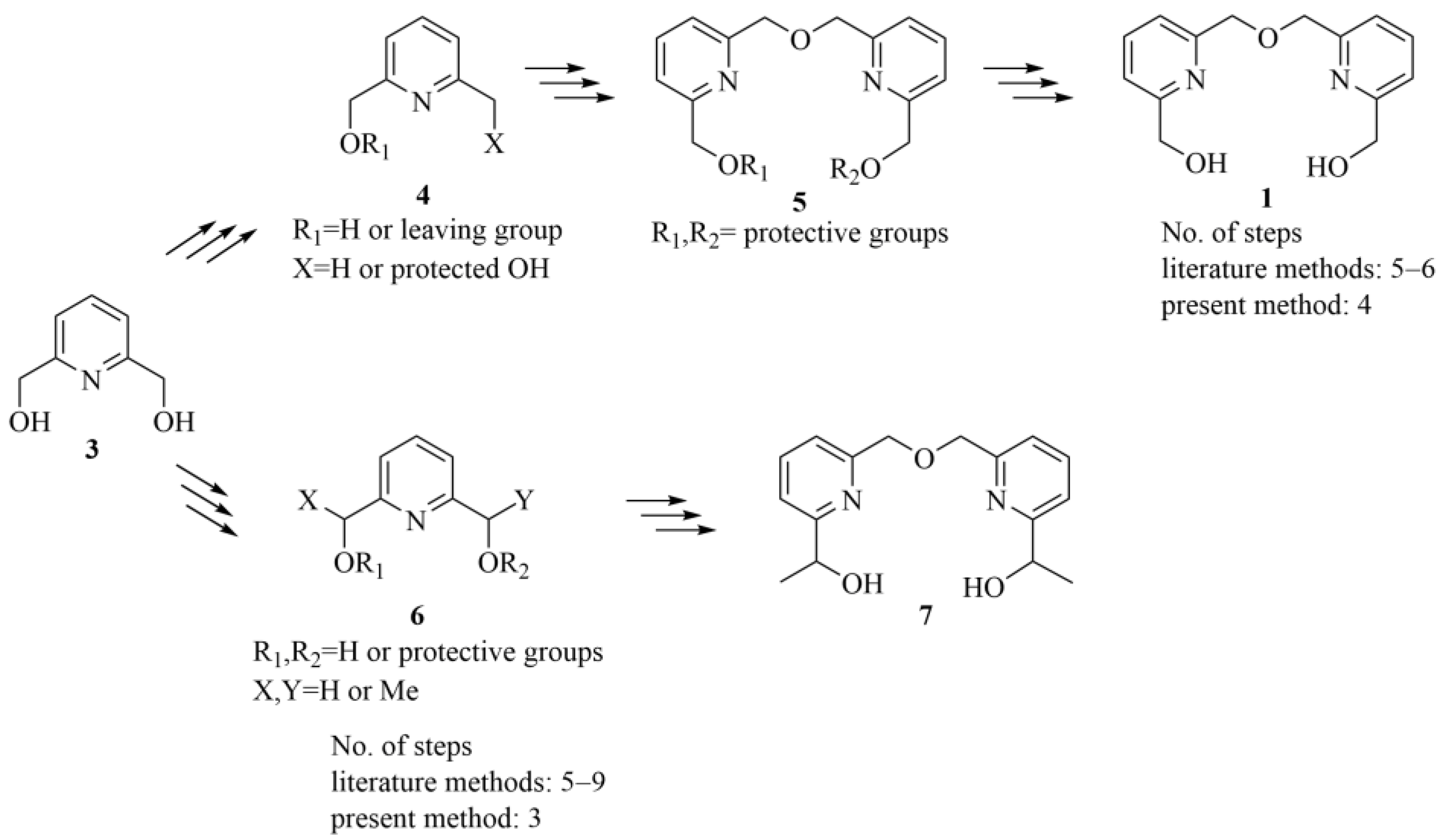
2.2. Synthesis
2.2.1. Preparation of {6-[(benzyloxy)methyl]pyridin-2-yl}methanol (15, Scheme 3)
Procedure ‘A’
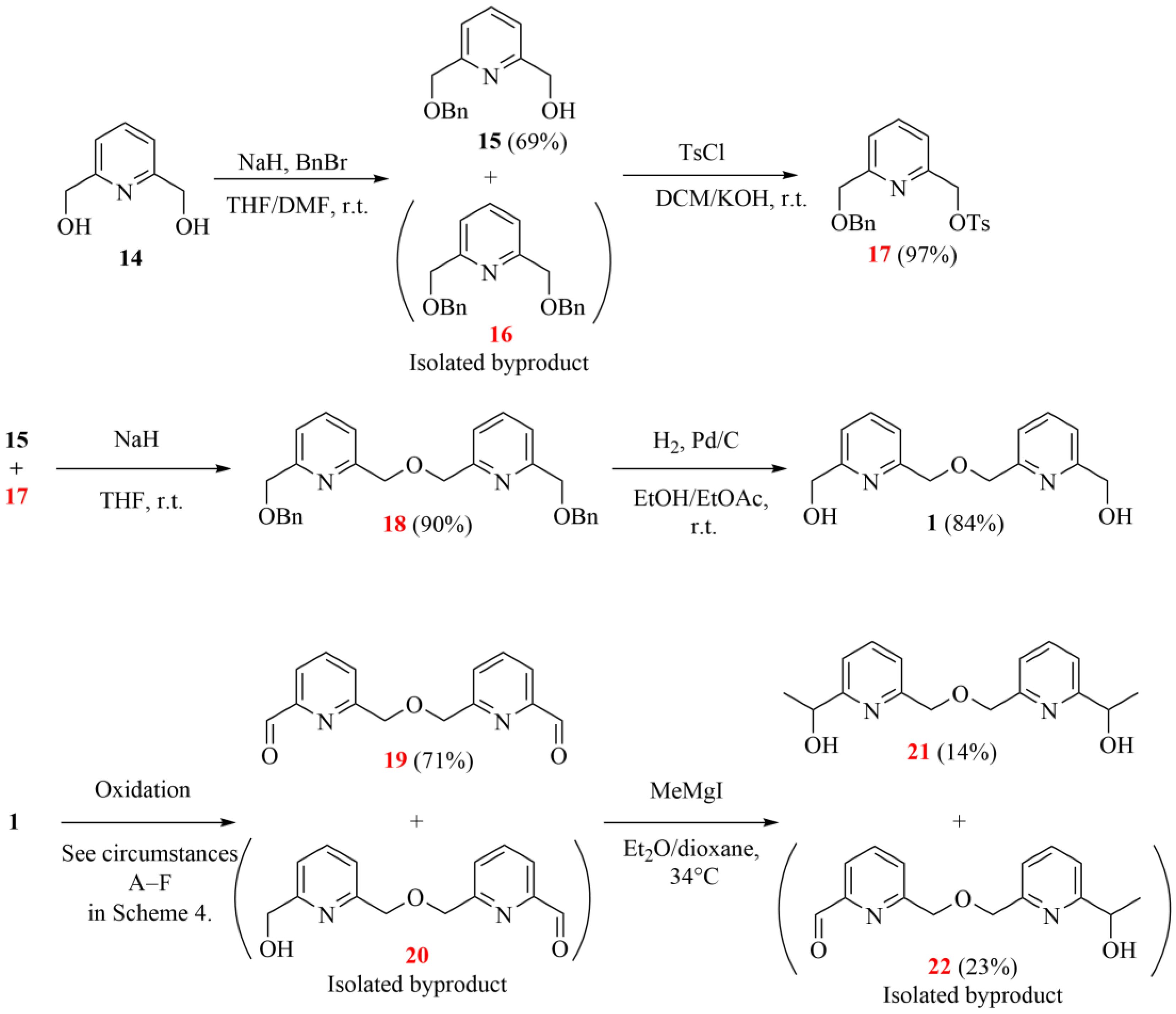
Pyridinedimethanol 14 (28.70 g, 0.21 mol) was dissolved in a mixture of 300 mL THF and 40 mL DMF and cooled down to 0 °C under an argon atmosphere. Then, sodium hydride (16.60 g, 60 w/w% dispersion in mineral oil, 0.42 mol) was added to this solution in small portions. This mixture was stirred for 15 min, and then benzyl bromide (24.5 mL, 35.23 g, 0.21 mol) was added to it dropwise. The mixture was stirred at room temperature for 24 h. Distilled water (5 mL) was added to the reaction mixture, and the volatile components were evaporated. Water (400 mL) and ethyl acetate (400 mL) were added to the residue, and the aqueous phase was extracted with ethyl acetate (4 × 100 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated. The crude product was purified by column chromatography on a silica gel adsorbent using a mixture of ethyl acetate/hexane = 3:2 as an eluent to produce title compound 15.
Monobenzylated pyridine derivative
15 is a pale-yellow oil, 32.5 g (69%). The product was identical in every aspect to that reported in [
6].
Rf: 0.40 (SiO
2, ethyl acetate/hexane = 3:2).
Procedure ‘B’
Diol 14 (1.50 g, 10.78 mmol) was added to a stirred mixture of sodium hydride (0.69 g, 17.25 mmol) and DMSO (10 mL) under an argon atmosphere. Benzyl bromide (2.21 g, 12.94 mmol) in DMSO (10 mL) was added dropwise over 30 min and stirred for 1 day at room temperature. The reaction mixture was poured into ice-water (50 mL), and the mixture was extracted with diethyl ether (3 × 50 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated. The crude product was purified by column chromatography on a silica gel adsorbent using a mixture of propan-2-ol/cyclohexane = 1:10 as an eluent to produce title compound 15.
Product
15 was a pale-yellow oil (0.67 g, 27%). The product was identical in every aspect to that reported in [
6].
2.2.2. Preparation of 2,6-bis[(benzyloxy)methyl]pyridine (16, Scheme 3)
The dibenzylated pyridine derivative
16 is a major product; its synthesis is described above in the preparation of the monobenzylated derivative
8 (
Section 2.2.1). The dibenzylated derivative
16 from procedure ‘B’, 2.41 g (70%), is a white crystalline solid.
M.p.: 81 °C. Rf: 0.35 (SiO2, propane-2-ol/cyclohexane = 1:10). 1H-NMR (500 MHz, CDCl3) δ 7.71 (t, J = 7.7 Hz, 1H), 7.42–7.30 (m, 12H), 4.68 (s, 4H), 4.66 (s, 4H). 13C-NMR (125 MHz, CDCl3) δ 157.9, 138.0, 137.3, 128.4, 127.8, 127.7, 120.0, 73.1, 72.9. IR: νmax [cm−1] (KBr): 3074, 3061, 3027, 2921, 2881, 2819, 1592, 1580, 1469, 1454, 1443, 1389, 1344, 1305, 1229, 1075, 1027, 1018, 992, 916, 804, 787, 730, 694, 629, 465. HRMS (m/z): [MH+]: 320.1606, calculated [M+]: 319.1572.
2.2.3. Preparation of {6-[(benzyloxy)methyl]pyridin-2-yl}methyl 4-methylbenzenesulfonate (17, Scheme 3)
Monobenzyl derivative 15 (8.55 g, 37.30 mmol) was dissolved in 90 mL dichloromethane (DCM), and the solution was cooled to 0 °C. Potassium hydroxide (41.50 g, 0.73 mol) in 65 mL water was added to this solution under an argon atmosphere, and the mixture was stirred for 10 min at 0 °C. Tosyl chloride (7.11 g, 37.30 mol) in 90 mL DCM was added dropwise to the reaction mixture, and it was stirred for 1 day at room temperature. DCM (150 mL) and water (150 mL) were added to the mixture, and the phases were separated. The aqueous phase was extracted with DCM (3 × 100 mL). The combined organic phase was dried with anhydrous magnesium sulfate, filtered and evaporated. The crude product was purified by column chromatography on a silica gel adsorbent using a mixture of ethyl acetate/hexane = 1:3 as an eluent to give tosylate 17 (13.73 g, 97%) as a colorless oil.
Rf: 0.85 (SiO2, ethyl acetate/hexane = 3:2). 1H-NMR (500 MHz, CDCl3) δ 7.81 (d, J = 7.9 Hz, 2H), 7.68 (t, J = 8.1 Hz, 1H), 7.43–7.27 (m, 9H), 5.12 (s, 2H), 4.61 (s, 2H), 4.59 (s, 2H), 2.40 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 158.4, 153.0, 145.1, 137.6, 132.8, 129.9, 128.5, 128.1, 127.8, 121.0, 120.5, 73.0, 72.7, 71.8, 21.7. IR: νmax [cm−1]: 3086, 3062, 3031, 2943, 2869, 2827, 2584, 2033, 1722, 1654, 1624, 1541, 1496, 1453, 1362, 1228, 1159, 1117, 1030, 1008, 815, 742, 679, 561. HRMS (m/z): [MH+]: 384.1225, calculated [M+]: 383.1191.
2.2.4. Preparation of 6,6′-[oxybis(methylene)]bis{2-[(benzyloxy)methyl]pyridine} (18, Scheme 3)
Pyridinemethanol 15 (7.15 g, 31.00 mmol) was dissolved in THF (70 mL), and this solution was cooled to 0 °C under an argon atmosphere. Sodium hydride (60 w/w% dispersion in oil, 3.12 g, 78.00 mmol) was added to this solution in small portions, and the mixture was stirred for 15 min. Tosylate 17 (11.89 g, 31.00 mmol) in THF (70 mL) was added dropwise to this mixture, and it was stirred for 72 h at room temperature. Water (1.5 mL) was added to the reaction mixture. The solvent was then removed, and the solid residue was dissolved in a mixture of ethyl acetate (100 mL) and water (100 mL). The aqueous phase was extracted with ethyl acetate (4 × 70 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated. The crude product was recrystallized from ethyl acetate (200 mL) to give bipyridine derivative 18 (12.36 g, 90%) as a white crystalline solid.
M.p.: 104–106 °C. Rf: 0.50 (SiO2, ethyl acetate/hexane = 3:2). 1H-NMR (500 MHz, CDCl3) δ 7.71 (t, J = 7.8 Hz, 2H), 7.42–7.28 (m, 14H), 4.75 (s, 4H), 4.67 (s, 4H), 4.64 (s, 4H). 13C-NMR (125 MHz, DEPTQ-135, CDCl3) δ 158.1, 157.6, 138.0, 137.3, 128.4, 127.8, 127.8, 120.1, 120.0, 73.7, 73.1, 73.0. IR: νmax [cm−1]: 3086, 3058, 3030, 3006, 2924, 2887, 2873, 2852, 2782, 1591, 1581, 1496, 1468, 1445, 1381, 1345, 1331, 1307, 1239, 1207, 1131, 1071, 1028, 993, 913, 823, 785, 729, 695, 634, 508, 468. HRMS (m/z): [MH+]: 441.2133, calculated [M+]: 440.2100.
2.2.5. Preparation of [6-({[6-(hydroxymethyl)pyridin-2-yl]methoxy}methyl)pyridin-2-yl]methanol (1, Scheme 3)
Ethyl acetate (130 mL) was added to a mixture of dibenzyl derivative 18 (12.36 g, 28.00 mmol) and Pd/C catalyst (1.86 g, 15 w/w% Pd) under argon. Argon was replaced by hydrogen, and the mixture was stirred at 40 °C for 6 h under a hydrogen atmosphere (atm pressure). The mixture was filtered through folded filter paper, and the filtrate was evaporated. The crude product was purified by column chromatography on a silica gel adsorbent using a mixture of methanol/toluene = 1:5 as an eluent to give bipyridine diol 1 (6.13 g, 84%) as a pale-yellow crystalline solid. Rf: 0.25 (SiO2, methanol/toluene = 1:4).
This product was identical in every aspect to that reported in [
1,
14].
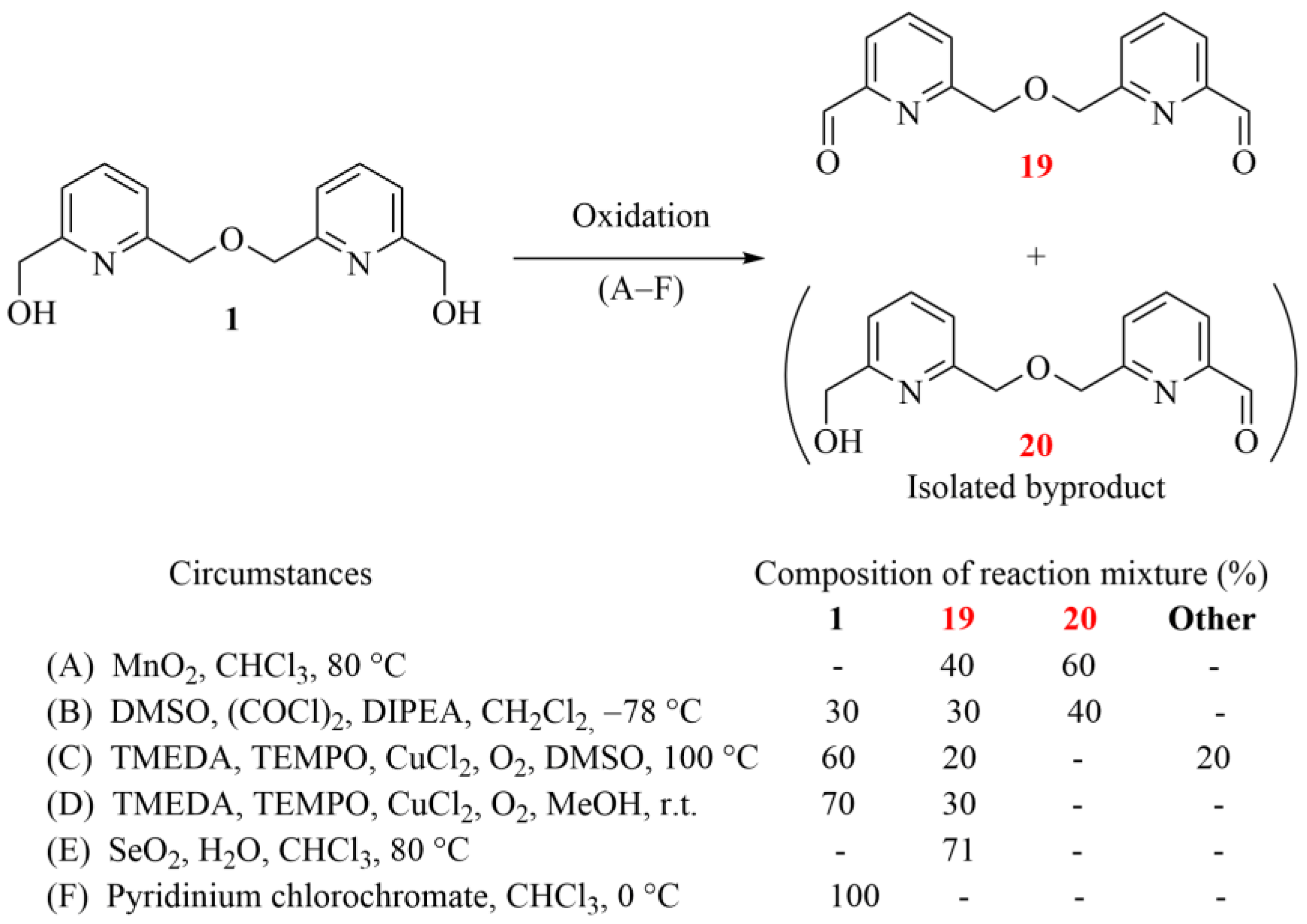
2.2.6. Preparation of 6,6′-[oxybis(methylene)]dipyridine-2-carbaldehyde (19, Schemes 3 and 5)
Procedure ‘A’
Bipyridine diol 1 (100 mg, 0.38 mmol) and manganese dioxide (134 mg, 1.54 mmol) were added to chloroform (1 mL). The mixture was stirred for 3 h at 80 °C in a pressure flask. The mixture was filtered and evaporated. The crude product was purified by column chromatography on a silica gel absorbent using a mixture of ethanol/toluene = 1:10 as an eluent. Bipyridine dialdehyde 19 (39 mg, 40%) is a white crystalline solid.
M.p.: 120–121 °C. Rf: 0.5 (SiO2, ethanol/toluene = 1:3), 0.3 (SiO2, ethanol/toluene = 1:10). 1H-NMR (500 MHz, CDCl3) δ 10.04 (s, 2H), 7.94–7.85 (m, 4H), 7.76 (dd, J = 7.1, 1.8 Hz, 2H), 4.89 (s, 4H). 13C-NMR (125 MHz, DEPTQ-135, CDCl3) δ 193.3, 158.9, 152.3, 137.8, 125.7, 120.7, 73.5. IR: νmax [cm−1]: 2847, 2372, 1707, 1624, 1592, 1522, 1445, 1390, 1350, 1263, 1243, 1212, 1169, 1112, 1039, 869, 802, 764, 664. HRMS (m/z): [MH+]: 257.0881, calculated [M+]: 256.0848.
Procedure ‘B’
Oxalyl chloride (410 mg, 3.23 mmol) was dissolved in DCM (1.4 mL) and cooled to 0 °C under an argon atmosphere. DMSO (417 mg, 5.08 mmol) was added to this solution dropwise. Then, diol 1 (100 mg, 0.39 mmol) in Hünig’s base (1.49 g, 11.50 mmol) and DCM (0.5 mL) was added dropwise, and the mixture was stirred for 45 min at 0 °C. The solution was evaporated, and the crude product was purified by column chromatography on a silica gel absorbent using a mixture of ethanol/toluene = 1:10 as an eluent. Bipyridine 19 (30 mg, 30%) is a white crystalline solid.
This product was identical in every aspect to that reported in Procedure ‘A’ in
Section 2.2.6.
Procedure ‘C’
Bipyridine diol 1 (100 mg, 0.38 mmol) was dissolved in DMSO (2 mL). The solution of CuCl2/TMEDA (30 µL, 0.003 mmol, 0.1 M in MeCN) and the solution of TEMPO (300 µL, 0.03 mmol, 0.1 M in DMSO) were added under an oxygen atmosphere (atm pressure). The mixture was stirred for 3 h at 100 °C and then evaporated, and the crude product was purified by column chromatography on a silica gel absorbent using a mixture of ethanol/toluene = 1:10 as an eluent to give bipyridine dialdehyde 19 (20 mg, 20%).
This product was identical in every aspect to that reported in Procedure ‘A’ in
Section 2.2.6.
Procedure ‘D’
Bipyridine diol 1 (100 mg, 0.38 mmol) was dissolved in methanol (2 mL). The solution of CuCl2/TMEDA (30 µL, 0.003 mmol, 0.1 M in MeCN) and the solution of TEMPO (300 µL, 0.03 mmol, 0.1 M in DMSO) were added under an oxygen atmosphere (atm pressure). The mixture was stirred for 3 h at room temperature. The solution was evaporated, and the crude product was purified by column chromatography on a silica gel absorbent using a mixture of ethanol/toluene = 1:10 as an eluent to give bipyridine dialdehyde 19 (30 mg, 30%).
This product was identical in every aspect to that reported in Procedure ‘A’ in
Section 2.2.6.
Procedure ‘E’
Chloroform (40 mL) was added to a mixture of bipyridine diol 1 (2.00 g, 7.68 mmol) and selenium dioxide (3.41 g, 30.70 mmol). A catalytic amount of water was added to this mixture, and it was stirred at 80 °C for 3 h in a pressure flask. The mixture was filtered, and the filtrate was extracted with aqueous hydrochloric acid solution (50 mL, 10 w/w%). The pH of the aqueous phase was set to 11, and it was extracted with ethyl acetate (3 × 50 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated to give bipyridine dialdehyde 19 (1.40 g, 71%).
This product was identical in every aspect to that reported in Procedure ‘A’ in
Section 2.2.6.
Procedure ‘F’
Pyridinium chlorochromate (332 mg, 1.54 mmol) was dissolved in chloroform (2 mL) and cooled to 0 °C under an argon atmosphere. Diol 1 (100 mg, 0.38 mmol) was added to this solution, and it was stirred for 3 h at 0 °C. The reaction was monitored by TLC, and it showed that the desired product did not form in this case.
2.2.7. Preparation of 6-({[6-(hydroxymethyl)pyridin-2-yl]methoxy}methyl)pyridine-2-carbaldehyde (20, Schemes 3 and 5)
Bipyridine derivative
20 is a byproduct; its synthesis is described above in
Section 2.2.6. Bipyridine derivative
20 is a white crystalline solid, and it had the highest yields in procedures ‘A’ (60 mg, 40%) and ‘B’ (40 mg, 60%).
M.p.: 83–84 °C. Rf: 0.44 (SiO2, ethanol/toluene = 1:4). 1H-NMR (500 MHz, CDCl3) δ 10.03 (s, 1H), 7.93–7.82 (m, 2H), 7.79–7.64 (m, 2H), 7.44–7.36 (m, 1H), 7.18–7.13 (m, 1H), 4.84 (s, 2H), 4.78 (s, 2H), 4.73 (s, 2H), 3.92 (broad s, 1H). 13C-NMR (125 MHz, DEPTQ-135, CDCl3) δ 193.3, 159.1, 158.6, 156.8, 152.2, 137.7, 137.4, 125.7, 120.6, 120.0, 119.4, 73.7, 73.2, 64.0. IR: νmax [cm−1]: 3174, 2955, 2918, 2884, 2850, 1711, 1596, 1581, 1458, 1435, 1340, 1309, 1262, 1209, 1113, 1081, 1050, 1023, 991, 896, 831, 776, 760, 668, 633, 554. HRMS (m/z): [MH+]: 259.1038, calculated [M+]: 258.1004.
2.2.8. Preparation of 1-[6-({[6-(1-hydroxyethyl)pyridin-2-yl]methoxy}methyl)pyridin-2-yl]ethan-1-ol (21, Scheme 3)
Methyl iodide (605 μL, 9.77 mmol) was added to activated magnesium (284 mg, 11.70 mmol) in diethyl ether (1 mL), and this mixture was refluxed under argon. After the reaction was completed, dialdehyde 19 (250 mg, 0.96 mmol) in dioxane (1 mL) was added dropwise, and the mixture was stirred for 48 h at room temperature. Water (10 mL) and ethyl acetate (10 mL) were added, and the aqueous phase was extracted with ethyl acetate (3 × 5 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated. The crude product was purified by column chromatography on a silica gel absorbent using a mixture of ethanol/toluene = 1:10 as an eluent to give diol 21 (40 mg, 14%) as a pale-yellow crystalline solid.
M.p.: >300 °C (decomposition). Rf: 0.12 (SiO2, ethanol/toluene = 1:3), 0.20 (SiO2, ethanol/toluene = 1:3). 1H-NMR (500 MHz, CDCl3) δ 7.66 (t, J = 7.6 Hz, 2H), 7.13 (d, J = 7.7 Hz, 2H), 7.05 (d, J = 7.7 Hz, 2H), 5.26 (s, 2H), 4.92 (s, 2H), 4.91–4.89 (m, 2H), 4.88 (s, 2H), 1.27–1.22 (m, 6H). 13C-NMR (125 MHz, DMSO-d6) δ 148.5, 146.4, 142.4, 128.7, 127.9, 72.5, 61.1, 20.7. IR: νmax [cm−1]: 3445, 3339, 2962, 2925, 2853, 1735, 1597, 1582, 1450, 1410, 1365, 1259, 1085, 1014, 864, 794, 703, 661, 399. HRMS (m/z): [MH+]: 289.1507, calculated [M+]: 288.1474.
2.2.9. Preparation of 6-(((6-(1-hydroxyethyl)pyridin-2-yl)methoxy)methyl)picolinaldehyde (22, Scheme 3)
Bipyridine
22 is a byproduct; its synthesis is described in
Section 2.2.8. It is a white crystalline solid, and its yield was 60 mg (23%).
M.p.: >300 °C (decomposition). Rf: 0.58 (SiO2, ethanol/toluene = 1:3). 1H-NMR (500 MHz, CDCl3) δ 9.99 (s, 1H), 7.88 (d, J = 7.3 Hz, 1H), 7.76 (dd, J = 7.5, 1.8 Hz, 1H), 7.71 (t, J = 7.7 Hz, 2H), 7.42 (d, J = 7.7 Hz, 1H), 7.16 (d, J = 7.8 Hz, 1H), 4.86 (s, 2H), 4.85–4.84 (m, 1H), 4.81 (s, 2H), 1.21–1.16 (m, 3H). 13C-NMR (125 MHz, DMSO-d6) δ 192.4, 148.2, 146.2, 142.6, 128.7, 128.3, 128.2, 128.0, 127.6, 127.1, 81.0, 72.7, 60.7, 21.6. IR: νmax [cm−1]: 3262, 2922, 2850, 1711, 1594, 1578, 1465, 1341, 1311, 1261, 1209, 1113, 1083, 1057, 1023, 992, 908, 791, 776, 759, 668, 633, 554. HRMS (m/z): [MH+]: 273.3097, calculated [M+]: 272.3040.
2.2.10. Preparation of 6-((benzyloxy)methyl)pyridine-2-carbaldehyde (23, Scheme 5)
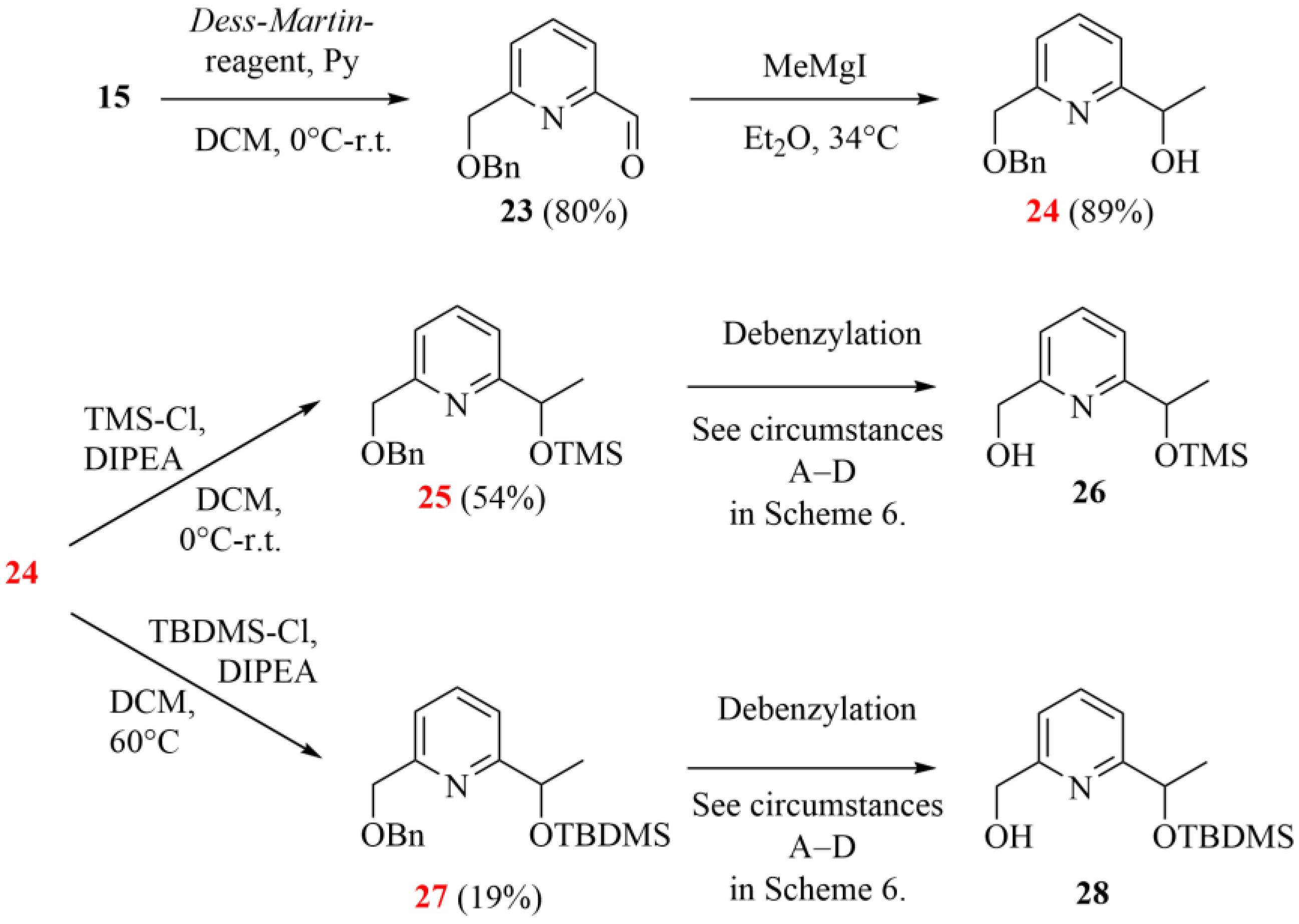
The monobenzyl derivative of pyridinedimethanol (15, 2.35 g, 10.25 mmol) was dissolved in a mixture of DCM (65 mL), pyridine (2.56 mL) and water (2.75 mL). Dess–Martin reagent (5.65 g, 13.32 mmol) was added in small portions to this mixture, and it was stirred for 1 day at room temperature under an argon atmosphere. Silica gel (5.0 g) was added to the mixture, and the volatile components were evaporated. The crude product was purified by column chromatography on a silica gel adsorbent using a mixture of propane-2-ol/hexane = 1:10 as an eluent to give aldehyde 23 (1.91 g, 80%) as a pale-yellow viscous oil. Rf: 0.6 (SiO2, propane-2-ol/hexane = 1:10).
This product was identical in every aspect to that reported in [
35].
2.2.11. Preparation of 1-{6-[(benzyloxy)methyl]pyridin-2-yl}ethan-1-ol (24, Scheme 5)
Aldehyde 23 (500 mg, 2.20 mmol) in diethyl ether (1 mL) was added dropwise to a stirred solution of methylmagnesium iodide (498 mg, 3.00 mmol) in 10 mL diethyl ether under an argon atmosphere for 10 min at room temperature. The mixture was refluxed for 2 h and then cooled to room temperature. Saturated aqueous NH4Cl solution (20 mL) was added to the mixture, and then the aqueous phase was extracted with diethyl ether (3 × 30 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated. The crude product was purified by column chromatography on a silica gel adsorbent using a mixture of propan-2-ol/cyclohexane = 1:10 as an eluent to give alcohol 24 (476 mg, 89%) as a pale-yellow viscous oil.
Rf: 0.3 (SiO2, propane-2-ol/cyclohexane = 1:10). 1H-NMR (500 MHz, CDCl3) δ 7.62 (t, J = 7.7 Hz, 1H), 7.34–7.21 (m, 6H), 7.09 (d, J = 7,7 Hz, 1H), 4.79 (q, J = 6.6 Hz, 1H), 4.61 (s, 2H), 4.56 (s, 2H), 4.42 (broad s, 1H), 1.41 (d, J = 6.6 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 162.3, 157.1, 137.9, 137.7, 128.5, 127.8, 127.8, 119.8, 118.5, 73.0, 72.7, 68.5, 24.2. IR: νmax [cm−1]: 3402 (broad), 3074, 3061, 3027, 2921, 2882, 2817, 1590, 1470, 1454, 1443, 1389, 1344, 1305, 1230, 1075, 1027, 916, 804, 735, 464. HRMS (m/z): [MH+]: 244.1293, calculated [M+]: 243.1259.
2.2.12. Preparation of 2-[(benzyloxy)methyl]-6-{1-[(trimethylsilyl)oxy]ethyl}pyridine (25, Scheme 5)
Alcohol 24 (500 mg, 2.06 mmol) was dissolved in a mixture of DCM (10 mL) and DIPEA (1.07 g, 8.28 mmol). Trimethylsilyl chloride (448 mg, 4.12 mmol) in 1 mL DCM was added dropwise to the mixture for 5 min at 0 °C under an argon atmosphere. The mixture was allowed to warm up to room temperature and stirred for 1 day. Water (50 mL) was added to the reaction mixture, and the aqueous phase was extracted with DCM (3 × 50 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated. The crude product was purified by column chromatography on a silica gel adsorbent using a mixture of acetone/hexane = 1:30 as an eluent to give 25 (350 mg, 54%) as a pale-yellow viscous oil.
Rf: 0.3 (SiO2, acetone/hexane = 1:30). 1H-NMR (500 MHz, CDCl3) δ 7.70 (t, J = 7.8 Hz, 1H), 7.45–7.27 (m, 7H), 4.95 (q, J = 6.5 Hz, 1H), 4.68 (s, 2H), 4.65 (s, 2H), 1.47 (d, J = 6.5 Hz, 3H), 0.12 (s, 9H). 13C-NMR (125 MHz, CDCl3) δ 164.9, 157.2, 138.1, 137.5, 128.5, 127.9, 127.8, 119.6, 118.1, 73.2, 73.0, 71.8, 25.6, 0.1. IR: νmax [cm−1] (film): 3079, 3057, 3030, 2917, 2874, 2825, 1592, 1578, 1453, 1397, 1360, 1346, 1250, 1152, 1115, 1099, 1078, 1028, 992, 958, 890, 841, 800, 736, 697, 622. HRMS (m/z): [MH+]: 316.1688, calculated [M+]: 315.1655.
2.2.13. Preparation of 2-[(benzyloxy)methyl]-6-{1-[(tert-butyldimethylsilyl)oxy]ethyl}pyridine (27, Scheme 5)
Alcohol 24 (110 mg, 0.45 mmol) was dissolved in a mixture of THF (2.5 mL) and DIPEA (234 mg, 1.81 mmol). Tert-butyldimethylsilyl chloride (136 mg, 0.90 mmol) in 0.5 mL THF was added dropwise to the mixture, and it was heated to 60 °C and stirred at this temperature for 1 day. After the reaction was complete, the volatile components were evaporated. The crude product was purified by PTLC on a silica gel adsorbent using a mixture of acetone/hexane = 1:10 as an eluent to give 27 (30 mg, 19%) as a pale-yellow viscous oil.
Rf: 0.6 (SiO2, acetone/hexane = 1:10). 1H-NMR (500 MHz, CDCl3) δ 7.76 (t, J = 7.7 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.42–7.27 (m, 6H), 5.03–4.99 (m, 1H), 4.73 (s, 2H), 4.68 (s, 2H), 1.48 (d, J = 6,4 Hz, 3H), 0.94 (s, 9H), 0.11 (s, 3H), 0.04 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 165.1, 156.8, 138.0, 137.7, 128.4, 127.9, 127.8, 119.6, 118.1, 73.0, 72.7, 71.7, 25.9, 25.8, 18.2, −4.8, −4.9. IR: νmax [cm−1]: 2953, 2928, 2884, 1594, 1578, 1455, 1390, 1361, 1253, 1115, 1080, 1020, 736, 622, 606. HRMS (m/z): [MH+]: 358.2158, calculated [M+]: 357.2124.
2.2.14. Preparation of (6-{[(tert-butyldimethylsilyl)oxy]methyl}pyridin-2-yl)methanol (29, Scheme 7)
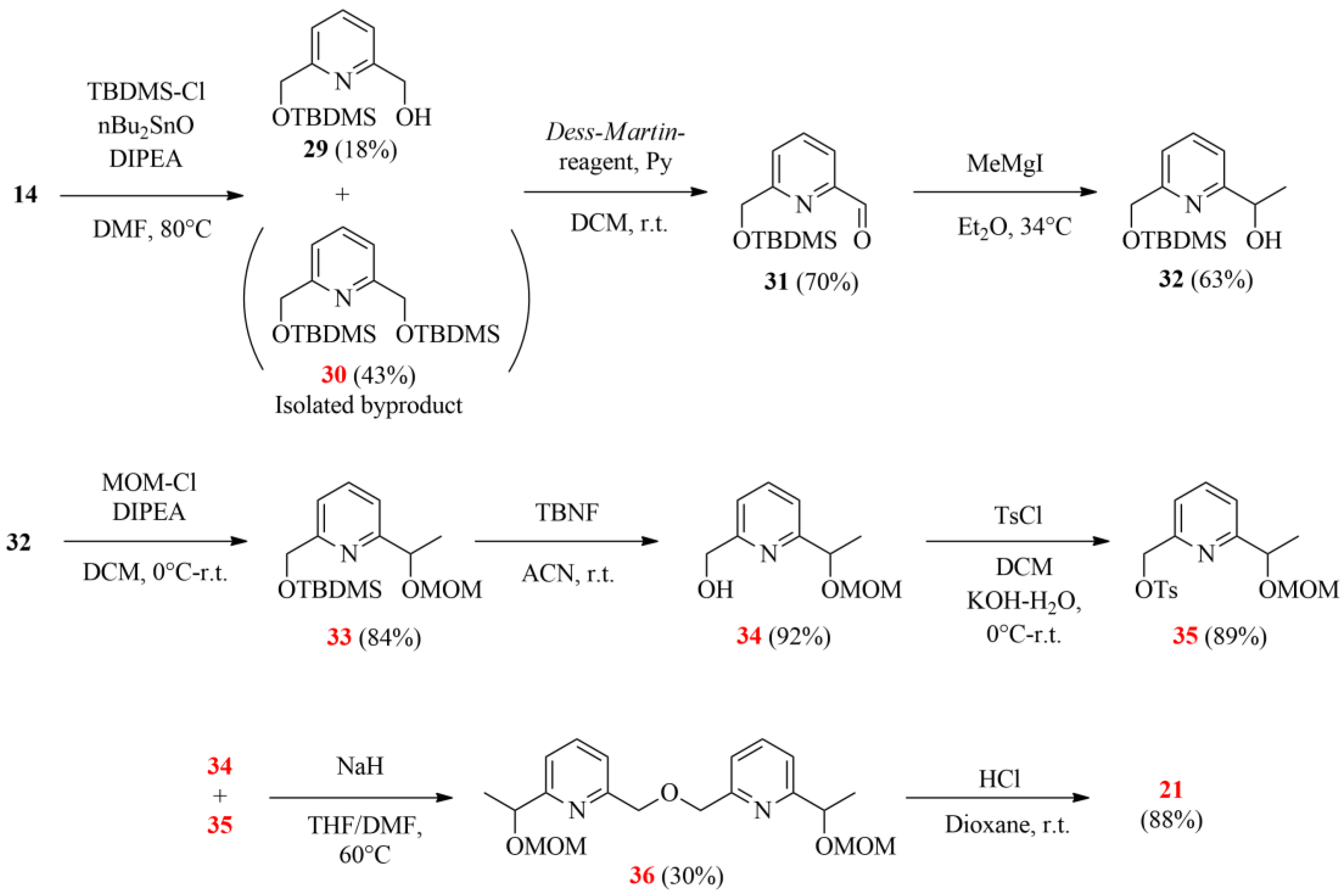
Diol 14 (3.78 g, 27.20 mmol) was dissolved in DMF (112 mL). Tert-butyldimethylsilyl chloride (4.52 g, 30.00 mmol) and dibutyltin oxide (6.72 g, 27.00 mmol) were added to the solution, and then the resulting mixture was heated to 80 °C and stirred under argon at this temperature. After 2 days, the volatile components were evaporated. The residue was taken up in DCM (200 mL) and extracted with aqueous NaOH solution (100 mL, 10 w/w%). The precipitate was filtered, and the organic phase was dried with magnesium sulfate, filtered and evaporated. The crude product was purified by column chromatography on a silica gel absorbent using a gradient elution of a propan-2-ol/cyclohexane mixture (1→20 V/V% propane-2-ol) to give monoprotected pyridinemethanol 29 (1.24 g, 18%) as a pale-yellow viscous oil. Rf: 0.35 (SiO2, propane-2-ol/hexane = 1:10).
This product was identical in every aspect to that reported in [
36].
2.2.15. Preparation of 2,6-bis{[(tert-butyldimethylsilyl)oxy]methyl}pyridine (30, Scheme 7)
Double-protected pyridine derivative
30 is a byproduct; its synthesis is described in
Section 2.2.14. Its yield was 4.29 g (43%). It is a pale-yellow viscous oil.
Rf: 0.65 (SiO2, propane-2-ol/hexane = 1:10), 0.6 (SiO2, propane-2-ol/hexane = 1:20), 0.4 (SiO2, propane-2-ol/hexane = 1:50). 1H-NMR (500 MHz, CDCl3) δ 7.72 (t, J = 7.7 Hz, 1H), 7.37 (d, J = 7.7 Hz, 2H), 4.80 (s, 4H), 0.96 (s, 18H), 0.12 (s, 12H). 13C-NMR (125 MHz, CDCl3) δ 160.2, 137.3, 118.0, 66.1, 25.9, 18.4, −5.3, −5.4. IR: νmax [cm−1]: 2954, 2929, 2885, 2856, 1593, 1579, 1471, 1461, 1450, 1389, 1362, 1254, 1112, 1006, 909, 835, 776, 736, 668, 606. HRMS (m/z): [MH+]: 368.2396, calculated [M+]: 367.2363.
2.2.16. Preparation of 6-{[(tert-butyldimethylsilyl)oxy]methyl}pyridine-2-carbaldehyde (31, Scheme 7)
Alcohol 29 (1.22 g, 4.81 mmol) was dissolved in a mixture of DCM (65 mL), pyridine (2.6 mL) and water (2.8 mL). Dess–Martin reagent (3.06 g, 7.22 mmol) was added to this solution in small portions, and the resulting mixture was stirred for 1 day at room temperature under an argon atmosphere. When the reaction was complete, hexane (25 mL) was added to the reaction mixture, and it was filtered. The filtrate was evaporated, and the crude product was purified by column chromatography on a silica gel adsorbent using a mixture of propane-2-ol/cyclohexane = 1:50 as an eluent to give 31 (0.85 g, 70%) as a pale-yellow viscous oil. Rf: 0.1 (SiO2, propane-2-ol/cyclohexane = 1:100), 0.4 (SiO2, propane-2-ol/cyclohexane = 1:50).
This product was identical in every aspect to that reported in [
15].
2.2.17. Preparation of 1-(6-{[(tert-butyldimethylsilyl)oxy]methyl}pyridin-2-yl)ethan-1-ol (32, Scheme 7)
Aldehyde 31 (815 mg, 3.24 mmol) in diethyl ether (1.5 mL) was added dropwise to a stirred solution of methylmagnesium iodide (747 mg, 4.50 mmol in 10 mL diethyl ether) under an argon atmosphere at room temperature for 10 min. The mixture was refluxed for 30 min and then cooled to room temperature. Saturated aqueous NH4Cl solution (20 mL) was added to the mixture, and the aqueous phase was extracted with diethyl ether (3 × 20 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated. The product did not need purification. Alcohol 32 (550 mg, 63%) is a pale-yellow viscous oil. Rf: 0.3 (SiO2, propane-2-ol/cyclohexane = 1:20).
This product was identical in every aspect to that reported in [
15].
2.2.18. Preparation of 2-{[(tert-butyldimethylsilyl)oxy]methyl}-6-[1-(methoxymethoxy)ethyl]pyridine (33, Scheme 7)
Alcohol 32 (490 mg, 1.83 mmol) was added to a mixture of DCM (10 mL) and DIPEA (1040 mg, 8.05 mmol). Methoxymethylene chloride (440 mg, 5.47 mmol) in 10 mL DCM was added dropwise to this solution for 20 min at 0 °C under an argon atmosphere. The mixture was warmed up to room temperature and stirred for 3 days. The volatile components were evaporated, and the crude product was purified by column chromatography on a silica gel adsorbent using a mixture of propane-2-ol/hexane = 1:70 as an eluent to give 33 (480 mg, 84%) as a pale-yellow viscous oil.
Rf: 0.5 (SiO2, propane-2-ol/hexane = 1:70). 1H-NMR (500 MHz, CDCl3) δ 7.69 (t, J = 7.7 Hz, 1H), 7.40 (d, J = 7.5 Hz, 1H), 7.27 (d, J = 7.8 Hz, 1H), 4.84–4.76 (m, 3H), 4.64 (dd, J = 43.7, 28.1 Hz, 2H), 3.35 (s, 3H), 1.48 (d, J = 6.6 Hz, 3H), 0.94 (s, 9H), 0.11 (s, 6H). 13C-NMR (125 MHz, CDCl3) δ 161.7, 160.7, 137.3, 118.5, 118.2, 94.9, 75.5, 66.1, 55.5, 25.9, 18.4, −5.3, −5.4. IR: νmax [cm−1]: 2954, 2929, 2887, 2857, 1593, 1579, 1462, 1362, 1255, 1216, 1156, 1102, 1081, 1035, 1005, 993, 920, 851, 835, 815, 798, 777, 671, 599. HRMS (m/z): [MH+]: 312.1950, calculated [M+]: 311.1917.
2.2.19. Preparation of {6-[1-(methoxymethoxy)ethyl]pyridin-2-yl}methanol (34, Scheme 7)
Tetrabutylammonium fluoride (604 mg, 2.31 mmol) in 2.5 mL acetonitrile was added dropwise to diprotected pyridine derivative 33 (480 mg, 1.54 mmol) in acetonitrile (7.5 mL) for 10 min. The mixture was stirred for 2 days at room temperature, and then the volatile components were evaporated. The residue was taken up in diethyl ether (20 mL) and water (20 mL). The aqueous phase was extracted with ether (3 × 15 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated. The crude product was purified by column chromatography on a silica gel adsorbent using a mixture of propane-2-ol/hexane = 1:6 as an eluent to give 34 (280 mg, 92%) as a pale-yellow viscous oil.
Rf: 0.4 (SiO2, propane-2-ol/hexane = 1:6), 0.15 (SiO2, propane-2-ol/hexane = 1:10). 1H-NMR (500 MHz, CDCl3) δ 7.67 (t, J = 7.7 Hz, 1H), 7.32 (d, J = 7.7 Hz, 1H), 7.14 (d, J = 7.7 Hz, 1H), 4.83 (q, J = 6.6 Hz, 1H), 4.72 (s, 2H), 4.63 (dd, J = 45.7, 32.0 Hz, 2H), 3.34 (s, 3H), 1.48 (d, J = 6.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 161.9, 158.5, 137.6, 119.4, 118.9, 95.0, 75.2, 64.0, 55.6, 22.2. IR: νmax [cm−1]: 3389 (wide), 2931, 2890, 2824, 1595, 1578, 1460, 1370, 1217, 1157, 1099, 1073, 1030, 993, 918, 841, 800, 754, 733, 611. HRMS (m/z): [MH+]: 198.1085, calculated [M+]: 197.1052.
2.2.20. Preparation of {6-[1-(methoxymethoxy)ethyl]pyridin-2-yl}methyl 4-methylbenzenesulfonate (35, Scheme 7)
Alcohol 34 (100 mg, 0.51 mmol) was added to a mixture of DCM (8 mL) and aqueous KOH solution (8 mL, 40 w/w%). Tosyl chloride (156 mg, 0.82 mmol) in 2 mL DCM was added dropwise to this mixture with vigorous stirring at 0 °C. The mixture was warmed up to room temperature and stirred for 3 days. After the reaction was complete, water (10 mL) and DCM (10 mL) were added to this mixture. The phases were separated, and the aqueous phase was extracted with DCM (3 × 15 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated. Tosylate 35 (159 mg, 89%) is a pale-yellow viscous oil.
Rf: 0.4 (SiO2, propane-2-ol/hexane = 1:10). 1H-NMR (500 MHz, CDCl3) δ 7.82 (d, J = 8.3 Hz, 2H), 7.68 (t, J = 7.8 Hz, 1H), 7.36–7.28 (m, 4H), 5.12 (s, 2H), 4.74 (q, J = 6.6 Hz, 1H), 4.61 (dd, J = 55.1, 41.7 Hz, 2H), 3.32 (s, 3H), 2.43 (s, 3H), 1.43 (d, J = 6.6 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 162.7, 153.2, 145.1, 137.7, 132.9, 130.0, 128.2, 120.4, 119.9, 95.0, 75.3, 71.9, 55.6, 22.2, 21.7. IR: νmax [cm−1]: 2930, 2888, 2823, 1596, 1578, 1494, 1461, 1361, 1308, 1213, 1189, 1175, 1158, 1097, 1074, 1032, 991, 953, 917, 836, 813, 753, 668, 545. HRMS (m/z): [MH+]: 352.1174, calculated [M+]: 351.1140.
2.2.21. Preparation of 6,6′-[oxybis(methylene)]bis{2-[1-(methoxymethoxy)ethyl]pyridine} (36, Scheme 7)
Alcohol 34 (86 mg, 0.44 mmol) in THF (3 mL) was added dropwise to a stirred suspension of sodium hydride (105 mg, 2.62 mmol) in THF (1.5 mL) under an argon atmosphere. The mixture was stirred for 30 min at room temperature and for 2 h at 60 °C. Tosylate 35 (153 mg, 0.44 mmol) in a mixture of THF (3 mL) and DMF (1 mL) was added dropwise to the reaction mixture, and it was stirred for 3 days at 60 °C. After the reaction was complete, the volatile components were evaporated. The residue was taken up in ice-water (20 mL) and DCM (20 mL). The phases were separated, and the aqueous phase was shaken with DCM (3 × 15 mL). The combined organic phase was dried with magnesium sulfate, filtered and evaporated. The crude product was purified by column chromatography on a silica gel adsorbent using a mixture of propane-2-ol/hexane = 1:15 as an eluent to give dipyridine 36 (50 mg, 30%) as a pale-yellow viscous oil.
Rf: 0.1 (SiO2, propane-2-ol/hexane = 1:15). 1H-NMR (500 MHz, CDCl3) δ 7.71 (t, J = 7.7 Hz, 2H), 7.43 (d, J = 7.7 Hz, 2H), 7.34 (d, J = 7.7 Hz, 2H), 4.83 (q, J = 6.7 Hz, 2H), 4.77 (s, 4H), 4.66 (dd, J = 42.8, 29.1 Hz, 4H), 3.37 (s, 6H), 1.51 (d, J = 6.6 Hz, 6H). 13C-NMR (125 MHz, CDCl3) δ 162.4, 157.7, 137.3, 119.9, 118.8, 94.9, 75.5, 73.8, 55.5, 22.3. IR: νmax [cm−1]: 2928, 2887, 2822, 1592, 1577, 1458, 1400, 1366, 1341, 1260, 1215, 1155, 1099, 1032, 992, 918, 843, 801, 753, 599. HRMS (m/z): [MH+]: 377.2032, calculated [M+]: 376.1998.
2.2.22. Preparation of 1,1′-{[oxybis(methylene)]bis(pyridine-6,2-diyl)}bis(ethan-1-ol) (21, Scheme 7)
Aqueous HCl solution (2 mL, 1.0 M) was added dropwise to a solution of MOM-protected dipyridine derivative 36 (100 mg, 0.27 mmol) in 2 mL dioxane. The resulting reaction mixture was stirred at room temperature for 24 h, and then the volatile components were removed. The residue was taken up in water (20 mL) and ethyl acetate (20 mL). The aqueous phase was saturated with sodium chloride, and the phases were separated. The aqueous phase was extracted with ethyl acetate (3 × 15 mL). The combined organic phase was dried over anhydrous sodium sulfate, filtered and evaporated. The residue was triturated with methanol to give the compound dipyridine diol 21 as yellow crystals (67 mg, 0.27 mmol, 88%).
The product was identical in every aspect to that reported in
Section 2.2.8.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}