Small Deviations in Geometries Affect Detonation Velocities and Pressures of Nitroaromatic Molecules


Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Calculations of Detonation Velocity and Detonation Pressure
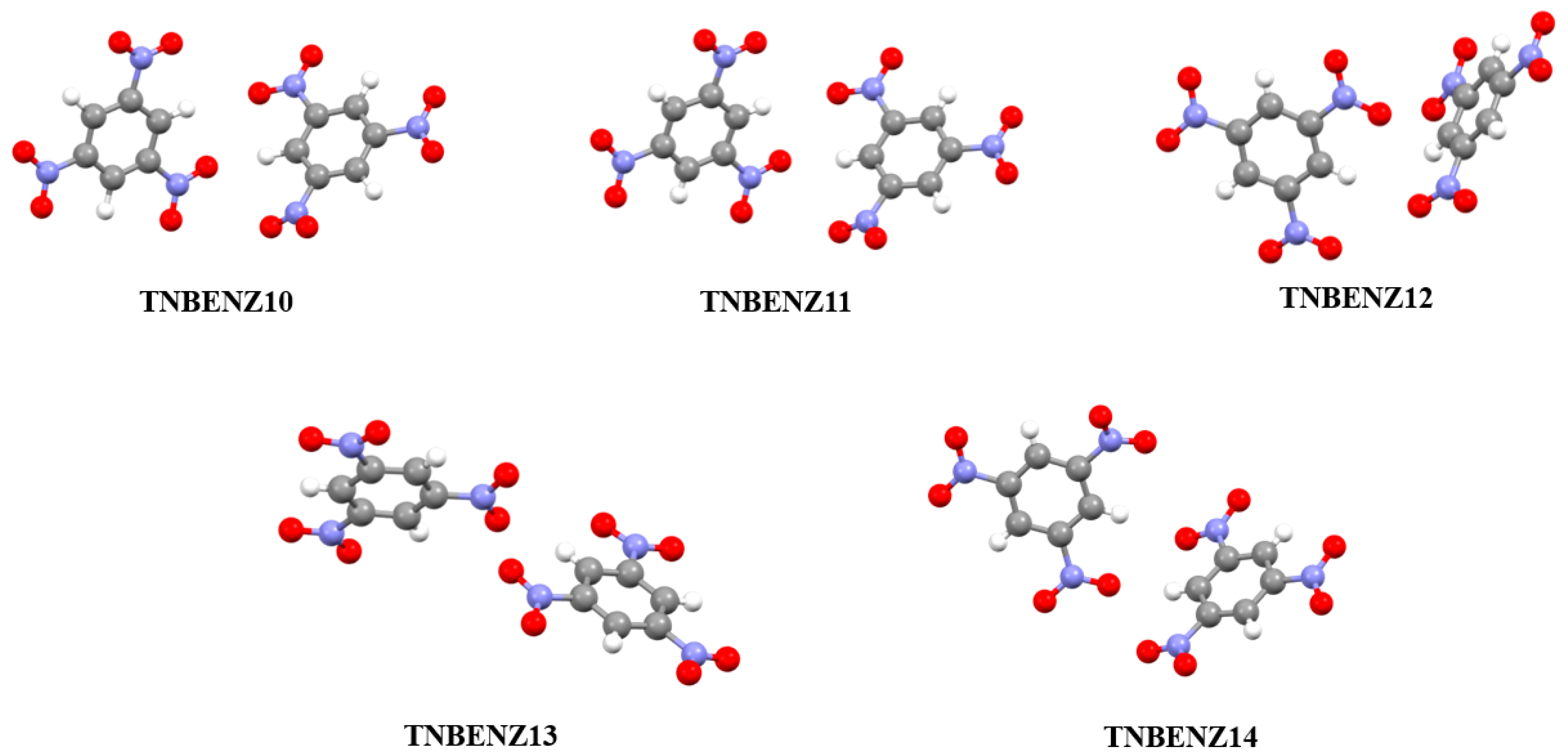
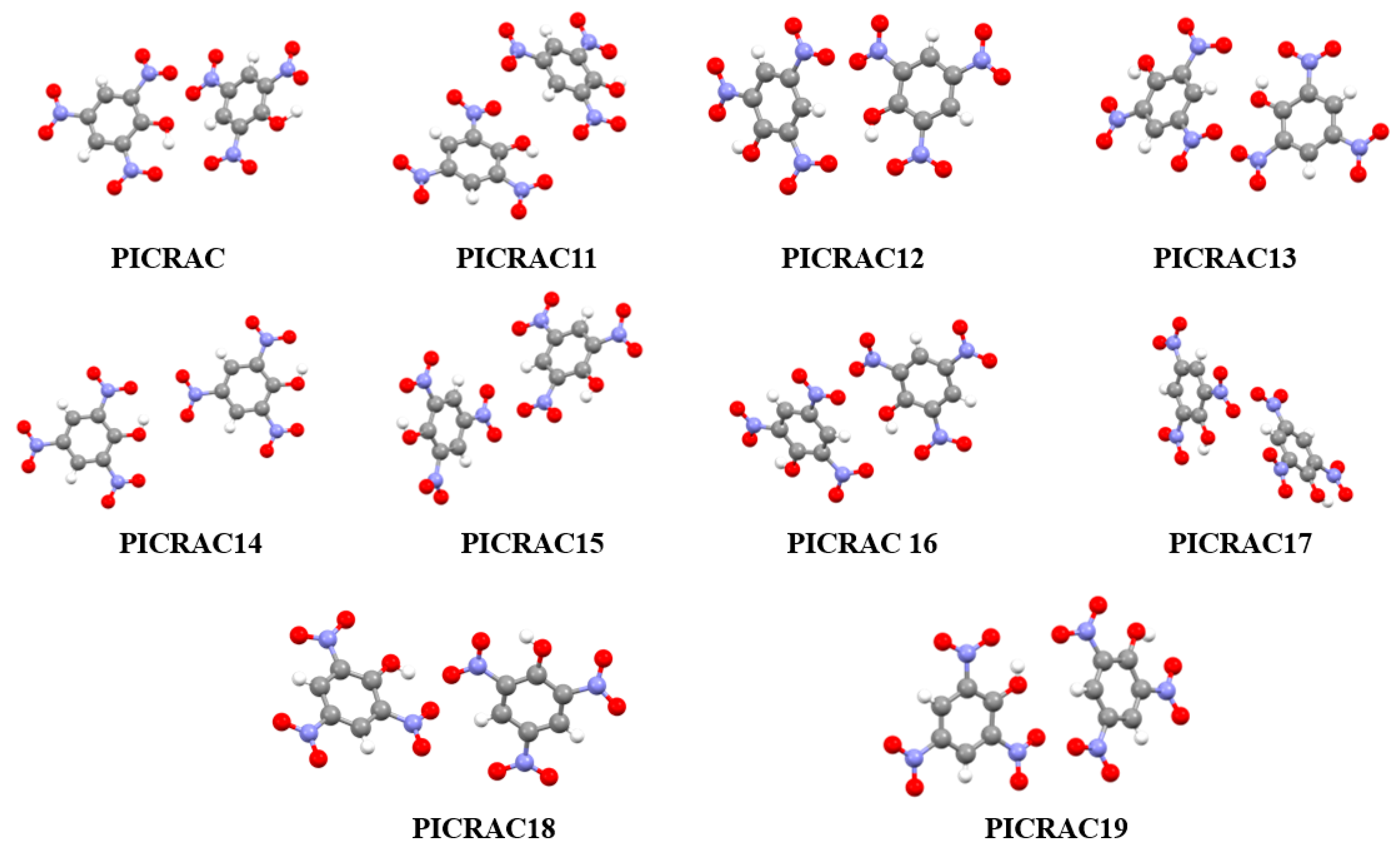
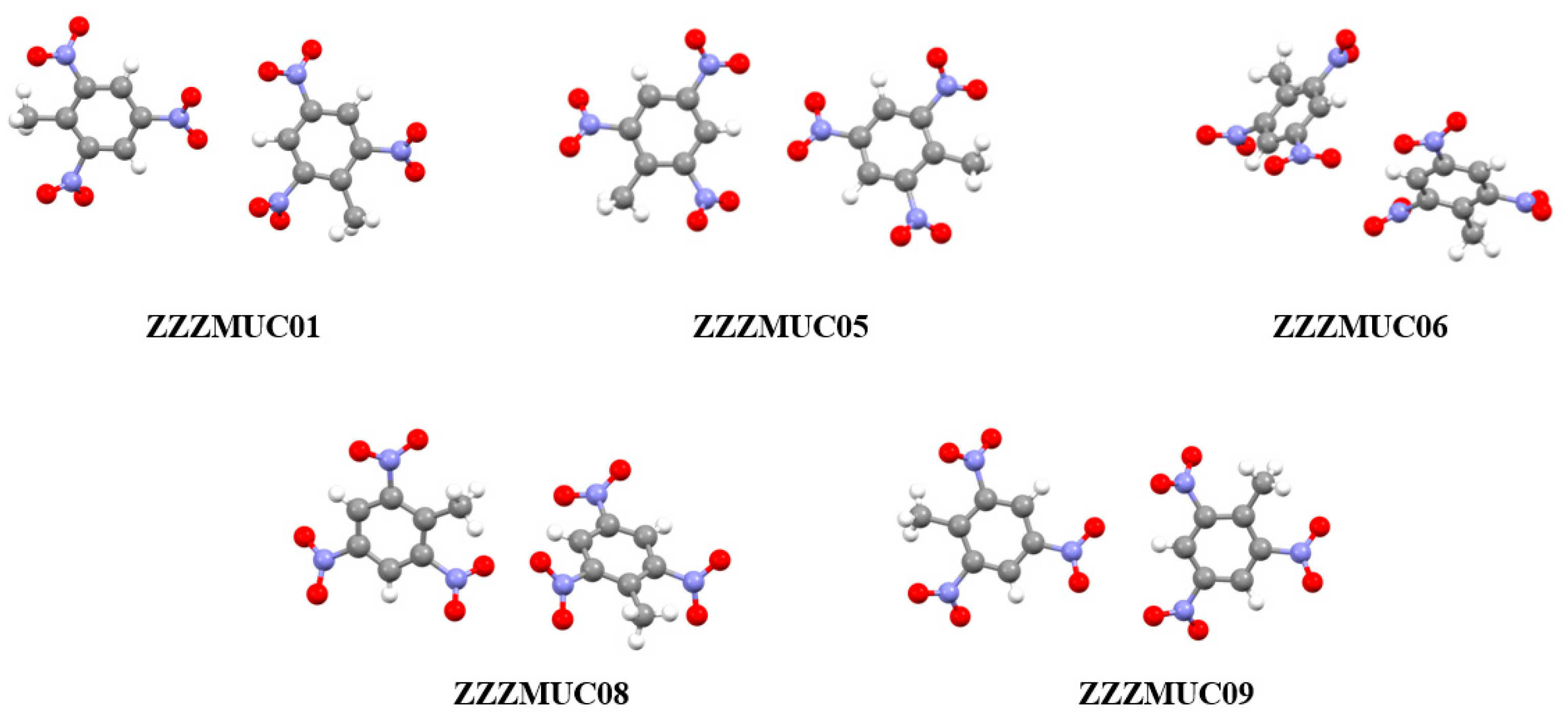
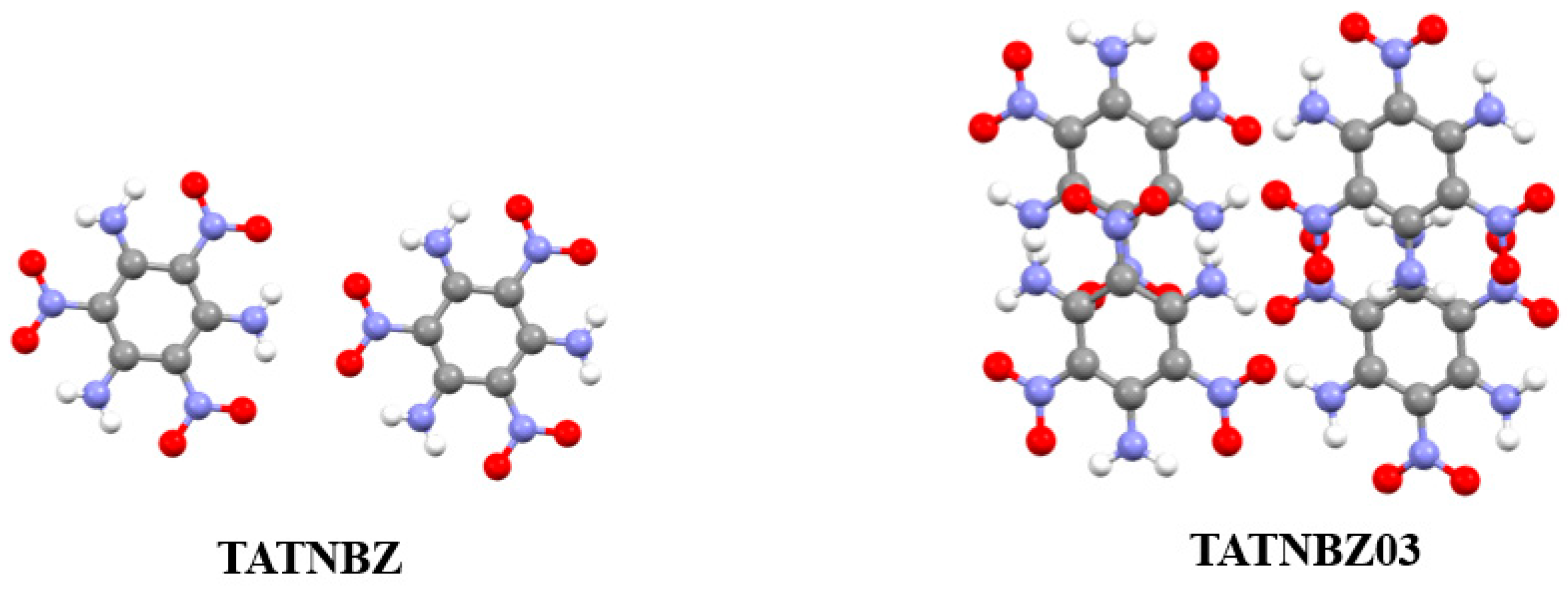
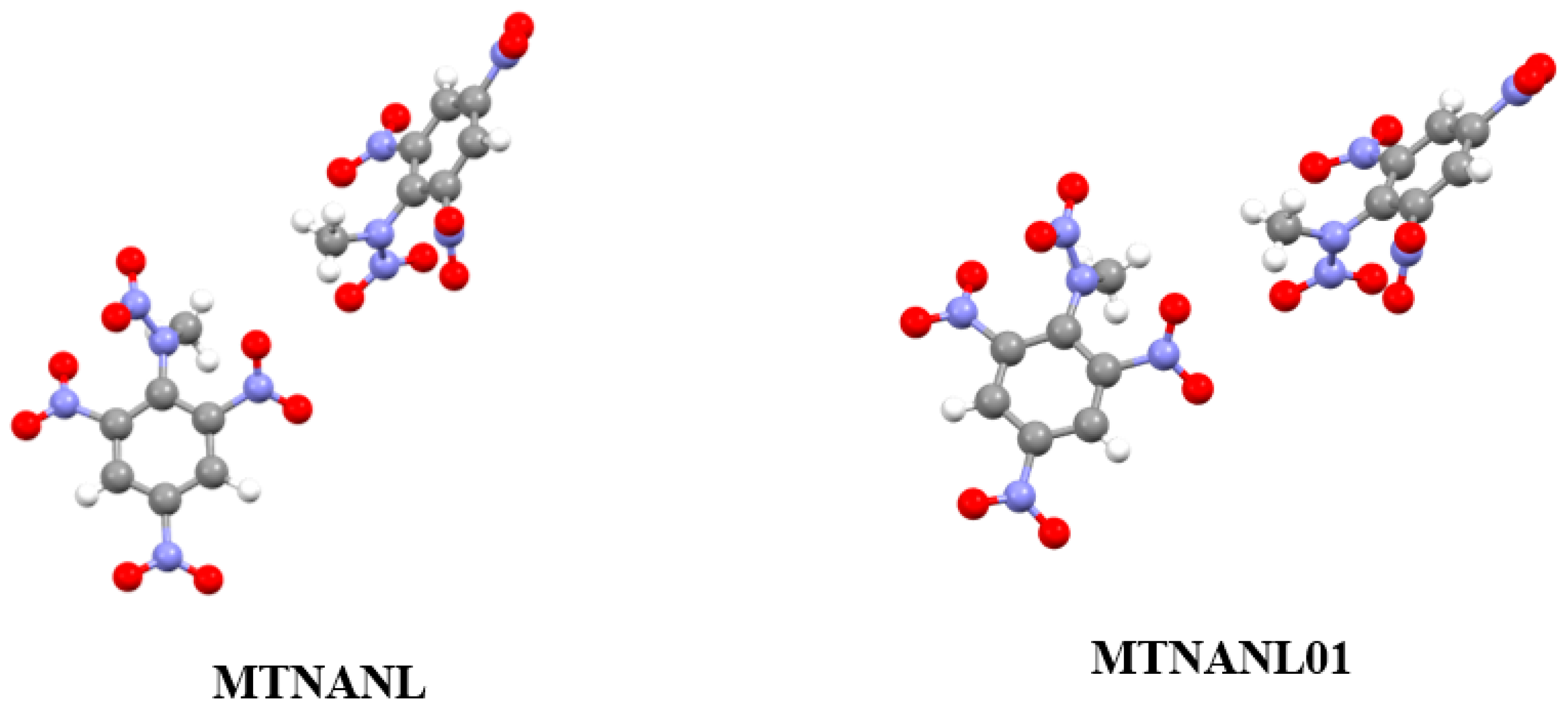
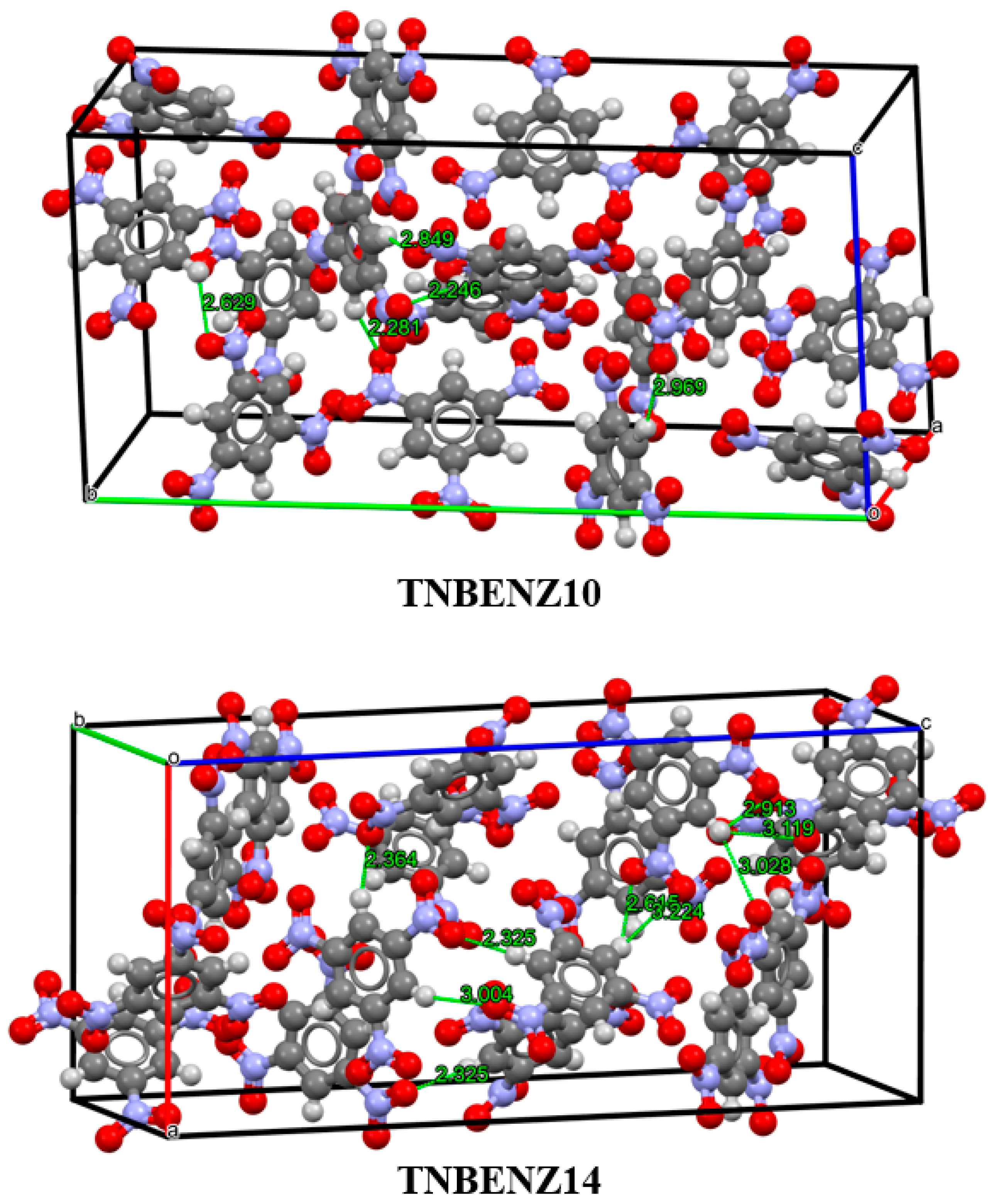
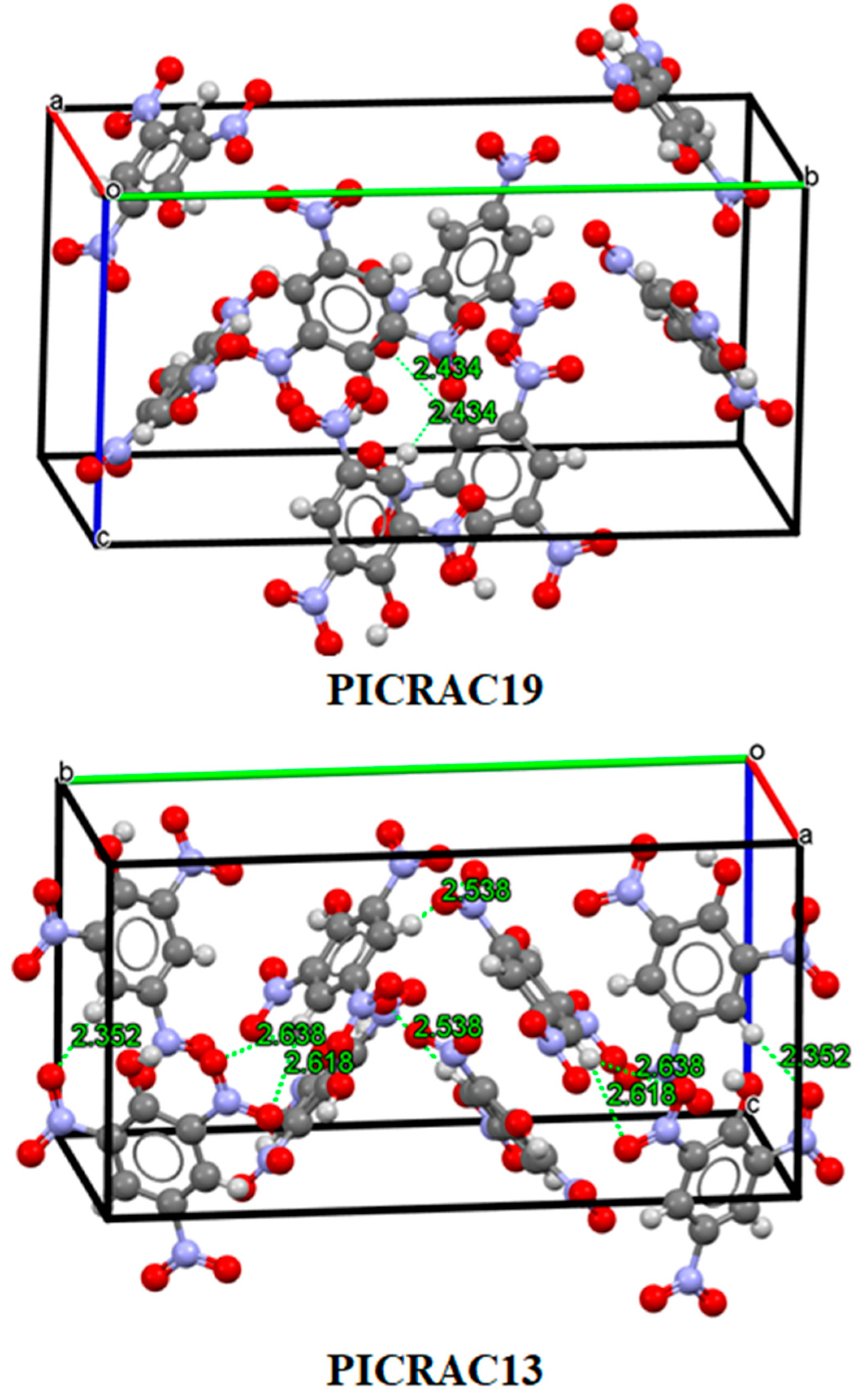
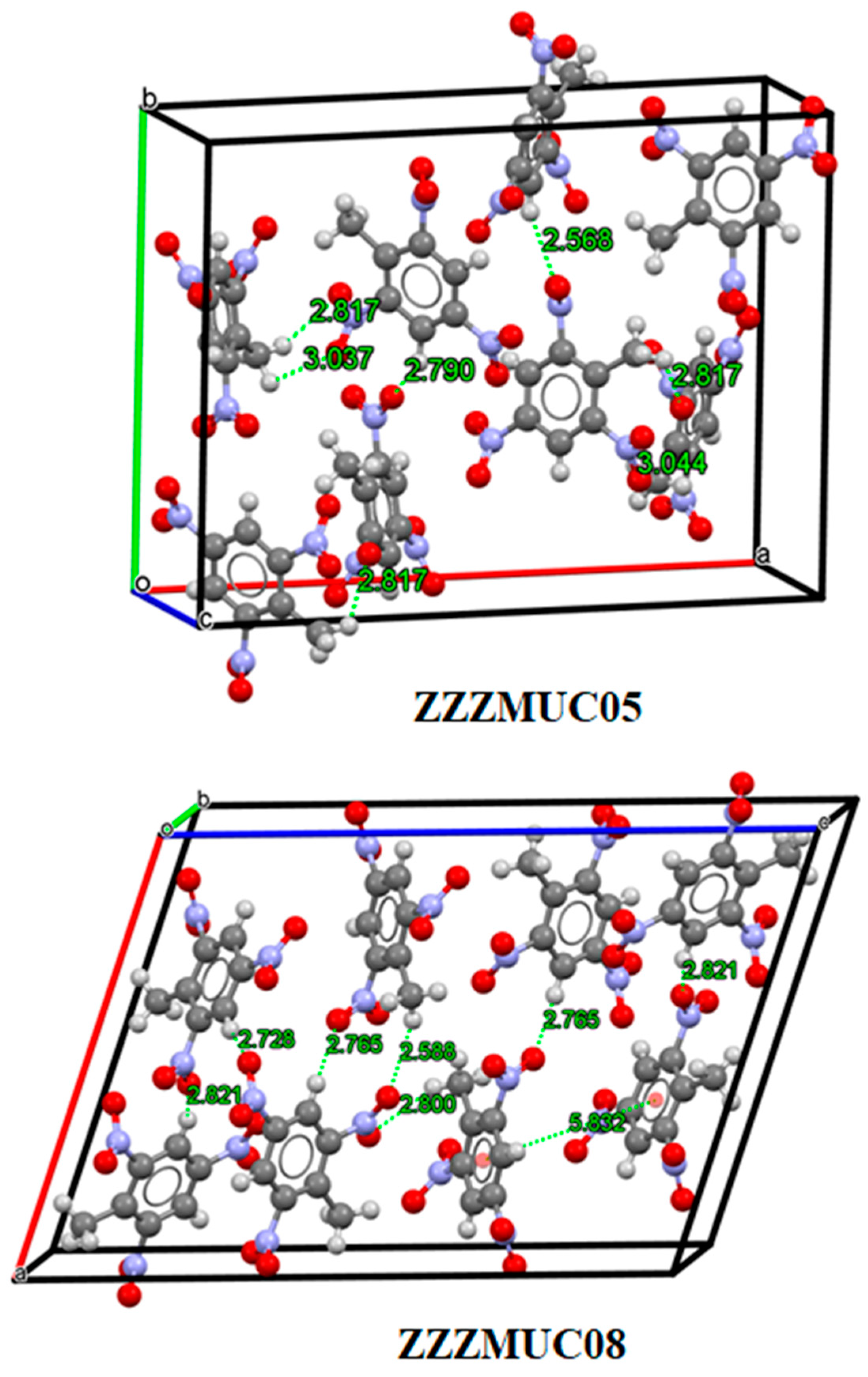
3.2. Analysis of Non-Covalent Interactions in Crystal Structures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Born, M.; Karaghiosoff, K.; Klapötke, T.M.; Voggenreiter, M. Oxetane Monomers Based on the Powerful Explosive LLM-116: Improved Performance, Insensitivity, and Thermostability. ChemPlusChem 2022, 87, e202200049. [Google Scholar] [CrossRef] [PubMed]
- Born, M.; Plank, J.; Klapötke, T.M. Energetic Polymers: A Chance for Lightweight Reactive Structure Materials? Prop. Explos. Pyrotech. 2022, 47, e202100368. [Google Scholar] [CrossRef]
- Agrawal, J.P.; Dodke, V.S. Some Novel High Energy Materials for Improved Performance. Z. Anorg. Allg. Chem. 2021, 647, 1856–1882. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. High Performance, Low Sensitivity: Conflicting or Compatible? Propellants Explos. Pyrotech. 2016, 41, 414–425. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Detonation Performance and Sensitivity: A Quest for Balance. In Advances in Quantum Chemistry; Academic Press: Cambridge, MA, USA, 2014; Volume 69, pp. 1–30. [Google Scholar]
- Nešić, J.; Cvijetić, I.N.; Bogdanov, J.; Marinković, A. Synthesis and Characterization of Azido Esters as Green Energetic Plasticizers. Prop. Explos. Pyrotech. 2021, 46, 1537–1546. [Google Scholar] [CrossRef]
- Kretić, D.S.; Veljković, I.S.; Veljković, D.Ž. Tris(3-nitropentane-2,4-dionato-κ2 O,O′) Complexes as a New Type of Highly Energetic Materials: Theoretical and Experimental Considerations. Chemistry 2023, 5, 1843–1854. [Google Scholar] [CrossRef]
- Kretić, D.S.; Medaković, V.B.; Veljković, D.Ž. How Do Small Differences in Geometries Affect Electrostatic Potentials of High-Energy Molecules? Critical News from Critical Points. Crystals 2022, 12, 1455. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Jacobs, S.J. Chemistry of Detonations. I. A Simple Method for Calculating Detonation Properties of C–H–N–O Explosives. J. Chem. Phys. 1968, 48, 23–35. [Google Scholar]
- Politzer, P.; Murray, J.S. The Kamlet-Jacobs Parameter φ: A Measure of Intrinsic Detonation Potential. Propellants Explos. Pyrotech. 2019, 44, 844–849. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Su, H.; Li, S.; Zhang, S.; Pang, S. A Simple Method for the Prediction of the Detonation Performances of Metal-Containing Explosives. J. Phys. Chem. A 2014, 118, 4575–4581. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Perspectives on the crystal densities and packing coefficients of explosive compounds. Struct. Chem. 2016, 27, 401–408. [Google Scholar] [CrossRef]
- Kretić, D.S.; Radovanović, J.I.; Veljković, D.Ž. Can the sensitivity of energetic materials be tuned by using hydrgen bonds? Another look at the role of hydrogen bonding in the design of high energetic compounds. Phys. Chem. Chem. Phys. 2021, 23, 7472–7479. [Google Scholar] [CrossRef]
- Shoaf, A.L.; Bayse, C.A. Trigger Bond Analysis of Nitroaromatic Energetic Materials Using Wiberg Bond Indices. J. Comput. Chem. 2018, 39, 1236–1248. [Google Scholar] [CrossRef]
- Liu, G.; Gou, R.; Li, H.; Zhang, C. Polymorphism of Energetic Materials: A Comprehensive Study of Molecular Conformers, Crystal Packing, and the Dominance of Their Energetics in Governing the Most Stable Polymorph. Cryst. Growth Des. 2018, 18, 4174–4186. [Google Scholar] [CrossRef]
- Aina, A.A.; Misquitta, A.J.; Phipps, M.J.S.; Price, S.L. Charge Distributions of Nitro Groups Within Organic Explosive Crystals: Effects on Sensitivity and Modeling. ACS Omega 2019, 4, 8614–8625. [Google Scholar] [CrossRef]
- Christopher, I.L.; Pulham, C.R.; Michalchuk, A.A.L.; Morrison, C.A. Is the impact sensitivity of RDX polymorph dependent? J. Chem. Phys. 2023, 158, 124115. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D.; Filippini, G.; Gavezzotti, A. A Statistical Study of Density and Packing Variations Among Crystalline Isomers. Tetrahedron 2000, 56, 6595–6601. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Armaković, S.; Armaković, S.J. Atomistica.online—Web Application for Generating input Files for ORCA Molecular Modelling Package Made with the Anvil Platform. Mol. Simul. 2022, 49, 117–123. [Google Scholar] [CrossRef]
- Armaković, S.; Armaković, S.J. Online and desktop graphical user interfaces for xtb programme from Atomistica.online platform. Mol. Simul. 2024, 50, 560–570. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.; Smith, A.E. Thermochemistry of Nitrophenols. V. Enthalpies of formation of 2,4- and 2,6-dinitrophenols. Thermochim. Acta 1983, 69, 375–378. [Google Scholar] [CrossRef]
- Rouse, P.E., Jr. Enthalpies of formation and calculated detonation properties of some thermally stable explosives. J. Chem. Eng. Data 1976, 21, 16–20. [Google Scholar] [CrossRef]
- Boddu, V.M.; Viswanath, D.S.; Ghosh, T.K.; Damavarapu, R. 2,4,6-Triamino-1,3,5-Trinitrobenzene (TATB) and TATB-based formulations: A review. J. Hazard. Mater. 2010, 181, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Krien, G.; Licht, H.H.; Zierath, J. Thermochemical investigation of nitramines. Thermochim. Acta 1973, 6, 465–472. [Google Scholar] [CrossRef]
- Veljković, D.Ž.; Janjić, G.V.; Zarić, S.D. Are C-H-O interactions linear? The case of aromatic CH donors. CrystEngComm 2011, 13, 5005–5010. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CSD REFCODE | ρ (g/cm³) | D (km/s) | P (kbar) |
|---|---|---|---|
| TNBENZ10 | 1.676 | 7.2789 | 224.9444 |
| TNBENZ12 | 1.688 | 7.3146 | 228.1770 |
| TNBENZ13 | 1.717 | 7.4009 | 236.0846 |
| TNBENZ11 | 1.729 | 7.4366 | 239.3961 |
| TNBENZ14 | 1.737 | 7.4575 | 241.3384 |
| CSD REFCODE | ρ (g/cm³) | D (km/s) | P (kbar) |
|---|---|---|---|
| PICRAC19 | 1.761 | 7.5504 | 249.5617 |
| PICRAC11 | 1.768 | 7.5713 | 251.5496 |
| PICRAC | 1.771 | 7.5803 | 252.4040 |
| PICRAC18 | 1.774 | 7.5892 | 253.2599 |
| PICRAC17 | 1.789 | 7.6340 | 257.5609 |
| PICRAC12 | 1.794 | 7.6489 | 259.0026 |
| PICRAC16 | 1.801 | 7.6698 | 261.0277 |
| PICRAC15 | 1.812 | 7.7026 | 264.2260 |
| PICRAC14 | 1.819 | 7.7235 | 266.2714 |
| PICRAC13 | 1.822 | 7.7324 | 267.1505 |
| CSD REFCODE | ρ (g/cm³) | D (km/s) | P (kbar) |
|---|---|---|---|
| ZZZMUC05 | 1.648 | 7.0024 | 205.9722 |
| ZZZMUC06 | 1.650 | 7.0082 | 206.4724 |
| ZZZMUC01 | 1.655 | 7.0227 | 207.7257 |
| ZZZMUC09 | 1.704 | 7.1646 | 220.2082 |
| ZZZMUC08 | 1.713 | 7.1907 | 222.5404 |
| CSD REFCODE | ρ (g/cm³) | D (km/s) | P (kbar) |
|---|---|---|---|
| TATNBZ | 1.937 | 8.0555 | 300.4390 |
| TATNBZ03 | 2.016 | 8.2907 | 325.4453 |
| CSD REFCODE | ρ (g/cm³) | D (km/s) | P (kbar) |
|---|---|---|---|
| MTNANL | 1.731 | 7.7101 | 257.5120 |
| MTNANL01 | 1.729 | 7.7040 | 256.9173 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretić, D.S.; Maslarević, M.I.; Veljković, D.Ž. Small Deviations in Geometries Affect Detonation Velocities and Pressures of Nitroaromatic Molecules. Organics 2025, 6, 17. https://doi.org/10.3390/org6020017
Kretić DS, Maslarević MI, Veljković DŽ. Small Deviations in Geometries Affect Detonation Velocities and Pressures of Nitroaromatic Molecules. Organics. 2025; 6(2):17. https://doi.org/10.3390/org6020017
Chicago/Turabian StyleKretić, Danijela S., Marija I. Maslarević, and Dušan Ž. Veljković. 2025. "Small Deviations in Geometries Affect Detonation Velocities and Pressures of Nitroaromatic Molecules" Organics 6, no. 2: 17. https://doi.org/10.3390/org6020017
APA StyleKretić, D. S., Maslarević, M. I., & Veljković, D. Ž. (2025). Small Deviations in Geometries Affect Detonation Velocities and Pressures of Nitroaromatic Molecules. Organics, 6(2), 17. https://doi.org/10.3390/org6020017