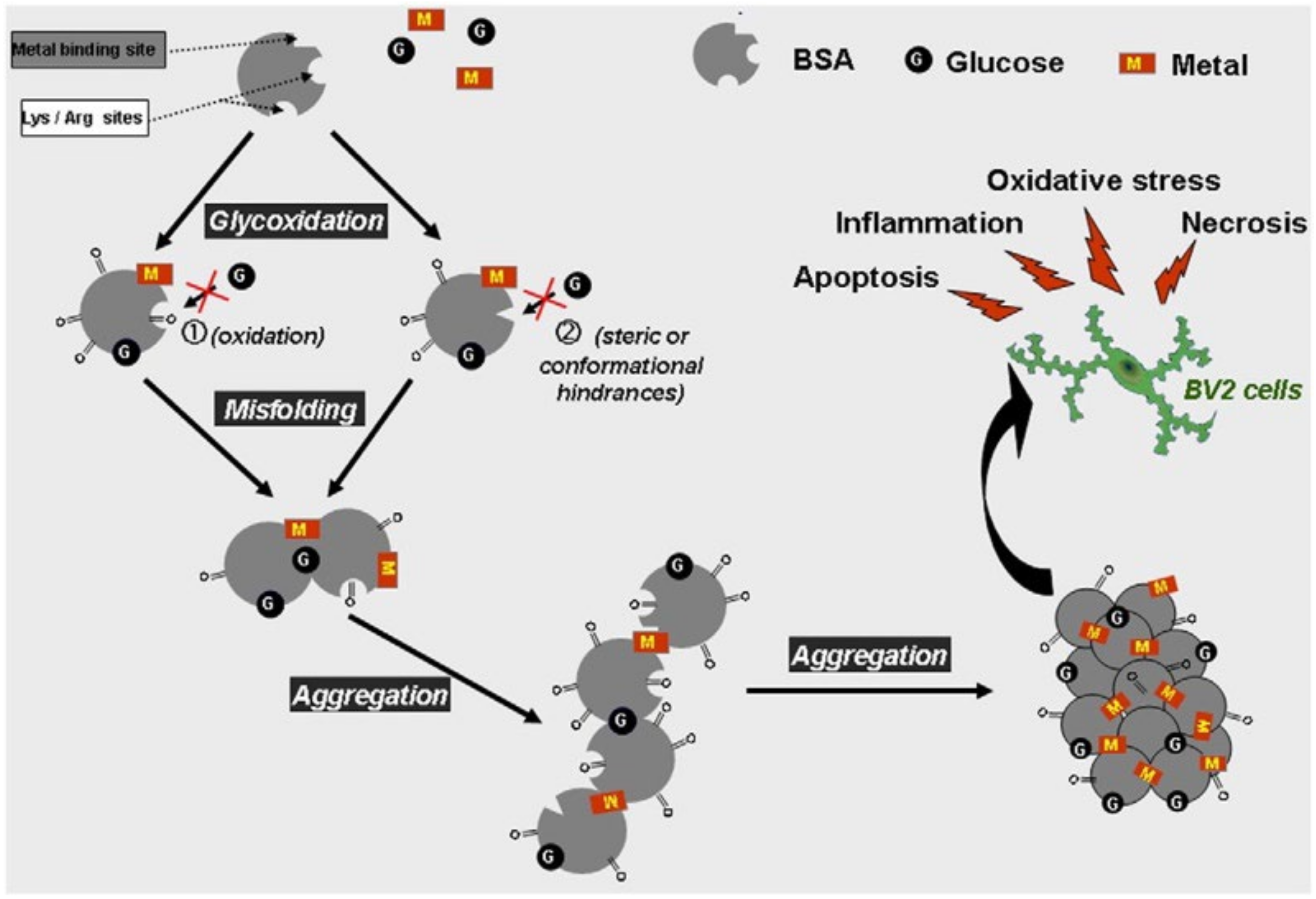
2.2. The Role of Glycation in Protein Aggregation
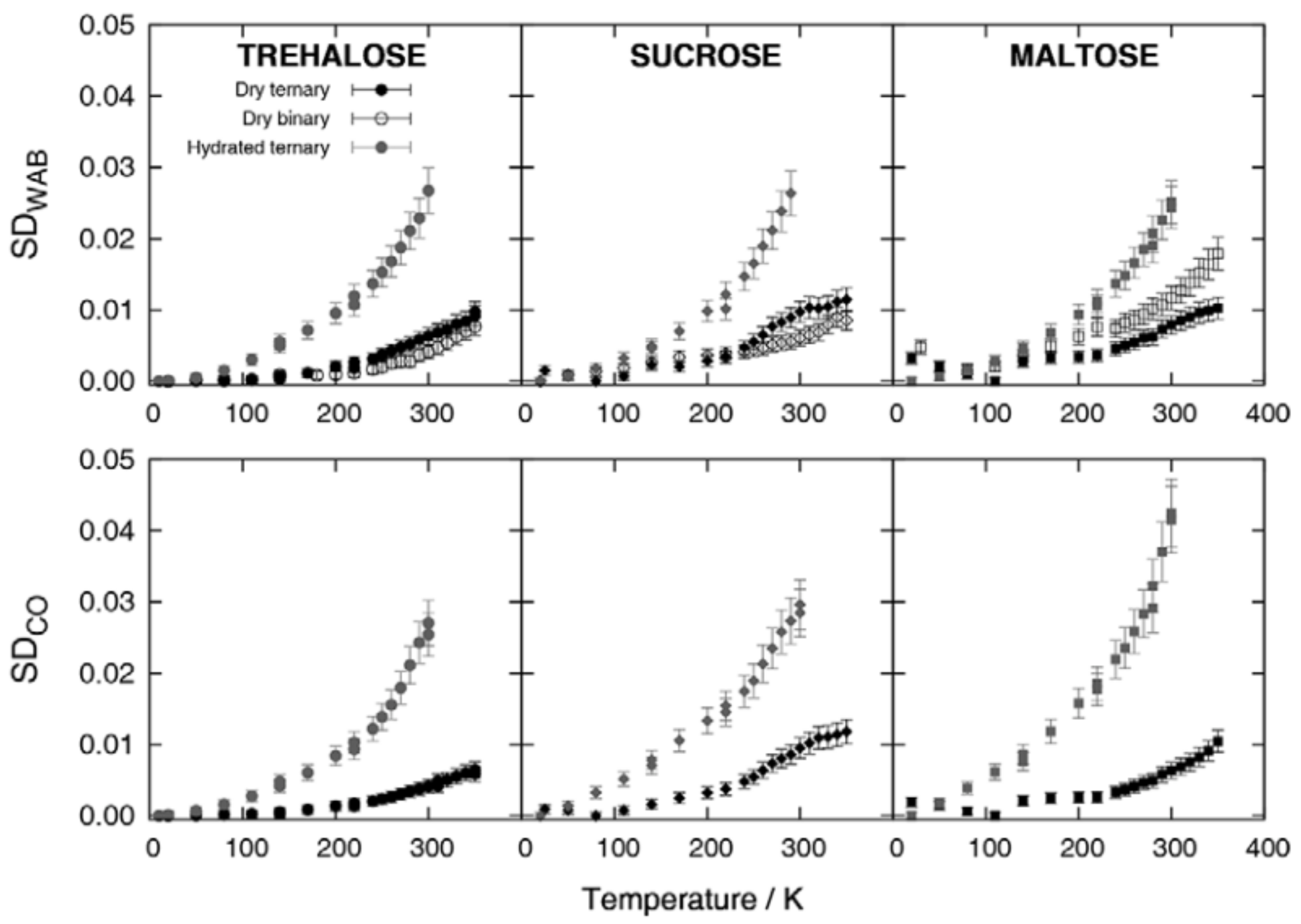
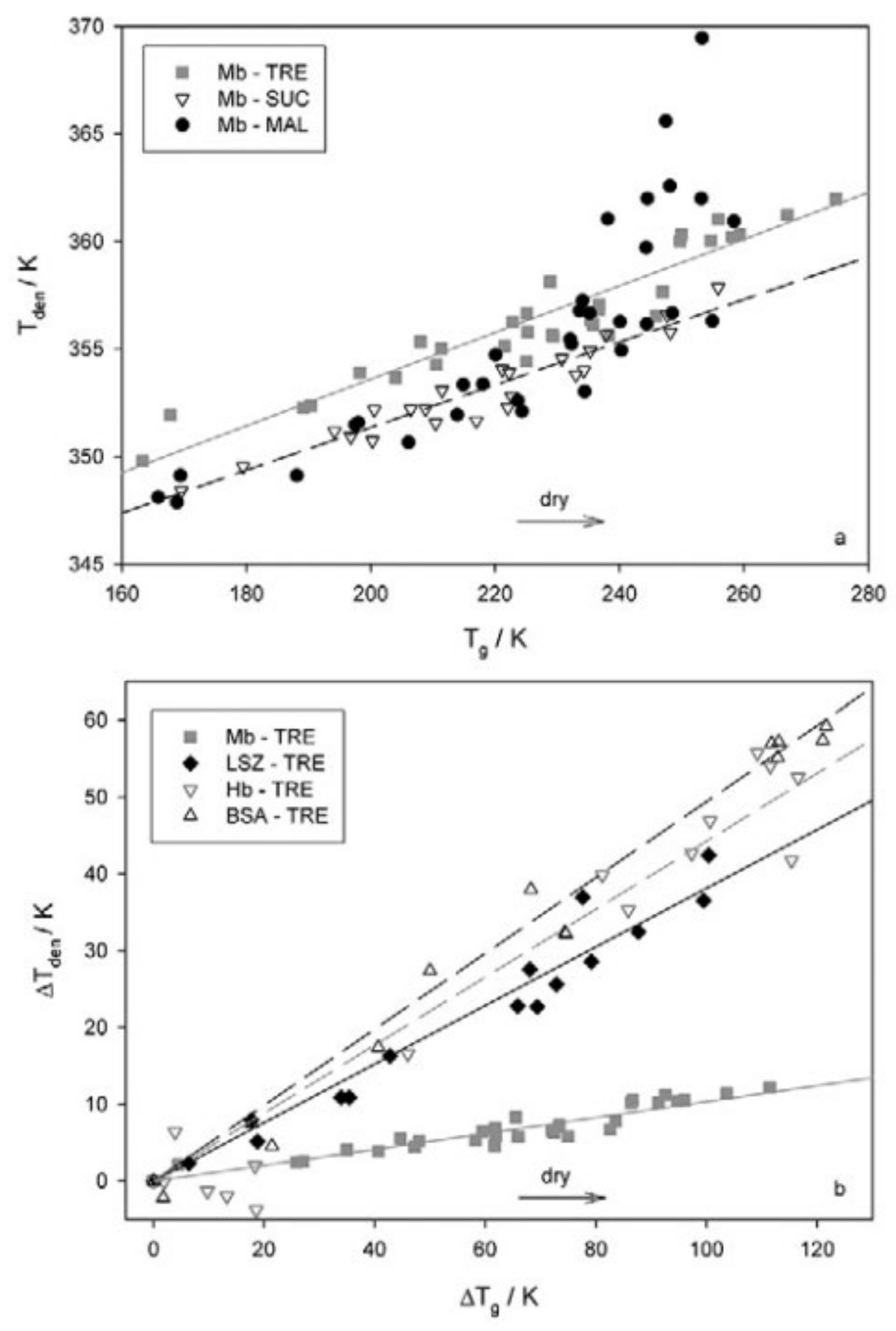
3. Proteins in Extreme Conditions: Bioprotection by Saccharides and the Trehalose Peculiarity
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- San Biagio, P.L.; Vitrano, E.; Cupane, A.; Madonia, F. Temperature induced difference spectra of Oxy and Deoxy hemoglobin in the near IR, visible and soret regions. Biochem. Biophys. Res. Commun. 1977, 77, 1158–1165. [Google Scholar] [CrossRef]
- Leone, M.; Cupane, A.; Vitrano, E.; Cordone, L. Dynamic properties of oxy-and carbonmonoxyhemoglobin probed by optical spectroscopy in the temperature range of 300–20 K. Biopolym. Orig. Res. Biomol. 1987, 26, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Cordone, L.; Cupane, A.; Leone, M.; Vitrano, E.; Bulone, D. Interaction between external medium and haem pocket in myoglobin probed by low-temperature optical spectroscopy. J. Mol. Biol. 1988, 199, 213–218. [Google Scholar] [CrossRef]
- Cupane, A.; Leone, M.; Vitrano, E.; Cordone, L. Optical absorption spectra of azurin and stellacyanin in glycerol/water and ethylene glycol/water solutions in the temperature range 290–20 K. Biophys. Chem. 1990, 38, 213–224. [Google Scholar] [CrossRef]
- Cupane, A.; Leone, M.; Militello, V.; Stroppolo, M.E.; Polticelli, F.; Desideri, A. Low-temperature optical spectroscopy of native and azide-reacted bovine Cu, Zn superoxide dismutase. A structural dynamics study. Biochemistry 1994, 33, 15103–15109. [Google Scholar] [CrossRef]
- Vitrano, E.; Cupane, A.; Leone, M.; Militello, V.; Cordone, L.; Salvato, B.; Beltramini, M.; Bubacco, L.; Rocco, G.P. Low temperature optical spectroscopy of cobalt-substituted hemocyanin from Carcinus maenas. Eur. Biophys. J. 1993, 22, 157–167. [Google Scholar] [CrossRef]
- Di Pace, A.; Cupane, A.; Leone, M.; Vitrano, E.; Cordone, L. Protein dynamics. Vibrational coupling, spectral broadening mechanisms, and anharmonicity effects in carbonmonoxy heme proteins studied by the temperature dependence of the Soret band lineshape. Biophys. J. 1992, 63, 475–484. [Google Scholar] [CrossRef][Green Version]
- Cupane, A.; Cammarata, M.; Cordone, L.; Leone, M.; Vitrano, E.; Engler, N.; Parak, F. Spectral broadening of the Soret band in myoglobin: An interpretation by the full spectrum of low-frequency modes from a normal modes analysis. Eur. Biophys. J. 2005, 34, 881–889. [Google Scholar] [CrossRef]
- Cupane, A.; Leone, M.; Vitrano, E. Protein dynamics: Conformational disorder, vibrational coupling and anharmonicity in deoxy-hemoglobin and myoglobin. Eur. Biophys. J. 1993, 21, 385–391. [Google Scholar] [CrossRef]
- Cupane, A.; Leone, M.; Vitrano, E.; Cordone, L. Low temperature optical absorption spectroscopy: An approach to the study of stereodynamic properties of hemeproteins. Eur. Biophys. J. 1995, 23, 385–398. [Google Scholar] [CrossRef]
- Frauenfelder, H.; Petsko, G.A.; Tsernoglou, D. Temperature-dependent x-ray diffraction as a probe of protein structural dynamics. Nature 1979, 280, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Melchers, B.; Knapp, E.W.; Parak, F.; Cordone, L.; Cupane, A.; Leone, M. Structural fluctuations of myoglobin from normal-modes, Mössbauer, Raman, and absorption spectroscopy. Biophys. J. 1996, 70, 2092–2099. [Google Scholar] [CrossRef][Green Version]
- Di Iorio, E.E.; Hiltpold, U.R.; Filipovic, D.; Winterhalter, K.H.; Gratton, E.; Vitrano, E.; Cupane, A.; Leone, M.; Cordone, L. Protein dynamics. Comparative investigation on heme-proteins with different physiological roles. Biophys. J. 1991, 59, 742–754. [Google Scholar] [CrossRef][Green Version]
- Cupane, A.; Leone, M.; Vitrano, E.; Cordone, L.; Hiltpold, U.R.; Winterhalter, K.H.; Yu, W.; Di Iorio, E.E. Structure-dynamics-function relationships in Asian elephant (Elephas maximus) myoglobin. An optical spectroscopy and flash photolysis study on functionally important motions. Biophys. J. 1993, 65, 2461–2472. [Google Scholar] [CrossRef]
- Boffi, A.; Verzili, D.; Chiancone, E.; Leone, M.; Cupane, A.; Militello, V.; Vitrano, E.; Cordone, L.; Yu, W.; Di Iorio, E.E. Stereodynamic properties of the cooperative homodimeric Scapharca inaequivalvis hemoglobin studied through optical absorption spectroscopy and ligand rebinding kinetics. Biophys. J. 1994, 67, 1713–1723. [Google Scholar] [CrossRef]
- Falconi, M.; Desideri, A.; Cupane, A.; Leone, M.; Ciccotti, G.; Peterson, E.S.; Friedman, J.M.; Gambacurta, A.; Ascoli, F. Structural and Dynamic Properties of the Homodimeric Hemoglobin from Scapharca inaequivalvis Thr-72 → Ile Mutant: Molecular Dynamics Simulation, Low Temperature Visible Absorption Spectroscopy, and Resonance Raman Spectroscopy Studies. Biophys. J. 1998, 75, 2489–2503. [Google Scholar] [CrossRef]
- Militello, V.; Cupane, A.; Leone, M.; Brinigar, W.S.; Lu, A.L.; Fronticelli, C. Dynamic properties of some β-chain mutant hemoglobins. Proteins Struct. Funct. Bioinform. 1995, 22, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Militello, V.; Cupane, A.; Leone, M.; Vitrano, E. Dynamic and functional properties of a β-β crosslinked derivative of human hemoglobin. Phys. Med. 1993, 9, 43–46. [Google Scholar]
- Cupane, A.; Leone, M.; Militello, V.; Friedman, F.K.; Koley, A.P.; Vasquez, G.B.; Brinigar, W.S.; Karavitis, M.; Fronticelli, C. Modification of α-Chain or β-Chain Heme Pocket Polarity by Val (E11) → Thr Substitution Has Different Effects on the Steric, Dynamic, and Functional Properties of Human Recombinant Hemoglobin: Deoxy Derivatives. J. Biol. Chem. 1997, 272, 26271–26278. [Google Scholar] [CrossRef]
- Karavitis, M.; Fronticelli, C.; Brinigar, W.S.; Vasquez, G.B.; Militello, V.; Leone, M.; Cupane, A. Properties of Human Hemoglobins with Increased Polarity in the α-or β-Heme Pocket: Carbonmonoxy Derivatives. J. Biol. Chem. 1998, 273, 23740–23749. [Google Scholar] [CrossRef]
- Militello, V.; Leone, M.; Cupane, A.; Santucci, R.; Desideri, A. Local dynamic properties of the heme pocket in native and solvent-induced molten-globule-like states of cytochrome c. Biophys. Chem. 2002, 97, 121–128. [Google Scholar] [CrossRef]
- Levantino, M.; Huang, Q.; Cupane, A.; Laberge, M.; Hagarman, A.; Schweitzer-Stenner, R. The importance of vibronic perturbations in ferrocytochrome c spectra: A reevaluation of spectral properties based on low-temperature optical absorption, resonance Raman, and molecular-dynamics simulations. J. Chem. Phys. 2005, 123, 054508. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer-Stenner, R.; Levantino, M.; Cupane, A.; Wallace, C.; Laberge, M.; Huang, Q. Functionally relevant electric-field induced perturbations of the prosthetic group of yeast ferrocytochrome c mutants obtained from a vibronic analysis of low-temperature absorption spectra. J. Phys. Chem. B 2006, 110, 12155–12161. [Google Scholar] [CrossRef]
- Schweitzer-Stenner, R.; Cupane, A.; Leone, M.; Lemke, C.; Schott, J.; Dreybrodt, W. Anharmonic protein motions and heme deformations in myoglobin cyanide probed by absorption and resonance Raman spectroscopy. J. Phys. Chem. B 2000, 104, 4754–4764. [Google Scholar] [CrossRef]
- Cupane, A.; Leone, M.; Unger, E.; Lemke, C.; Beck, M.; Dreybrodt, W.; Schweitzer-Stenner, R. Dynamics of various metal-octaethylporphyrins in solution studied by resonance Raman and low-temperature optical absorption spectroscopies. Role of the central metal. J. Phys. Chem. B 1998, 102, 6612–6620. [Google Scholar] [CrossRef]
- Fomina, M.; Schirò, G.; Cupane, A. Hydration dependence of myoglobin dynamics studied with elastic neutron scattering, differential scanning calorimetry and broadband dielectric spectroscopy. Biophys. Chem. 2014, 185, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Doster, W.; Cusack, S.; Petry, W. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature 1989, 337, 754–756. [Google Scholar] [CrossRef]
- Schiro, G.; Sclafani, M.; Natali, F.; Cupane, A. Hydration dependent dynamics in sol–gel encapsulated myoglobin. Eur. Biophys. J. 2008, 37, 543–549. [Google Scholar] [CrossRef]
- Schiro, G.; Sclafani, M.; Caronna, C.; Natali, F.; Plazanet, M.; Cupane, A. Dynamics of myoglobin in confinement: An elastic and quasi-elastic neutron scattering study. Chem. Phys. 2008, 345, 259–266. [Google Scholar] [CrossRef]
- Schiro, G.; Cupane, A.; Vitrano, E.; Bruni, F. Dielectric relaxations in confined hydrated myoglobin. J. Phys. Chem. B 2009, 113, 9606–9613. [Google Scholar] [CrossRef]
- Schiró, G.; Caronna, C.; Natali, F.; Cupane, A. Direct evidence of the amino acid side chain and backbone contributions to protein anharmonicity. J. Am. Chem. Soc. 2010, 132, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Schiró, G.; Caronna, C.; Natali, F.; Cupane, A. Molecular origin and hydration dependence of protein anharmonicity: An elastic neutron scattering study. Phys. Chem. Chem. Phys. 2010, 12, 10215–10220. [Google Scholar] [CrossRef] [PubMed]
- Schiró, G.; Natali, F.; Cupane, A. Physical origin of anharmonic dynamics in proteins: New insights from resolution-dependent neutron scattering on homomeric polypeptides. Phys. Rev. Lett. 2012, 109, 128102. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Fomina, M.; Cupane, A. Communication: Protein dynamical transition vs. liquid-liquid phase transition in protein hydration water. J. Chem. Phys. 2013, 139, 121102. [Google Scholar] [CrossRef]
- Cupane, A.; Levantino, M.; Santangelo, M.G. Near-infrared spectra of water confined in silica hydrogels in the temperature interval 365–5 K. J. Phys. Chem. B 2002, 106, 11323–11328. [Google Scholar] [CrossRef]
- Cammarata, M.; Levantino, M.; Cupane, A.; Longo, A.; Martorana, A.; Bruni, F. Structure and dynamics of water confined in silica hydrogels: X-ray scattering and dielectric spectroscopy studies. Eur. Phys. J. E 2003, 12, 63–66. [Google Scholar] [CrossRef]
- Santangelo, G.; Di Matteo, A.; Müller-Plathe, F.; Milano, G. From mesoscale back to atomistic models: A fast reverse-mapping procedure for vinyl polymer chains. J. Phys. Chem. B 2007, 111, 2765–2773. [Google Scholar] [CrossRef]
- De Michele, V.; Romanelli, G.; Cupane, A. Dynamics of supercooled confined water measured by deep inelastic neutron scattering. Front. Phys. 2018, 13, 138205. [Google Scholar] [CrossRef]
- Cupane, A.; Fomina, M.; Schirò, G. The boson peak of deeply cooled confined water reveals the existence of a low-temperature liquid-liquid crossover. J. Chem. Phys. 2014, 141, 18C510. [Google Scholar] [CrossRef]
- De Michele, V.; Levantino, M.; Cupane, A. Hysteresis in the temperature dependence of the IR bending vibration of deeply cooled confined water. J. Chem. Phys. 2019, 150, 224509. [Google Scholar] [CrossRef]
- Cupane, A.; Fomina, M.; Piazza, I.; Peters, J.; Schirò, G. Experimental evidence for a liquid-liquid crossover in deeply cooled confined water. Phys. Rev. Lett. 2014, 113, 215701. [Google Scholar] [CrossRef] [PubMed]
- Piazza, I.; Cupane, A.; Barbier, E.L.; Rome, C.; Collomb, N.; Ollivier, J.; Gonzalez, M.A.; Natali, F. Dynamical properties of water in living cells. Front. Phys. 2018, 13, 138301. [Google Scholar] [CrossRef]
- Natali, F.; Dolce, C.; Peters, J.; Stelletta, C.; Demé, B.; Ollivier, J.; Leduc, G.; Cupane, A.; Barbier, E.L. Brain lateralization probed by water diffusion at the atomic to micrometric scale. Sci. Rep. 2019, 9, 14694. [Google Scholar] [CrossRef]
- Natali, F.; Dolce, C.; Peters, J.; Stelletta, C.; Demé, B.; Ollivier, J.; Boehm, M.; Leduc, G.; Piazza, I.; Cupane, A.; et al. Anomalous water dynamics in brain: A combined diffusion magnetic resonance imaging and neutron scattering investigation. J. R. Soc. Interface 2019, 16, 20190186. [Google Scholar] [CrossRef] [PubMed]
- Levantino, M.; Cupane, A.; Zimányi, L.; Ormos, P. Different relaxations in myoglobin after photolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 14402–14407. [Google Scholar] [CrossRef]
- Levantino, M.; Cupane, A.; Zimányi, L. Quaternary structure dependence of kinetic hole burning and conformational substates interconversion in hemoglobin. Biochemistry 2003, 42, 4499–4505. [Google Scholar] [CrossRef]
- Schiro, G.; Cammarata, M.; Levantino, M.; Cupane, A. Spectroscopic markers of the T↔R quaternary transition in human hemoglobin. Biophys. Chem. 2005, 114, 27–33. [Google Scholar] [CrossRef]
- Schirò, G.; Cupane, A. Quaternary Relaxations in Sol–Gel Encapsulated Hemoglobin Studied via NIR and UV Spectroscopy. Biochemistry 2007, 46, 11568–11576. [Google Scholar] [CrossRef]
- Cammarata, M.; Levantino, M.; Schotte, F.; Anfinrud, P.A.; Ewald, F.; Choi, J.; Cupane, A.; Wulff, M.; Ihee, H. Tracking the structural dynamics of proteins in solution using time-resolved wide-angle X-ray scattering. Nat. Methods 2008, 5, 881–886. [Google Scholar] [CrossRef]
- Cammarata, M.; Levantino, M.; Wulff, M.; Cupane, A. Unveiling the timescale of the R–T transition in human hemoglobin. J. Mol. Biol. 2010, 400, 951–962. [Google Scholar] [CrossRef]
- Spilotros, A.; Levantino, M.; Schiro, G.; Cammarata, M.; Wulff, M.; Cupane, A. Probing in cell protein structural changes with time-resolved X-ray scattering. Soft Matter 2012, 8, 6434–6437. [Google Scholar] [CrossRef][Green Version]
- Levantino, M.; Spilotros, A.; Cammarata, M.; Schirò, G.; Ardiccioni, C.; Vallone, B.; Brunori, M.; Cupane, A. The Monod-Wyman-Changeux allosteric model accounts for the quaternary transition dynamics in wild type and a recombinant mutant human hemoglobin. Proc. Natl. Acad. Sci. USA 2012, 109, 14894–14899. [Google Scholar] [CrossRef] [PubMed]
- Levantino, M.; Schiro, G.; Lemke, H.T.; Cottone, G.; Glownia, J.M.; Zhu, D.; Chollet, M.; Ihee, H.; Cupane, A.; Cammarata, M. Ultrafast myoglobin structural dynamics observed with an X-ray free-electron laser. Nat. Commun. 2015, 6, 6772. [Google Scholar] [CrossRef] [PubMed]
- Levantino, M.; Lemke, H.T.; Schirò, G.; Glownia, M.; Cupane, A.; Cammarata, M. Observing heme doming in myoglobin with femtosecond X-ray absorption spectroscopy. Struct. Dyn. 2015, 2, 041713. [Google Scholar] [CrossRef] [PubMed]
- Elber, R.; Karplus, M. Enhanced sampling in molecular dynamics: Use of the time-dependent Hartree approximation for a simulation of carbon monoxide diffusion through myoglobin. J. Am. Chem. Soc. 1990, 112, 9161–9175. [Google Scholar] [CrossRef]
- Schotte, F.; Lim, M.; Jackson, T.A.; Smirnov, A.V.; Soman, J.; Olson, J.S.; Phillips, G.N., Jr.; Wulff, M.; Anfinrud, P.A. Watching a protein as it functions with 150-ps time-resolved x-ray crystallography. Science 2003, 300, 1944–1947. [Google Scholar] [CrossRef]
- Brunori, M.; Gibson, Q.H. Cavities and packing defects in the structural dynamics of myoglobin. EMBO Rep. 2001, 2, 674–679. [Google Scholar] [CrossRef]
- Maragliano, L.; Cottone, G.; Ciccotti, G.; Vanden-Eijnden, E. Mapping the network of pathways of CO diffusion in myoglobin. J. Am. Chem. Soc. 2010, 132, 1010–1017. [Google Scholar] [CrossRef]
- Cottone, G.; Lattanzi, G.; Ciccotti, G.; Elber, R. Multiphoton absorption of myoglobin–nitric oxide complex: Relaxation by D-NEMD of a stationary state. J. Phys. Chem. B 2012, 116, 3397–3410. [Google Scholar] [CrossRef]
- Ciccotti, G.; Jacucci, G. Direct computation of dynamical response by molecular dynamics: The mobility of a charged Lennard-Jones particle. Phys. Rev. Lett. 1975, 357, 789. [Google Scholar] [CrossRef]
- San Biagio, P.L.; Martorana, V.; Emanuele, A.; Vaiana, S.M.; Manno, M.; Bulone, D.; Palma-Vittorelli, M.B.; Palma, M.U. Interacting processes in protein coagulation. Proteins Struct. Funct. Genet. 1999, 37, 116–120. [Google Scholar] [CrossRef]
- Manno, M.; Martorana, V.; Emanuele, A.; Bulone, D.; San Biagio, P.L.; Palma-Vittorelli, M.B.; Palma, M.U. Multiple-path interactions and fractal structure kinetics in supramolecular self-assembly. Biophys. J. 1998, 74, A282. [Google Scholar]
- Bulone, D.; Martorana, V.; San Biagio, P.L. Effects of intermediates on aggregation of native bovine serum albumin. Biophys. Chem. 2001, 91, 61–69. [Google Scholar] [CrossRef]
- Vaiana, S.M.; Emanuele, A.; Palma-Vittorelli, M.B.; Palma, M.U. Irreversible formation of intermediate BSA oligomers requires and induces conformational changes. Proteins 2004, 1, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Manno, M.; Craparo, E.F.; Bulone, D.; Martorana, V.; San Biagio, P.L. Kinetics of Insulin Aggregation: Disentanglement of Amyloid Fibrillation from Large-Size Cluster Formation. Biophys. J. 2006, 90, 585–4591. [Google Scholar] [CrossRef] [PubMed]
- Militello, V.; Vetri, V.; Leone, M. Conformational changes involved in thermal aggregation processes of Bovine Serum Albumin. Biophys. Chem. 2003, 105, 133–141. [Google Scholar] [CrossRef]
- Pullara, F.; Emanuele, A.; Palma-Vittorelli, M.B.; Palma, M.U. Protein Aggregation/Crystallization and Minor Structural Changes: Universal versus Specific Aspects. Biophys. J. 2007, 93, 3271–3278. [Google Scholar] [CrossRef]
- Moriyama, Y.; Ohta, D.; Hachiya, K.; Mitsui, Y.; Takeda, K. Fluorescence behaviour of tryptophan residues of bovine and human serum albumins in ionic surfactant solutions: A comparative study of the two and one tryptophan(s) of bovine and human Albumins. J. Protein Chem. 1996, 15, 265–271. [Google Scholar] [CrossRef]
- Lakovicz, J.R. Principles of Fluorescence Spectroscopy; Plenum Press: New York, NY, USA, 1983. [Google Scholar]
- Vetri, V.; Librizzi, F.; Leone, M.; Militello, V. Thermal aggregation of bovine serum albumin at different pH: Comparison with human serum albumin. Eur. Biophys. J. 2007, 36, 717–725. [Google Scholar] [CrossRef]
- Vetri, V.; D’Amico, M.; Foderà, V.; Leone, M.; Ponzoni, A.; Sberveglieri, G.; Militello, V. Bovine Serum Albumin protofibril-like aggregates formation: Solo but not simple mechanism. Arch. Biochem. Biophys. 2011, 508, 13–24. [Google Scholar] [CrossRef]
- Militello, V.; Casarino, C.; Emanuele, A.; Giostra, A.; Pullara, F.; Leone, M. Aggregation kinetics of Bovine Serum Albumin studied by FTIR spectroscopy and light Scattering. Biophys. Chem. 2004, 107, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Oligomers on the brain: The emerging role of soluble protein aggregates in neurodegeneration. Protein Pept. Lett. 2004, 11, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Protein misfolding diseases. Annu. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef]
- Choi, M.L.; Gandhi, S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer’s and Parkinson’s diseases. FEBS J. 2018, 285, 3631–3644. [Google Scholar] [CrossRef]
- Kulenkampff, K.; Wolf Perez, A.M.; Sormanni, P.; Habchi, J.; Vendruscolo, M. Quantifying misfolded protein oligomers as drug targets and biomarkers in Alzheimer and Parkinson diseases. Nat. Rev. Chem. 2021, 5, 277–294. [Google Scholar] [CrossRef]
- Vetri, V.; Foderà, V. The route to protein aggregate superstructures: Particulates and amyloid-like spherulites. FEBS Lett. 2015, 589, 2448–2463. [Google Scholar] [CrossRef]
- Vetri, V.; Piccirilli, F.; Krausser, J.; Buscarino, G.; Łapińska, U.; Vestergaard, B.; Zaccon, A.; Foderà, V. Ethanol controls the self-assembly and mesoscopic properties of human insulin amyloid spherulites. J. Phys. Chem. B 2018, 122, 3101–3112. [Google Scholar] [CrossRef]
- Fennema Galparsoro, D.; Zhou, X.; Jaaloul, A.; Piccirilli, F.; Vetri, V.; Foderà, V. Conformational transitions upon maturation rule surface and pH-responsiveness of α-lactalbumin microparticulates. ACS Appl. Bio Mater. 2021, 4, 1876–1887. [Google Scholar] [CrossRef]
- Librizzi, F.; Rischel, C. The kinetic behavior of insulin fibrillation is determined by heterogeneous nucleation pathways. Protein Sci. 2005, 14, 3129–3134. [Google Scholar] [CrossRef]
- Fodera, V.; Librizzi, F.; Groenning, M.; van de Weert, M.; Leone, M. Secondary nucleation and accessible surface in insulin amyloid fibril formation. J. Phys. Chem. B 2008, 112, 3853–3858. [Google Scholar] [CrossRef] [PubMed]
- Fodera, V.; Cataldo, S.; Librizzi, F.; Pignataro, B.; Spiccia, P.; Leone, M. Self-organization pathways and spatial heterogeneity in insulin amyloid fibril formation. J. Phys. Chem. B 2009, 113, 10830–10837. [Google Scholar] [CrossRef] [PubMed]
- LeVine, H. Thioflavine T interaction with amyloid β-sheet structures. Amyloid 1995, 2, 1–6. [Google Scholar] [CrossRef]
- Foderà, V.; Groenning, M.; Vetri, V.; Librizzi, F.; Spagnolo, S.; Cornett, C.; Olsen, L.; Van De Weert, M.; Leone, M. Thioflavin T hydroxylation at basic pH and its effect on amyloid fibril detection. J. Phys. Chem. B 2008, 112, 15174–15181. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.; Di Carlo, M.G.; Groenning, M.; Militello, V.; Vetri, V.; Leone, M. Thioflavin T promotes Aβ(1-40) amyloid fibrils formation. J. Phys. Chem. Lett. 2012, 3, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, M.G.; Minicozzi, V.; Foderà, V.; Militello, V.; Vetri, V.; Morante, S.; Leone, M. Thioflavin T templates amyloid β(1-40) conformation and aggregation pathway. Biophys. Chem. 2015, 206, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Navarra, G.; Peres, C.; Contardi, M.; Picone, P.; San Biagio, P.L.; Di Carlo, M.; Giacomazza, D.; Militello, V. Heat- and pH-induced BSA conformational changes, hydrogels formation and their applications as 3D cell scaffold. Arch. Biochem. Biophys. 2016, 606, 134–142. [Google Scholar] [CrossRef] [PubMed]
- De Luca, G.; Galparsoro, D.F.; Sancataldo, G.; Leone, M.; Foderà, V.; Vetri, V. Probing ensemble polymorphism and single aggregate structural heterogeneity in insulin amyloid self-assembly. J. Colloid Interface Sci. 2020, 574, 229–240. [Google Scholar] [CrossRef]
- Zhou, X.; Galparsoro, D.F.; Madsen, A.Ø.; Vetri, V.; van de Weert, M.; Nielsen, H.M.; Foderà, V. Polysorbate 80 controls Morphology, structure and stability of human insulin Amyloid-Like spherulites. J. Colloid Interface Sci. 2022, 606, 1928–1939. [Google Scholar] [CrossRef]
- Van Maarschalkerweerd, A.; Vetri, V.; Langkilde, A.E.; Fodera, V.; Vestergaard, B. Protein/lipid coaggregates are formed during α-synuclein-induced disruption of lipid bilayers. Biomacromolecules 2014, 15, 3643–3654. [Google Scholar] [CrossRef]
- Rao, E.; Foderà, V.; Leone, M.; Vetri, V. Direct observation of alpha-lactalbumin, adsorption and incorporation into lipid membrane and formation of lipid/protein hybrid structures. Biochim. Biophys. Acta BBA Gen. Subj. 2019, 1863, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, S.; Sancataldo, G.; Mørck Nielsen, H.; Foderà, V.; Vetri, V. Peptide–membrane interactions monitored by fluorescence lifetime imaging: A study case of Transportan 10. Langmuir 2021, 37, 13148–13159. [Google Scholar] [CrossRef] [PubMed]
- Rondeau, P.; Navarra, G.; Militello, V.; Bourdon, E. Aggregation of albumin: Influence of the protein glycation. In Protein Aggregation; Series: Protein Science and Engineering Microbiology Research Advances; Stein, D.A., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2011; Chapter 5; pp. 139–159. [Google Scholar]
- Baraka-Vidot, J.; Navarra, G.; Leone, M.; Bourdon, E.; Militello, V.; Rondeau, P. Deciphering metal-induced oxidative damages on glycated albumin structure and function. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 1712–1724. [Google Scholar] [CrossRef] [PubMed]
- Baraka-Vidot, J.; Planesse, C.; Meilhac, O.; Militello, V.; van den Elsen, J.M.H.; Bourdon, E.; Rondeau, P. Glycation alters ligand-binding, enzymatic and pharmacological properties of human albumin. Biochemistry 2015, 54, 3051–3062. [Google Scholar] [CrossRef]
- Khan, M.W.; Rasheed, Z.; Khan, W.A.; Ali, R. Biochemical, biophysical, and thermodynamic analysis of in vitro glycated human serum albumin. Biochemistry 2007, 72, 146–152. [Google Scholar]
- Cohen, M.P.; Ziyadeh, F.N.; Chen, S. Amadori-modified glycated serum proteins and accelerated atherosclerosis in diabetes: Pathogenic and therapeutic implications. J. Lab. Clin. Med. 2006, 147, 211–219. [Google Scholar] [CrossRef]
- Tanaka, A.; Kaneto, H.; Miyatsuka, T.; Yamamoto, K.; Yoshiuchi, K.; Yamasaki, Y.; Shimomura, I.; Matsuoka, T.; Matsuhisa, M. Role of copper ion in the pathogenesis of type 2 diabetes. Endocr. J. 2009, 56, 699–706. [Google Scholar] [CrossRef]
- Al-Maroof, R.A.; Al-Sharbatti, S.S. Serum zinc levels in diabetic patients and effect of zinc supplementation on glycemic control of type 2 diabetics. Saudi Med. J. 2006, 27, 344–350. [Google Scholar]
- Rondeau, P.; Bourdon, E. The glycation of albumin: Structural and functional impacts. Biochimie 2011, 93, 645–658. [Google Scholar] [CrossRef]
- Rondeau, P.; Navarra, G.; Cacciabaudo, F.; Leone, M.; Bourdon, E.; Militello, V. Thermal aggregation of glycated bovine serum albumin. Biochim. Biophys. Acta Protein Proteom. 2010, 1804, 789–798. [Google Scholar] [CrossRef]
- Navarra, G.; Tinti, A.; Leone, M.; Militello, V.; Torreggiani, A. Influence of metal ions on thermal aggregation of bovine serum albumin: Aggregation kinetics and structural changes. J. Inorg. Biochem. 2009, 103, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Cordone, L.; Cottone, G.; Giuffrida, S.; Palazzo, G.; Venturoli, G.; Viappiani, C. Internal dynamics and protein–matrix coupling in trehalose-coated proteins. Biochim. Biophys. Acta Proteins Proteom. 2005, 1749, 252–281. [Google Scholar] [CrossRef] [PubMed]
- Cordone, L.; Cottone, G.; Cupane, A.; Emanuele, A.; Giuffrida, S.; Levantino, M. Proteins in Saccharides Matrices and the Trehalose Peculiarity: Biochemical and Biophysical Properties. Curr. Org. Chem. 2015, 19, 1684–1706. [Google Scholar] [CrossRef]
- Giuffrida, S.; Cottone, G.; Bellavia, G.; Cordone, L. Proteins in Amorphous Saccharide Matrices: Structural and Dynamical Insights on Bioprotection. Eur. Phys. J. E 2013, 36, 7. [Google Scholar] [CrossRef]
- Cordone, L.; Cottone, G.; Giuffrida, S.; Librizzi, F. Thermal evolution of the CO stretching band in carboxy-myoglobin in the light of neutron scattering and molecular dynamics simulations. Chem. Phys. 2008, 345, 275–282. [Google Scholar] [CrossRef]
- Giuffrida, S.; Cottone, G.; Cordone, L. Role of solvent on protein-matrix coupling in MbCO embedded in water-saccharide systems: A Fourier transform infrared spectroscopy study. Biophys. J. 2006, 91, 968–980. [Google Scholar] [CrossRef]
- Cottone, G.; Giuffrida, S.; Bettati, S.; Bruno, S.; Campanini, B.; Marchetti, M.; Abruzzetti, S.; Viappiani, C.; Cupane, A.; Mozzarelli, A.; et al. More than a Confinement: “Soft” and “Hard” Enzyme Entrapment Modulates Biological Catalyst Function. Catalysts 2019, 9, 1024. [Google Scholar] [CrossRef]
- Da Silva, F.L.B.; Carloni, P.; Cheung, D.; Cottone, G.; Donnini, S.; Foegeding, E.A.; Gulzar, M.; Jacquier, J.C.; Lobaskin, V.; MacKernan, D.; et al. Understanding and Controlling Food Protein Structure and Function in Foods: Perspectives from Experiments and Computer Simulations. Annu. Rev. Food Sci. Technol. 2020, 11, 365–387. [Google Scholar] [CrossRef]
- Crowe, J.H.; Crowe, L.M.; Wolkers, W.F.; Oliver, A.E.; Ma, X.; Auh, J.H.; Tang, M.; Zhu, S.; Norris, J.; Tablin, F. Stabilization of Dry Mammalian Cells: Lessons from Nature. Integr. Comp. Biol. 2005, 45, 810–820. [Google Scholar] [CrossRef]
- Carpenter, J.F.; Crowe, J.H. An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry 1989, 28, 3916–3922. [Google Scholar] [CrossRef]
- Xie, G.; Timasheff, S.N. The thermodynamic mechanism of protein stabilization by trehalose. Biophys. Chem. 1997, 64, 25–43. [Google Scholar] [CrossRef]
- Belton, P.S.; Gil, A.M. IR and Raman spectroscopic studies of the interaction of trehalose with hen egg white lysozyme. Biopolymers 1994, 34, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Sampedro, J.G.; Uribe, S. Trehalose-enzyme interactions result in structure stabilization and activity inhibition. The role of viscosity. Mol. Cell. Biochem. 2004, 256, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Green, J.L.; Angell, C.A. Phase relations and vitrification in saccharide-water solutions and the trehalose anomaly. J. Phys. Chem. 1989, 93, 2880–2882. [Google Scholar] [CrossRef]
- Chiantia, S.; Giannola, L.; Cordone, L. Lipid phase transition in saccharide-coated cholate-containing liposomes: Coupling to the surrounding matrix. Langmuir 2005, 122, 4309–4317. [Google Scholar] [CrossRef]
- Panzica, M.; Emanuele, A.; Cordone, L. Thermal Aggregation of Bovine Serum Albumin in Trehalose and Sucrose Aqueous Solutions. J. Phys. Chem. B 2012, 116, 11829–11836. [Google Scholar] [CrossRef]
- Cordone, L.; Ferrand, M.; Vitrano, E.; Zaccai, G. Harmonic behavior of trehalose-coated carbon-monoxy-myoglobin at high temperature. Biophys. J. 1999, 76, 1043–1047. [Google Scholar] [CrossRef][Green Version]
- Cordone, L.; Galajda, P.; Vitrano, E.; Gassmann, A.; Ostermann, A.; Parak, F. A reduction of protein specific motions in co-ligated myoglobin embedded in a trehalose glass. Eur. Biophys. J. 1998, 27, 173–176. [Google Scholar] [CrossRef]
- Librizzi, F.; Vitrano, E.; Cordone, L. Dehydration and crystallization of trehalose and sucrose glasses containing carbonmonoxy-myoglobin. Biophys. J. 1999, 76, 2727–2734. [Google Scholar] [CrossRef]
- Librizzi, F.; Viappiani, C.; Abbruzzetti, S.; Cordone, L. Residual water modulates the dynamics of the protein and of the external matrix in “trehalose coated” MbCO: An infrared and flash-photolysis study. J. Chem. Phys. 2002, 116, 1193–1200. [Google Scholar] [CrossRef]
- Vojtěchovský, J.; Chu, K.; Berendzen, J.; Sweet, R.M.; Schlichting, I. Crystal Structures of Myoglobin-Ligand Complexes at Near-Atomic Resolution. Biophys. J. 1999, 77, 2153–2174. [Google Scholar] [CrossRef]
- Cottone, G.; Cordone, L.; Ciccotti, G. Molecular dynamics simulation of carboxy-myoglobin embedded in a trehalose-water matrix. Biophys. J. 2001, 80, 931–938. [Google Scholar] [CrossRef]
- Cottone, G.; Ciccotti, G.; Cordone, L. Protein–trehalose–water structures in trehalose coated carboxy-myoglobin. J. Chem. Phys. 2022, 117, 9862–9866. [Google Scholar] [CrossRef]
- Cottone, G.; Giuffrida, S.; Ciccotti, G.; Cordone, L. Molecular dynamics simulation of sucrose-and trehalose-coated carboxy-myoglobin. Proteins 2005, 59, 291–302. [Google Scholar] [CrossRef]
- Giachini, L.; Francia, F.; Cordone, L.; Boscherini, F.; Venturoli, G. Cytochrome c in a dry trehalose matrix: Structural and dynamical effects probed by x-ray absorption spectroscopy. Biophys. J. 2006, 92, 1350–1360. [Google Scholar] [CrossRef]
- Cottone, G. A comparative study of carboxy myoglobin in saccharide-water systems by molecular dynamics simulation. J. Phys. Chem. B 2007, 111, 3563–3569. [Google Scholar] [CrossRef]
- Massari, A.M.; Finkestein, I.J.; McClain, B.L.; Goj, A.; Wen, X.; Bren, K.L.; Loring, R.F.; Fayer, M.D.J. The influence of aqueous versus glassy solvents on protein dynamics: Vibrational echo experiments and molecular dynamics simulations. J. Am. Chem. Soc. 2005, 127, 14279–14289. [Google Scholar] [CrossRef]
- Massari, A.M.; Finkelstein, I.J.; Fayer, M.D. Dynamics of proteins encapsulated in silica sol–gel glasses studied with IR vibrational echo spectroscopy. J. Am. Chem. Soc. 2006, 128, 3990–3997. [Google Scholar] [CrossRef]
- Katyal, N.; Deep, S. Revisiting the conundrum of trehalose stabilization. Phys. Chem. Chem. Phys. 2014, 16, 26746–26761. [Google Scholar] [CrossRef]
- Lerbret, A.; Bordat, P.; Affouard, F.; Hédoux, A.; Guinet, Y.; Descamps, M. How do trehalose, maltose, and sucrose influence some structural and dynamical properties of lysozyme? Insight from molecular dynamics simulations. J. Phys. Chem. B 2007, 111, 9410–9420. [Google Scholar] [CrossRef]
- Lerbret, A.; Affouard, F.; Hédoux, A.; Krenzlin, S.; Siepmann, J.; Bellissent-Funel, M.C.; Descamps, M. How Strongly Does Trehalose Interact with Lysozyme in the Solid State? Insights from Molecular Dynamics Simulation and Inelastic Neutron Scattering. J. Phys. Chem. B 2012, 116, 11103–11116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Liu, F.; Dong, X.; Sun, Y. Molecular Insight into the Counteraction of Trehalose on Urea-Induced Protein Denaturation Using Molecular Dynamics Simulation. J. Phys. Chem. B 2012, 116, 7040–7047. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.; Stott, S.L.; Toner, M. Exploring Dynamics and Structure of Biomolecules, Cryoprotectants, and Water Using Molecular Dynamics Simulations: Implications for Biostabilization and Biopreservation. Annu. Rev. Biomed. Eng. 2019, 21, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Olsson, C.; Jansson, H.; Swenson, J. The Role of Trehalose for the Stabilization of Proteins. J. Phys. Chem. B 2016, 120, 4723–4731. [Google Scholar] [CrossRef]
- Olsson, C.; Genheden, S.; Sakai, V.G.; Swenson, J. Mechanism of Trehalose-Induced Protein Stabilization from Neutron Scattering and Modeling. Phys. Chem. Chem. Phys. 2019, 123, 3679–3697. [Google Scholar] [CrossRef] [PubMed]
- Olsson, C.; Zangana, R.; Swenson, J. Stabilization of proteins embedded in sugars and water as studied by dielectric spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 21197–21207. [Google Scholar] [CrossRef] [PubMed]
- Di Gioacchino, M.; Bruni, F.; Ricci, M.A. Protection against Dehydration: A Neutron Diffraction Study on Aqueous Solutions of a Model Peptide and Trehalose. J. Phys. Chem. B 2018, 122, 10291–10295. [Google Scholar] [CrossRef]
- Di Gioacchino, M.; Bruni, F.; Ricci, M.A. N-Methylacetamide Aqueous Solutions: A Neutron Diffraction Study. J. Phys. Chem. B 2019, 123, 1808–1814. [Google Scholar] [CrossRef]
- Giuffrida, S.; Cottone, G.; Librizzi, F.; Cordone, L. Coupling between the thermal evolution of the heme pocket and the external matrix structure in trehalose coated carboxymyoglobin. J. Phys. Chem. B 2003, 107, 13211–13217. [Google Scholar] [CrossRef]
- Giuffrida, S.; Cottone, G.; Cordone, L. Structure– Dynamics Coupling between Protein and External Matrix in Sucrose-Coated and in Trehalose-Coated MbCO: An FTIR Study. J. Phys. Chem. B 2004, 108, 15415–15421. [Google Scholar] [CrossRef]
- Cordone, L.; Cottone, G.; Giuffrida, S. Role of residual water hydrogen bonding in sugar/water/biomolecule systems: A possible explanation for trehalose peculiarity. J. Phys. Condens. Matter 2007, 19, 205110. [Google Scholar] [CrossRef]
- Giuffrida, S.; Cottone, G.; Vitrano, E.; Cordone, L. A FTIR study on low hydration saccharide amorphous matrices: Thermal behaviour of the Water Association Band. J. Non-Cryst. Solids 2011, 357, 677–682. [Google Scholar] [CrossRef]
- Francia, F.M.; Dezi, A.; Mallardi, G.; Palazzo, L.; Cordone, L.; Venturoli, G. Protein–matrix coupling/uncoupling in “Dry” systems of photosynthetic reaction center embedded in trehalose/sucrose: The origin of trehalose peculiarity. J. Am. Chem. Soc. 2008, 130, 10240–10246. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, A.; Malferrari, M.; Francia, F.; Venturoli, G.; Möbius, K. Bacterial Photosynthetic Reaction Centers in Trehalose Glasses: Coupling between Protein Conformational Dynamics and Electron-Transfer Kinetics as Studied by Laser-Flash and High-Field EPR Spectroscopies. J. Phys. Chem. B 2010, 114, 12729–12743. [Google Scholar] [CrossRef]
- Malferrari, M.; Nalepa, A.; Venturoli, G.; Francia, F.; Lubitz, W.; Möbius, K.; Savitsky, A. Structural and Dynamical Characteristics of Trehalose and Sucrose Matrices at Different Hydration Levels as Probed by FTIR and High-Field EPR. Phys. Chem. Chem. Phys. 2014, 16, 9831–9848. [Google Scholar] [CrossRef]
- Nalepa, A.; Marco Malferrari, M.; Lubitz, W.; Venturoli, G.; Möbius, K.; Savitsky, A. Local water sensing: Water exchange in bacterial photosynthetic reaction centers embedded in a trehalose glass studied using multiresonance EPR. Phys. Chem. Chem. Phys. 2017, 19, 28388–28400. [Google Scholar] [CrossRef]
- Giuffrida, S.; Cottone, G.; Cordone, L. The Water Association Band as a Marker of Hydrogen Bonds in Trehalose Amorphous Matrices. Phys. Chem. Chem. Phys. 2017, 19, 4251–4265. [Google Scholar] [CrossRef]
- Verma, P.K.; Kundu, A.; Puretz, M.S.; Dhoonmoon, C.; Chegwidden, O.S.; Londergan, C.H.; Cho, M. The Bend + Libration Combination Band Is an Intrinsic, Collective, and Strongly Solute-Dependent Reporter on the Hydrogen Bonding Network of Liquid Water. J. Phys. Chem. B 2018, 122, 2587–2599. [Google Scholar] [CrossRef]
- Giuffrida, S.; Cupane, A.; Cottone, G. Water Association Band in Saccharide Amorphous Matrices: Role of Residual Water on Bioprotection. Int. J. Mol. Sci. 2021, 22, 2496. [Google Scholar] [CrossRef]
- Librizzi, F.; Vitrano, E.; Paciaroni, A.; Cordone, L. Elastic neutron scattering of dry and rehydrated trehalose coated carboxy-myoglobin. Chem. Phys. 2008, 345, 283–288. [Google Scholar] [CrossRef]
- Giuffrida, S.; Cordone, L.; Cottone, G. Bioprotection Can Be Tuned with a Proper Protein/Saccharide Ratio: The Case of Solid Amorphous Matrices. J. Phys. Chem. B 2018, 122, 8642–8653. [Google Scholar] [CrossRef] [PubMed]
- Longo, A.; Giuffrida, S.; Cottone, G.; Cordone, L. Myoglobin Embedded in Saccharide Amorphous Matrices: Water-Dependent Domains Evidenced by Small Angle X-ray Scattering. Phys. Chem. Chem. Phys. 2010, 12, 6852–6858. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, S.; Panzica, M.; Giordano, F.; Longo, A. SAXS Study on Myoglobin Embedded in Amorphous Saccharide Matrices. Eur. Phys. J. E 2011, 34, 87. [Google Scholar] [CrossRef]
- Bellavia, G.; Cottone, G.; Giuffrida, S.; Cupane, A.; Cordone, L. Thermal Denaturation of Myoglobin in Water-Disaccharide Matrixes: Relation with the Glass Transition of the System. J. Phys. Chem. B 2009, 113, 11543–11549. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, G.; Giuffrida, S.; Cottone, G.; Cupane, A.; Cordone, L. Protein thermal denaturation and matrix glass transition in different protein–trehalose–water systems. J. Phys. Chem. B 2011, 115, 6340–6346. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, S.; Troia, R.; Schiraldi, C.; D’Agostino, A.; De Rosa, M.; Cordone, L. MbCO Embedded in Trehalosyldextrin Matrices: Thermal Effects and Protein–Matrix Coupling. Food Biophys. 2011, 6, 217–226. [Google Scholar] [CrossRef]
- Semeraro, E.F.; Giuffrida, S.; Cottone, G.; Cupane, A. Biopreservation of Myoglobin in Crowded Environment: A Comparison between Gelatin and Trehalose Matrixes. J. Phys. Chem. B 2017, 121, 8731–8741. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cottone, G.; Cupane, A.; Leone, M.; Vetri, V.; Militello, V. A Long Journey into the Investigation of the Structure–Dynamics–Function Paradigm in Proteins through the Activities of the Palermo Biophysics Group. Biophysica 2022, 2, 452-474. https://doi.org/10.3390/biophysica2040040
Cottone G, Cupane A, Leone M, Vetri V, Militello V. A Long Journey into the Investigation of the Structure–Dynamics–Function Paradigm in Proteins through the Activities of the Palermo Biophysics Group. Biophysica. 2022; 2(4):452-474. https://doi.org/10.3390/biophysica2040040
Chicago/Turabian StyleCottone, Grazia, Antonio Cupane, Maurizio Leone, Valeria Vetri, and Valeria Militello. 2022. "A Long Journey into the Investigation of the Structure–Dynamics–Function Paradigm in Proteins through the Activities of the Palermo Biophysics Group" Biophysica 2, no. 4: 452-474. https://doi.org/10.3390/biophysica2040040
APA StyleCottone, G., Cupane, A., Leone, M., Vetri, V., & Militello, V. (2022). A Long Journey into the Investigation of the Structure–Dynamics–Function Paradigm in Proteins through the Activities of the Palermo Biophysics Group. Biophysica, 2(4), 452-474. https://doi.org/10.3390/biophysica2040040