
Synthesis and Pharmacology of Clinical Drugs Containing Isoindoline Heterocycle Core


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
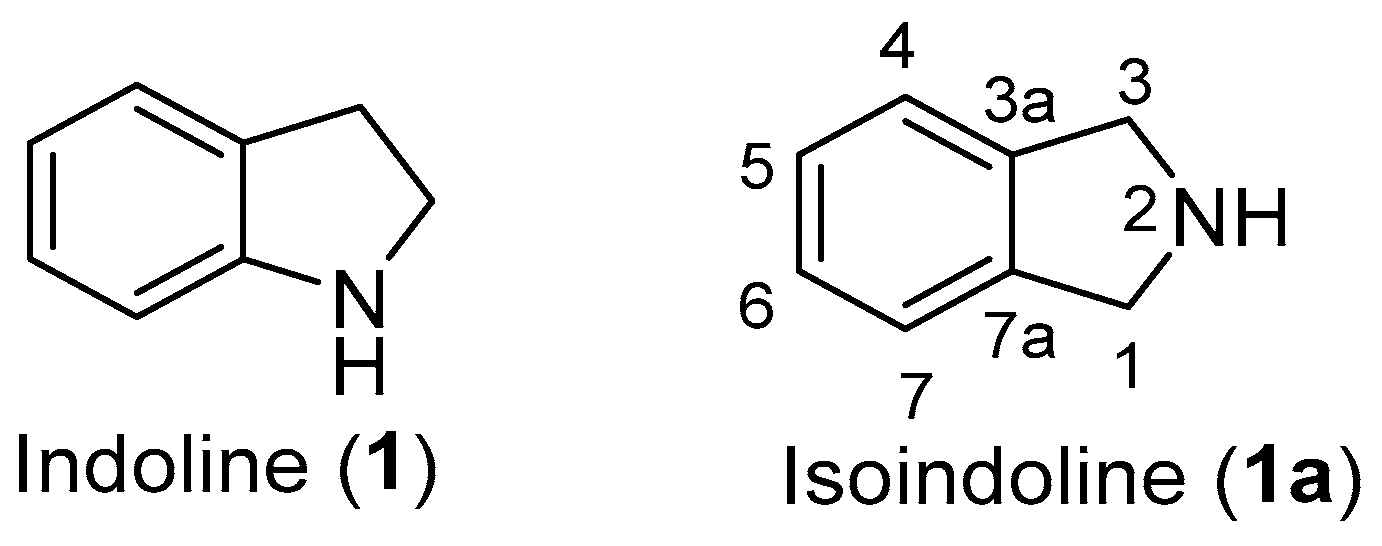
1. Introduction
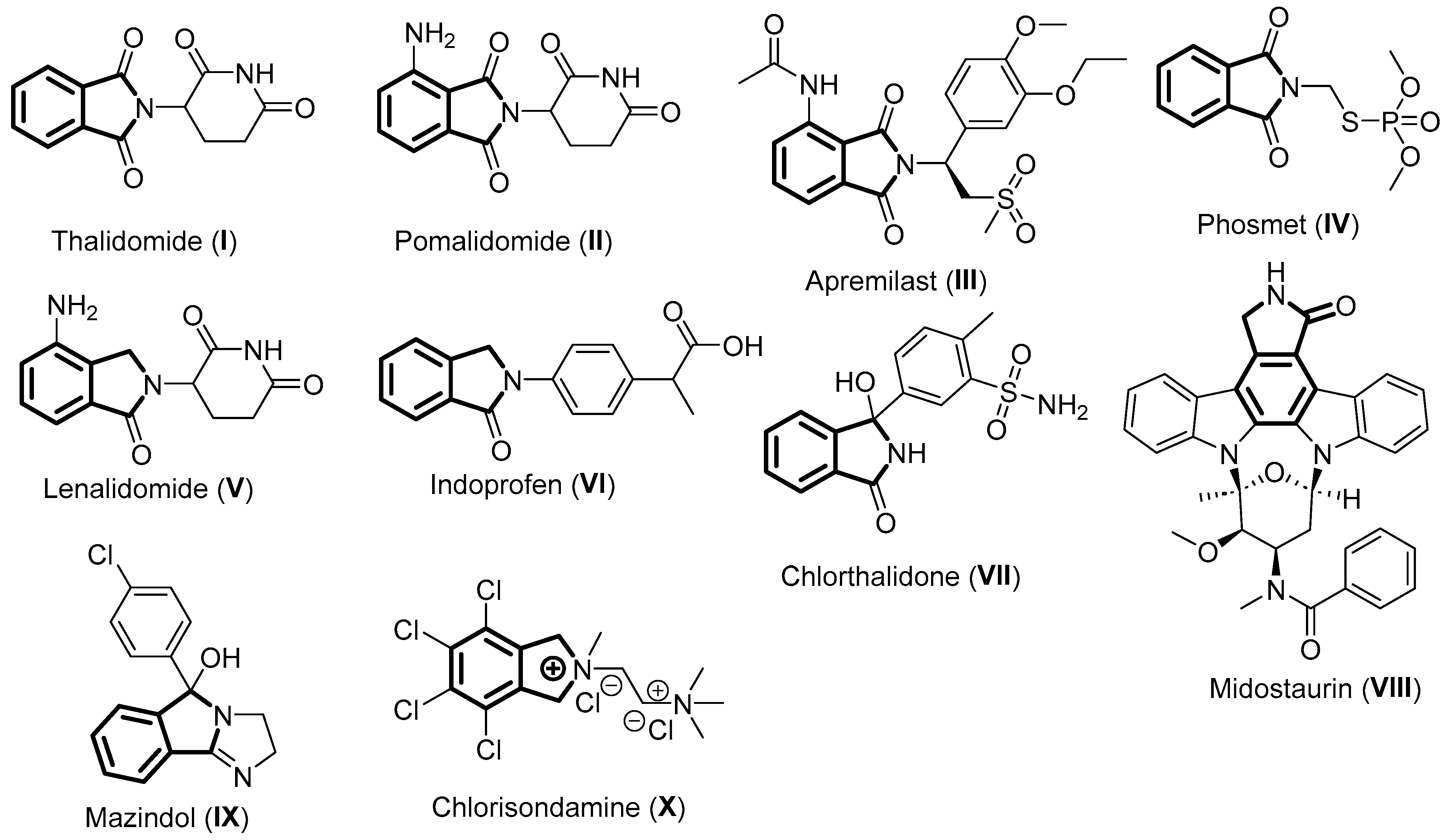
2. Thalidomide
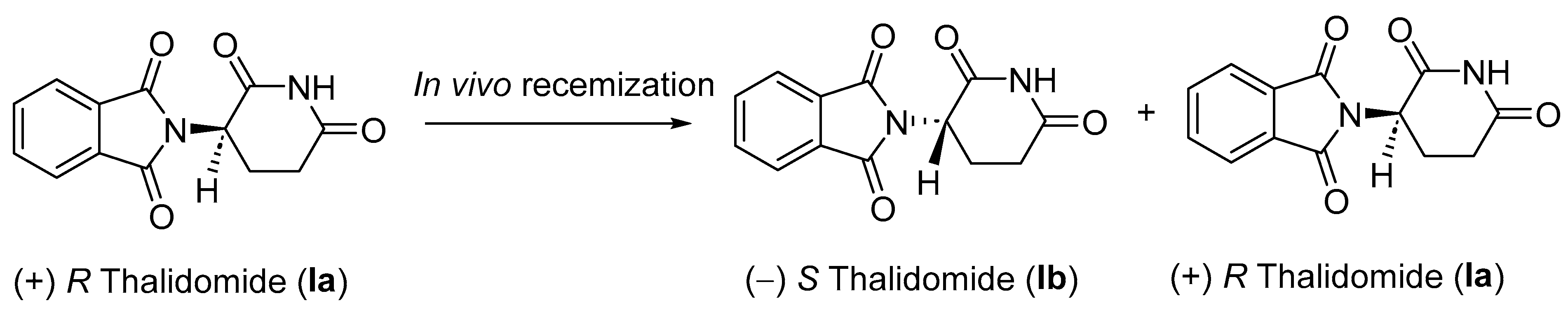
2.1. Pharmacology of Thalidomide
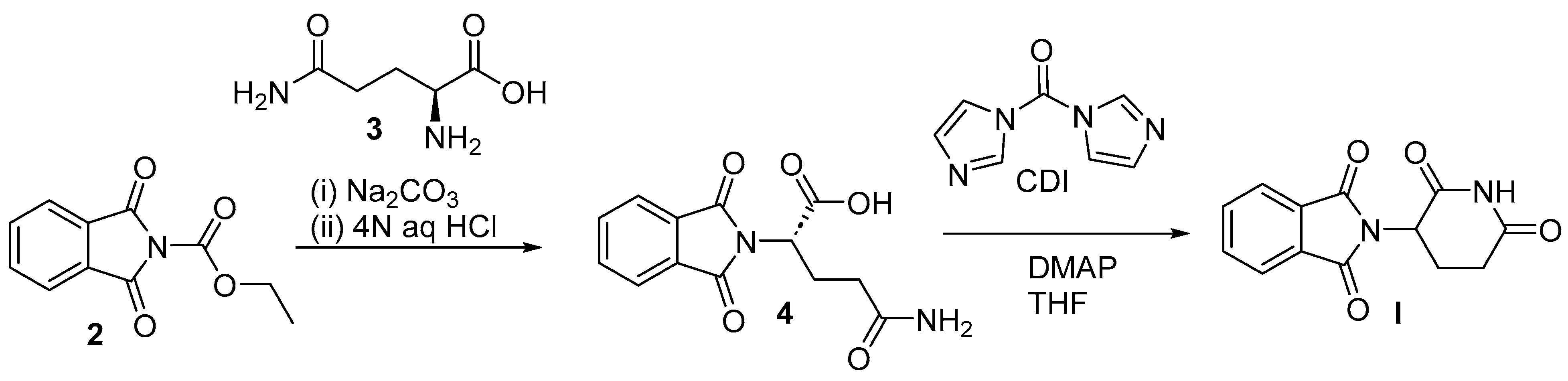
2.2. Chemical Synthesis of Thalidomide
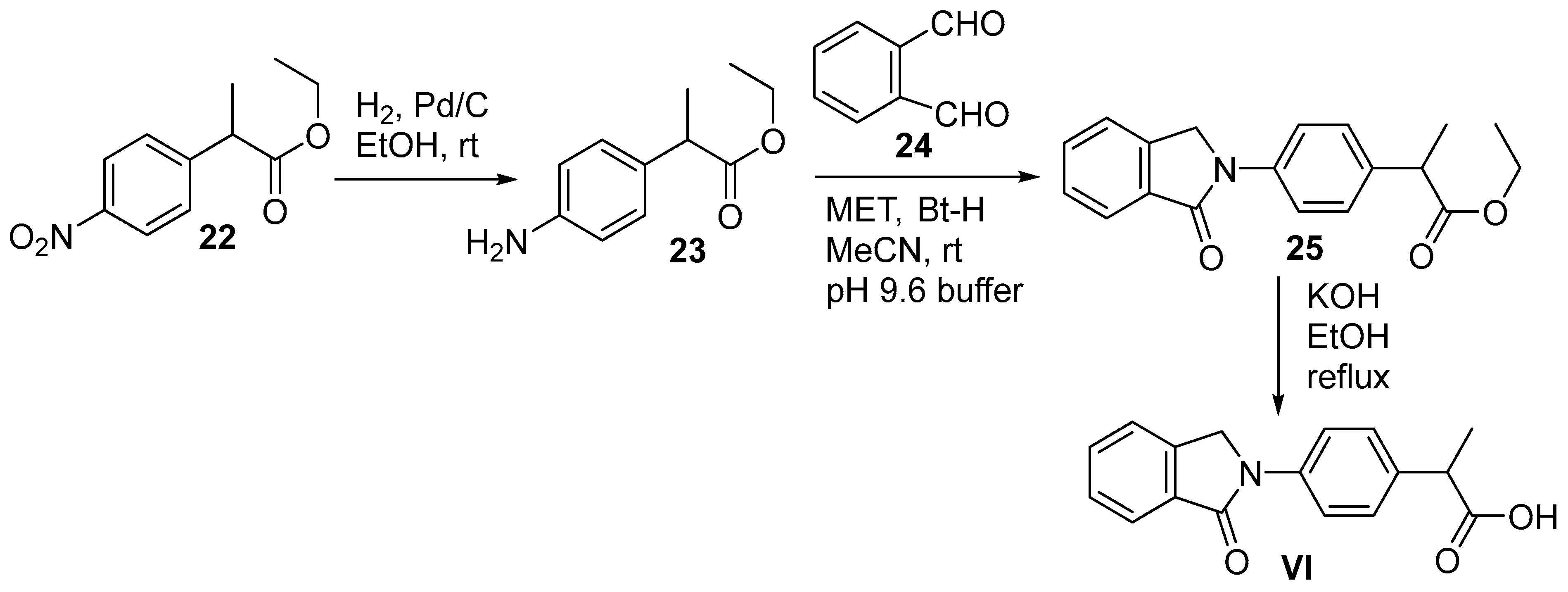
3. Pomalidomide
3.1. Pharmacology of Pomalidomide
3.2. Chemical Synthesis of Pomalidomide
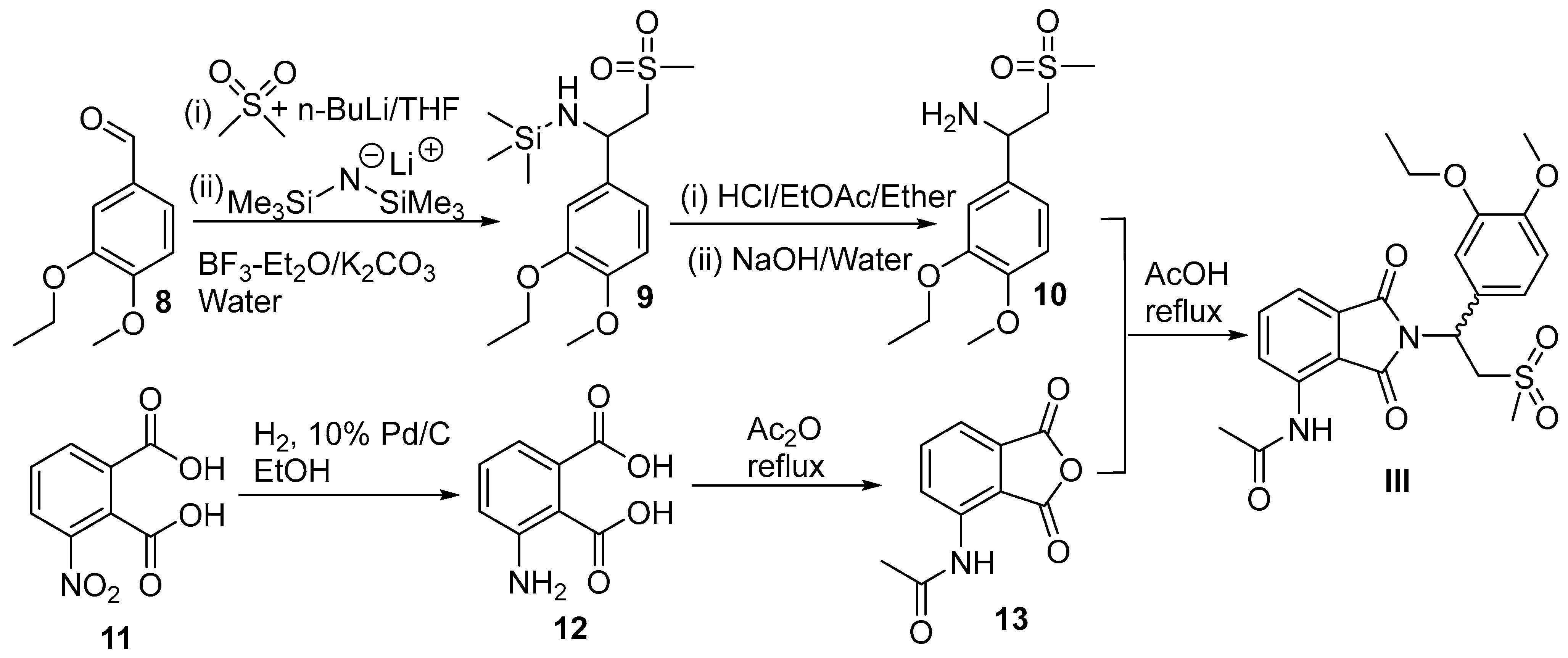
4. Apremilast
4.1. Pharmacology of Apremilast
4.2. Chemical Synthesis of Apremilast
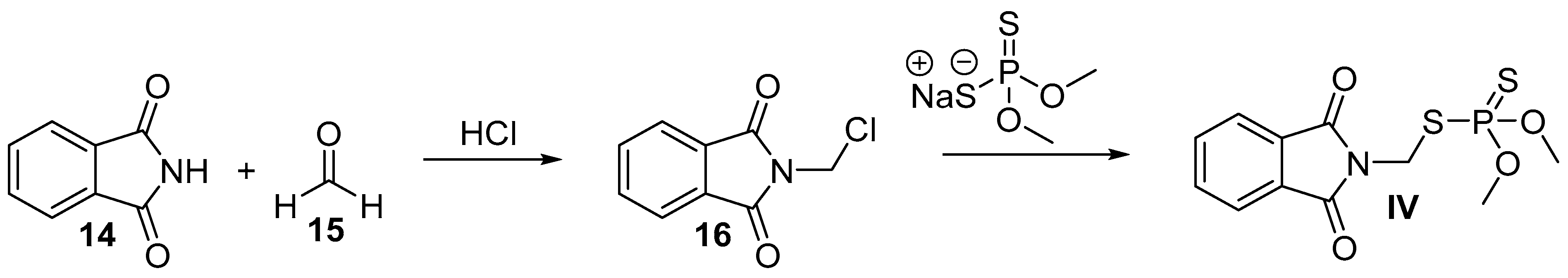
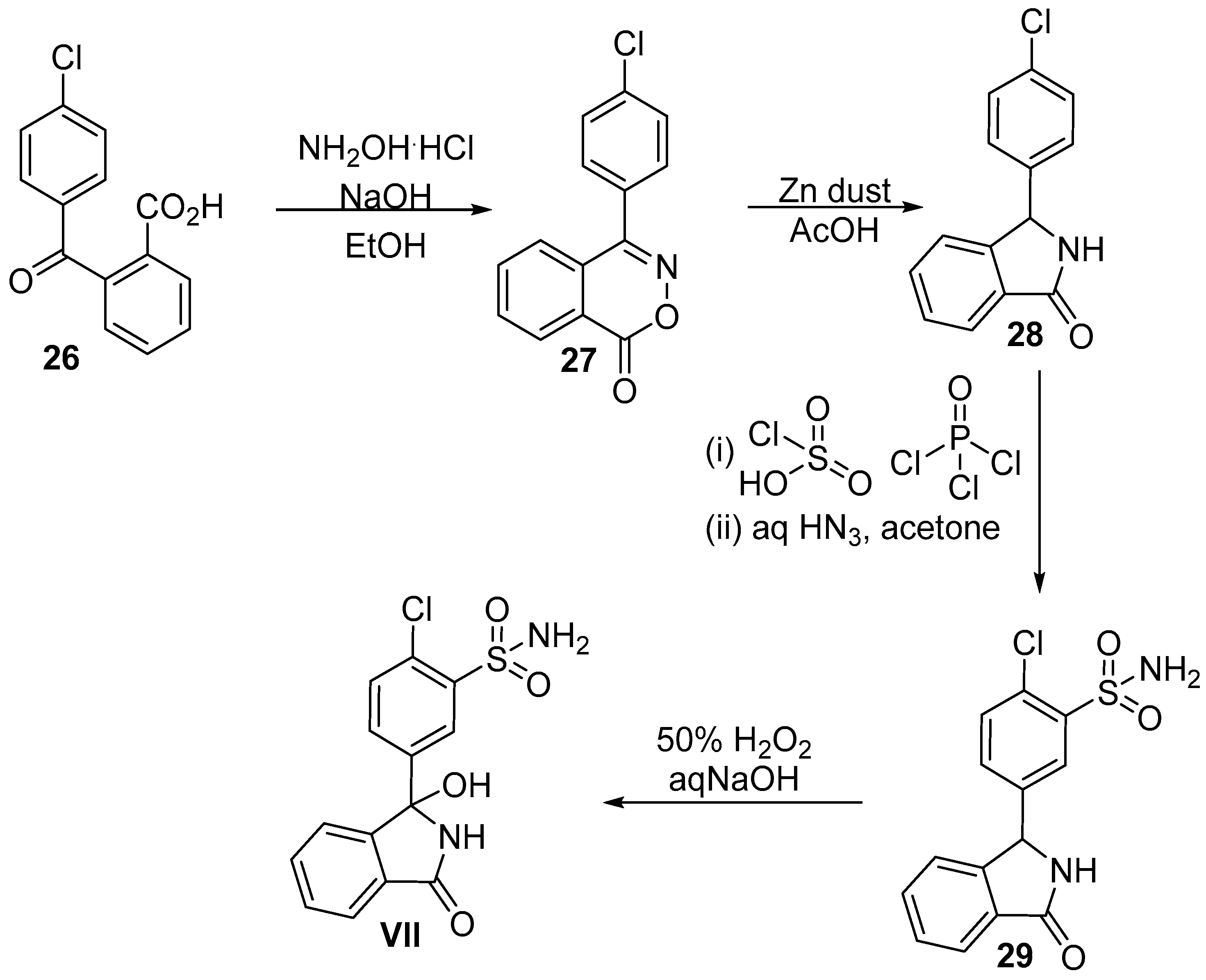
5. Phosmet
5.1. Pharmacology of Phosmet
5.2. Chemical Synthesis of Phosmet
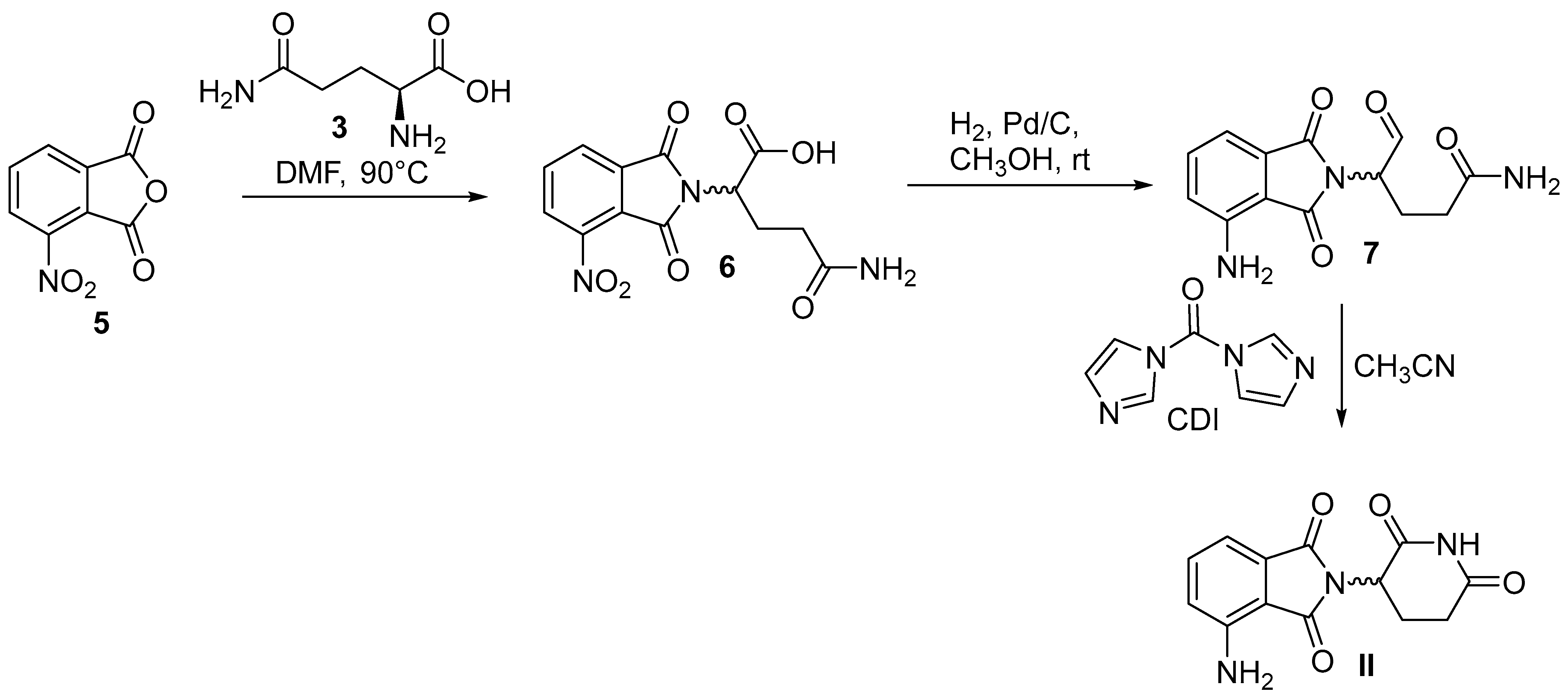
6. Lenalidomide
6.1. Pharmacology of Lenalidomide
6.2. Chemical Synthesis of Lenalidomide
7. Indoprofen
7.1. Pharmacology of Indoprofen
7.2. Chemical Synthesis of Indoprofen
8. Chlorthalidone
8.1. Pharmacology of Chlorthalidone
8.2. Chemical Synthesis of Chlorthalidone
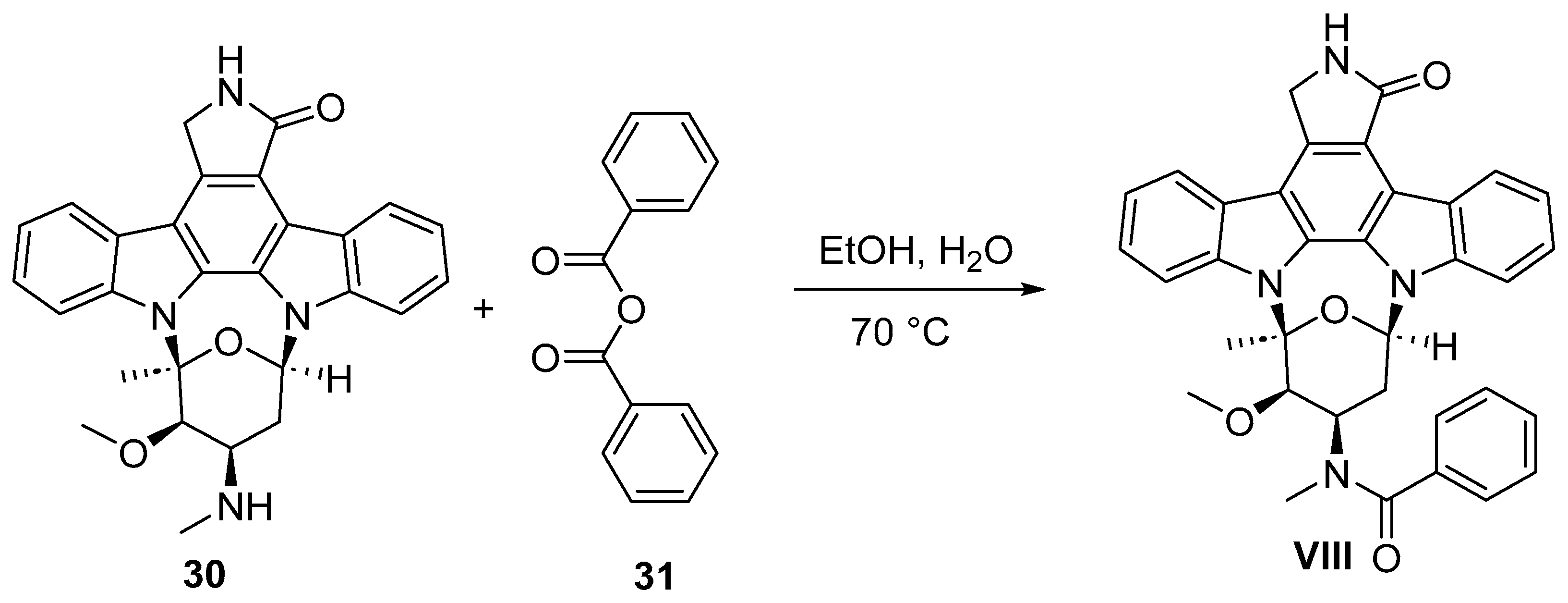
9. Midostaurin
9.1. Pharmacology of Midostaurin
9.2. Chemical Synthesis of Midostaurin
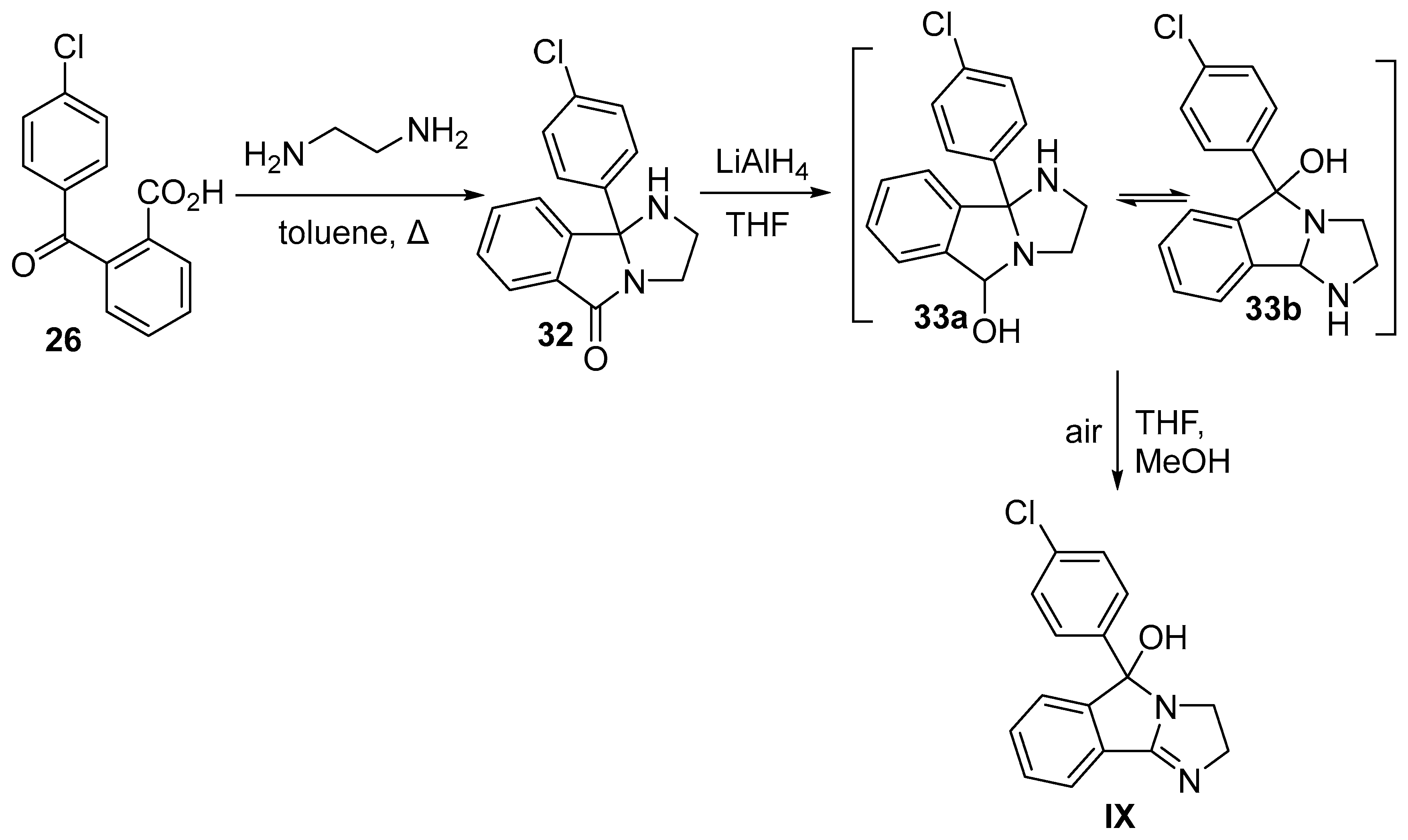
10. Mazindol
10.1. Pharmacology of Mazindol
10.2. Chemical Synthesis of Mazindol
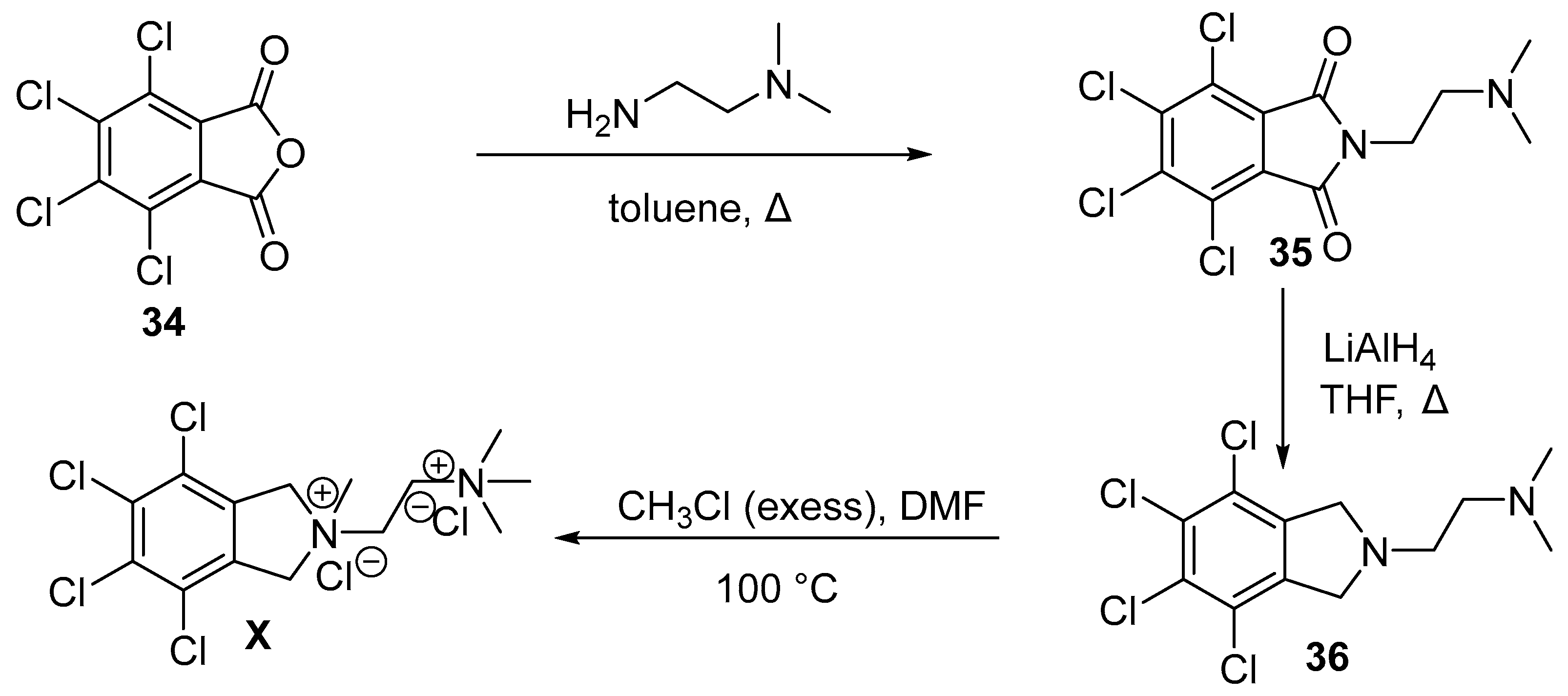
11. Chlorisondamine
11.1. Pharmacology of Chlorisondamine
11.2. Chemical Synthesis of Chlorisondamine
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Youssef, D.; Patel, R.; Mohapatra, P.; Jha, A. Pharmacology and synthesis of clinical drugs containing isoquinoline core. Trends Org. Chem. 2021, 21, 1–17. [Google Scholar]
- Kabir, E.; Uzzaman, M.A. A review on biological and medicinal impact of heterocyclic compounds. Results Chem. 2022, 4, 100606. [Google Scholar] [CrossRef]
- Ruiz, B.; Chávez, A.; Forero, A.; García-Huante, Y.; Romero, A.; Sánchez, M.; Rocha, D.; Sánchez, B.; Rodríguez-Sanoja, R.; Sánchez, S.; et al. Production of microbial secondary metabolites: Regulation by the carbon source. Crit. Rev. Microbiol. 2010, 36, 146–167. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Samota, M.K.; Choudhary, M.; Choudhary, M.; Pandey, A.K.; Sharma, A.; Thakur, J. How do plants defend themselves against pathogens-biochemical mechanisms and genetic interventions. Physiol. Mol. Biol. Plants 2022, 28, 485–504. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.T.; Jung, J.W.; Kim, N.J. recent advances in the synthesis of biologically active cinnoline, phthalazine and quinoxaline derivatives. Curr. Org. Chem. 2017, 21, 1265–1291. [Google Scholar] [CrossRef]
- Clary, K.N.; Parvez, M.; Back, T.G. Preparation of 1-aryl-substituted isoindoline derivatives by sequential Morita–Baylis–Hillman and intramolecular Diels–Alder reactions. Org. Biomol. Chem. 2009, 7, 1226–1230. [Google Scholar] [CrossRef]
- Thakur, A.; Singh, A.; Kaur, N.; Ojha, R.; Nepali, K. Steering the antitumor drug discovery campaign towards structurally diverse indolines. Bioorg. Chem. 2020, 94, 103436. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.P.; Thapa, P.; Sharma, R.; Sharma, M. 1-Isoindolinone Scaffold-Based Natural Products with a Promising Diverse Bioactivity. Fitoterapia 2020, 146, 104722. [Google Scholar] [CrossRef]
- Heras Martinez, H.M.; Barragan, E.; Marichev, K.O.; Chávez-Flores, D.; Bugarin, A. Phthalimides as Anti-Inflammatory Agents. Future Med. Chem. 2025, 17, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Starosotnikov, A.M.; Bastrakov, M.A. Cycloaddition reactions in the synthesis of isoindolines. Chem. Heterocl. Compd. 2017, 53, 1181–1183. [Google Scholar] [CrossRef]
- Williams, F.J.; Jarvo, E.R. Palladium-catalyzed cascade reaction for the synthesis of substituted isoindolines. Angew. Chem. Int. Ed. 2011, 50, 4459–4462. [Google Scholar] [CrossRef]
- Ward, S.P. Thalidomide and congenital abnormalities. Br. Med. J. 1962, 2, 646–647. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Weagle, R.; Corkum, D.; Kuanar, M.; Mohapatra, P.P.; Jha, A. Convenient access to 5-arylisoindolo[2,1-a]quinolin-11(6aH)-ones. Mol. Divers. 2017, 21, 455–462. [Google Scholar] [CrossRef]
- Jha, A.; Chou, T.; ALJaroudi, Z.; Ellis, B.D.; Cameron, T.S. Aza-Diels–Alder reaction between N-aryl-1-oxo-1H-isoindolium ions and tert-enamides: Steric effects on reaction outcome. Beilstein J. Org. Chem. 2014, 10, 848–857. [Google Scholar] [CrossRef]
- Al-Jaroudi, Z.; Mohapatra, P.P.; Cameron, T.S.; Jha, A. Expedient and diastereoselective synthesis of substituted 6,6a-dihydroisoindolo[2,1-a]quinolin-11(5H)-ones. Synthesis 2016, 48, 4477–4488. [Google Scholar]
- Al-Jaroudi, Z.; Mohapatra, P.P.; Jha, A. Facile synthesis of 3-substituted isoindolinones. Tetrahedron Lett. 2016, 57, 772–777. [Google Scholar] [CrossRef]
- Brandenburg, N.A.; Bwire, R.; Freeman, J.; Houn, F.; Sheehan, P.; Zeldis, J.B. Effectiveness of risk evaluation and mitigation strategies (REMS) for lenalidomide and thalidomide: Patient comprehension and knowledge retention. Drug Saf. 2017, 40, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Scialli, A.R. Thalidomide: The tragedy of birth defects and the effective treatment of disease. Toxicol. Sci. 2011, 122, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Wani, W.A.; Saleem, K.; Haque, A. Thalidomide: A banned drug resurged into future anticancer drug. Curr. Drug Ther. 2012, 7, 13–23. [Google Scholar] [CrossRef]
- Anderson, K.C. Lenalidomide and Thalidomide: Mechanisms of action—Similarities and differences. Semin. Hematol. 2005, 42, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.Y.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Sreedhara, R.C.; Banerjee, S.; Chatterjee, S. TNF-α signaling beholds thalidomide saga: A review of mechanistic role of TNF-α signaling under thalidomide. Curr. Top. Med. Chem. 2012, 12, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, P.P.; Cathers, B.E. Cereblon Modulators: Low molecular weight inducers of protein degradation. Drug Discov. Today Technol. 2019, 31, 29–34. [Google Scholar] [CrossRef]
- Muller, G.W.; Konnecke, W.E.; Smith, A.M.; Khetani, V.D. A concise two-step synthesis of thalidomide. Org. Process. Res. Dev. 1999, 3, 139–140. [Google Scholar] [CrossRef]
- Shibata, N.; Yamamoto, T.; Toru, T. Synthesis of thalidomide. In Bioactive Heterocycles II; Eguchi, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 73–97. [Google Scholar]
- Ribatti, D.; Vacca, A. Chapter 3—Anti-angiogenesis in multiple myeloma. In Anti-Angiogenesis Strategies in Cancer Therapeutics; Mousa, S.A., Davis, P.J., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 39–50. [Google Scholar]
- Jaeger, H.K.; Davis, D.A.; Nair, A.; Shrestha, P.; Stream, A.; Yaparla, A.; Yarchoan, R. Mechanism and therapeutic implications of pomalidomide-induced immune surface marker upregulation in EBV-positive lymphomas. Sci. Rep. 2023, 13, 11596. [Google Scholar] [CrossRef] [PubMed]
- Chanan-Khan, A.A.; Swaika, A.; Paulus, A.; Kumar, S.K.; Mikhael, J.R.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q. Pomalidomide: The new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer. J. 2013, 3, e143. [Google Scholar] [CrossRef]
- Ding, H.X.; Leverett, C.A.; Kyne, R.E.; Liu, K.K.; Fink, S.J.; Flick, A.C.; O’Donnell, C.J. Synthetic approaches to the 2013 new drugs. Bioorg. Med. Chem. 2015, 23, 1895–1922. [Google Scholar] [CrossRef]
- Fala, L. Otezla (Apremilast), an Oral PDE-4 inhibitor, receives fda approval for the treatment of patients with active psoriatic arthritis and plaque psoriasis. Am. Health. Drug Benefits 2015, 8, 105–110. [Google Scholar]
- Hatemi, G.; Mahr, A.; Takeno, M.; Kim, D.; Melikoğlu, M.; Cheng, S.; McCue, S.; Paris, M.; Chen, M.; Yazici, Y. Impact of apremilast on quality of life in Behçet’s syndrome: Analysis of the phase 3 RELIEF study. RMD Open 2022, 8, e002235. [Google Scholar] [CrossRef] [PubMed]
- Fertig, B.A.; Baillie, G.S. PDE4-mediated cAMP signalling. J. Cardiovasc. Dev. Dis. 2018, 5, 8. [Google Scholar] [CrossRef]
- Tong, B.N.; Liu, X.L.; Xiao, J.; Su, G.F. Immunopathogenesis of Behcet’s disease. Front. Immunol. 2019, 10, 665. [Google Scholar] [CrossRef]
- Narode, H.; Gayke, M.; Eppa, G.; Yadav, J.S. A review on synthetic advances toward the synthesis of apremilast, an anti-inflammatory drug. Org. Process Res. Dev. 2021, 25, 1512–1523. [Google Scholar] [CrossRef]
- Pohanish, R.P. (Ed.) Sittig’s Handbook of Pesticides and Agricultural Chemicals, 2nd ed.; William Andrew Publishing: Oxford, UK, 2015; pp. 629–724. [Google Scholar]
- Mensching, D.; Volmer, P.A. CHAPTER 125—Insecticides and Molluscicides. In Handbook of Small Animal Practice, 5th ed.; Morgan, R.V., Ed.; W.B. Saunders: Saint Louis, MO, USA, 2008; pp. 1197–1204. [Google Scholar]
- Müller, F.; Streibert, H.P.; Farooq, S. Acaricides. In Ullmann’s Encyclopedia of Industrial Chemistry; Woley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2009; pp. 91–190. [Google Scholar]
- Qiao, S.K.; Guo, X.N.; Ren, J.H.; Ren, H.Y. Efficacy and safety of lenalidomide in the treatment of multiple myeloma: A systematic review and meta-analysis of randomized controlled trials. Chin. Med. J. 2015, 128, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Gowda, S.; Tuscano, J. A comprehensive review of lenalidomide in B-cell non-Hodgkin lymphoma. Ther. Adv. Hematol. 2016, 7, 209–221. [Google Scholar] [CrossRef]
- Muller, G.W.; Chen, R.; Huang, S.; Corral, L.G.; Wong, L.M.; Patterson, R.T.; Chen, Y.; Kaplan, G.; Stirling, D.I. Amino-substituted thalidomide analogs: Potent inhibitors of TNF-α production. Bioorg. Med. Chem. Lett. 1999, 9, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Kotla, V.; Goel, S.; Nischal, S.; Heuck, C.; Vivek, K.; Das, B.; Verma, A. Mechanism of action of lenalidomide in hematological malignancies. J. Hematol. Oncol. 2009, 2, 36. [Google Scholar] [CrossRef]
- Hui, J.Y.; Fuchs, A.; Kumar, G. Embryo-fetal exposure and developmental outcome of lenalidomide following oral administration to pregnant cynomolgus monkeys. Reprod. Toxicolo. 2022, 114, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Galustian, C.; Dalgleish, A. Lenalidomide: A Novel Anticancer Drug with Multiple Modalities. Expert Opin. Pharmacother. 2009, 10, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J. Better drug discovery through better target identification. Biotechnol. Healthc. 2005, 2, 52–58. [Google Scholar] [PubMed]
- Love, A.J.; Ripley, S.H.; Brock-Utne, J.G.; Blake, G.T. Indoprofen, a non-steroidal anti-inflammatory analgesic which does not depress respiration in normal man. A study comparing indoprofen with morphine. S. Afr. Med. J. 1985, 68, 801–802. [Google Scholar]
- Cashman, J.N. The mechanisms of action of NSAIDs in analgesia. Drugs 1996, 52 (Suppl. S5), 13–23. [Google Scholar] [CrossRef]
- Kim, H.; Cho, S.C.; Jeong, H.; Lee, H.; Jeong, M.; Pyun, J.; Ryu, D.; Kim, M.; Lee, Y.; Kim, M.S.; et al. Indoprofen prevents muscle wasting in aged mice through activation of PDK1/AKT pathway. J. Cachexia Sarcopenia Muscle 2020, 11, 1070–1088. [Google Scholar] [CrossRef] [PubMed]
- Lunn, M.R.; Root, D.E.; Martino, A.M.; Flaherty, S.P.; Kelley, B.P.; Coovert, D.D.; Burghes, A.H.; Man, N.T.; Morris, G.E.; Zhou, J.H.; et al. Indoprofen upregulates the survival motor neuron protein through a cyclooxygenase-independent mechanism. Chem. Biol. 2004, 11, 1489–1493. [Google Scholar] [CrossRef]
- Carney, R.W.J.; de Stevens, G. Tertiary Aminoacids. U.S. Patent 4,316,850, 23 February 1982. [Google Scholar]
- Nannini, G.; Giraldi, P.N.; Molgora, G.; Biasoli, G.; Spinelli, F.; Logemann, W.; Dradi, E.; Zanni, G.; Buttinoni, A.; Tommasini, R. New analgesic-anti-inflammatory drugs. 1-Oxo-2-substituted isoindoline derivatives. Arzneimittelforschung 1973, 23, 1090–1100. [Google Scholar] [PubMed]
- Takahashi, I.; Kawakami, T.; Hirano, E.; Kimino, M.; Kamimura, S.; Miwa, T.; Tamura, T.; Tazaki, R.; Kitajima, H.; Hatanaka, M.; et al. Application of the mild-condition phthalimidine synthesis with use of 1,2,3-1H-benzotriazole and 2-mercaptoethanol as dual synthetic auxiliaries. Effective synthesis of phthalimidines possessing a variety of substituents at 2-position. Heterocycles 2016, 93, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.M.; Lee, C.H.; Chambers, G.K. Systematic review of antihypertensive therapies: Does the evidence assist in choosing a first-line drug? Can. Med. Assoc. J. CMAJ 1999, 161, 25–32. [Google Scholar]
- Shahin, M.H.; Johnson, J.A. Mechanisms and pharmacogenetic signals underlying thiazide diuretics blood pressure response. Curr. Opin. Pharmacol. 2016, 27, 31–37. [Google Scholar] [CrossRef]
- Graf, W.; Girod, E.; Schmid, E.; Stoll, W.G. Zur Konstitution von Benzophenon-2-carbonsäure-Derivaten. Helv. Chim. Acta 1959, 42, 1085–1101. [Google Scholar] [CrossRef]
- Wilfried, G.; Erich, S.; Stoll, W.G. New Isoindoline Derivatives. U.S. Patent 3,055,904, 25 September 1962. [Google Scholar]
- Golec, F.A., Jr.; Auerbach, J. Intermediates for the Synthesis of Phthalimidines. U.S. Patent 4331600, 25 May 1982. [Google Scholar]
- Kumar, A.; Singh, D.; Jadhav, A.; Pandya, D.N. An Efficient Industrial Process for 3-hydroxy-3(3′-sulfamyl-4′-chlorophenyl)phtalimidine. International Patent Application Publication WO2005065046, 21 July 2005. [Google Scholar]
- Gadakar, M.; Wagh, G.; Wakchaure, Y.; Thanasekaran, P.; Punde, D. Improved Process for the Preparation of Chlorthalidone. International Patent Application Publication WO2018158777, 7 September 2018. [Google Scholar]
- Manley, P.W.; Weisberg, E.; Sattler, M.; Griffin, J.D. Midostaurin, a Natural Product-Derived Kinase Inhibitor Recently Approved for the Treatment of Hematological Malignancies. Biochemistry 2018, 57, 477–478. [Google Scholar] [CrossRef]
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Yang, Q.; Wu, P.; He, C.; Yin, L.; Xu, F.; Yin, Z.; Yue, G.; Zou, Y.; Li, L.; et al. The synthesis review of the approved tyrosine kinase inhibitors for anticancer therapy in 2015–2020. Bioorg. Chem. 2021, 113, 105011. [Google Scholar] [CrossRef]
- Li, G.; Wu, D.; Xu, Y.; He, W.; Wang, D.; Zhu, W.; Wang, L. Synthesis and Antitumor Activity of Staurosporine Derivatives. Nat. Prod. Commun. 2022, 17, 1934578X2211030361. [Google Scholar] [CrossRef]
- Gonçalves, C.L.; Scaini, G.; Rezin, G.T.; Jeremias, I.C.; Bez, G.D.; Daufenbach, J.F.; Gomes, L.M.; Ferreira, G.K.; Zugno, A.I.; Streck, E.L. Effects of acute administration of mazindol on brain energy metabolism in adult mice. Acta Neuropsychiatr. 2014, 26, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S. Clinical Studies with Mazindol. Obes. Res. 1995, 3, 549S–552S. [Google Scholar] [CrossRef] [PubMed]
- Mazindol. DrugBank Online. Available online: https://go.drugbank.com/drugs/DB00579 (accessed on 11 March 2024).
- Seibyl, J.P.; Krystal, J.H.; Charney, D.S. Dopamine and Noradrenergic Reuptake Inhibitors in Treatment of Schizophrenia. U.S. Patent 5,447,948, 5 September 1995. [Google Scholar]
- Berger, S.P. Dopamine Uptake Inhibitors in Reducing Substance Abuse and/or Craving. U.S. Patent 5,217,987, 8 June 1993. [Google Scholar]
- Kovacs, B.; Pinegar, L. Use of Isoindoles for the Treatment of Neurobehavioral Disorders. International Patent Application Publication WO2009155139, 12 April 2009. [Google Scholar]
- Aeberli, P.; Eden, P.; Gogerty, J.H.; Houlihan, W.J.; Penberthy, C. 5-aryl-2,3-dihydro-5H-imidazo[2,1-a]isoindol-5-ols. novel class of anorectic agents. J. Med. Chem. 1975, 18, 177–182. [Google Scholar] [PubMed]
- Bakke, J.L.; Darvill, F.T. Chlorisondamine (ecolid) chloride in medical treatment of severe hypertension. J. Am. Med. Assoc. 1957, 163, 429–436. [Google Scholar]
- Souza, L.A.; Cooper, S.G.; Worker, C.J.; Thakore, P.; Feng Earley, Y. Use of chlorisondamine to assess the neurogenic contribution to blood pressure in mice: An evaluation of method. Physiol. Rep. 2021, 9, e14753. [Google Scholar] [CrossRef] [PubMed]
- Rosen, W.E.; Toohey, V.P.; Shabica, A.C. Tetrachloroisoindolines and related systems. alkylation reactions and inductive Effects. J. Am. Chem. Soc. 1957, 79, 3167–3174. [Google Scholar] [CrossRef]
- Zezula, J.; Wang, H.J.; Woods, A.S.; Wise, R.A.; Jacobson, A.E.; Rice, K.C. The high specific activity tritium labeling of the ganglion-blocking nicotinic antagonist chlorisondamine. J. Label Compd. Radiopharm. 2006, 49, 471–478. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jha, M.; Youssef, D.; Sheehy, H.; Jha, A. Synthesis and Pharmacology of Clinical Drugs Containing Isoindoline Heterocycle Core. Organics 2025, 6, 3. https://doi.org/10.3390/org6010003
Jha M, Youssef D, Sheehy H, Jha A. Synthesis and Pharmacology of Clinical Drugs Containing Isoindoline Heterocycle Core. Organics. 2025; 6(1):3. https://doi.org/10.3390/org6010003
Chicago/Turabian StyleJha, Mukund, Dani Youssef, Haley Sheehy, and Amitabh Jha. 2025. "Synthesis and Pharmacology of Clinical Drugs Containing Isoindoline Heterocycle Core" Organics 6, no. 1: 3. https://doi.org/10.3390/org6010003
APA StyleJha, M., Youssef, D., Sheehy, H., & Jha, A. (2025). Synthesis and Pharmacology of Clinical Drugs Containing Isoindoline Heterocycle Core. Organics, 6(1), 3. https://doi.org/10.3390/org6010003