
Polycyclic Tetramate Macrolactams and Their Potential as Anticancer Agents


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Availability and Inducibility
2.1. Availability
2.2. Inducibility
3. Chemical Configuration, Biosynthetic Pathway, and Anticancer Properties
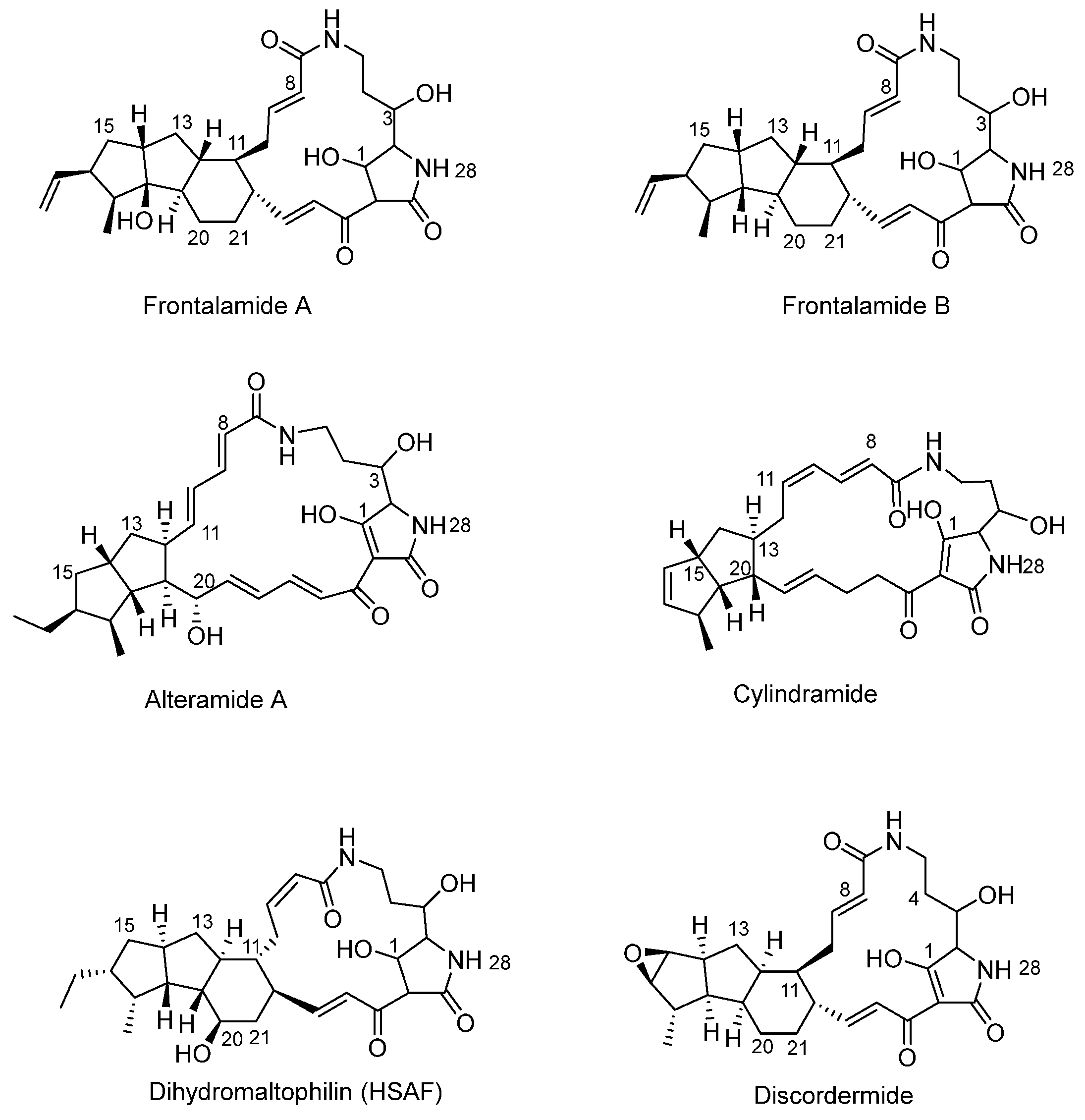
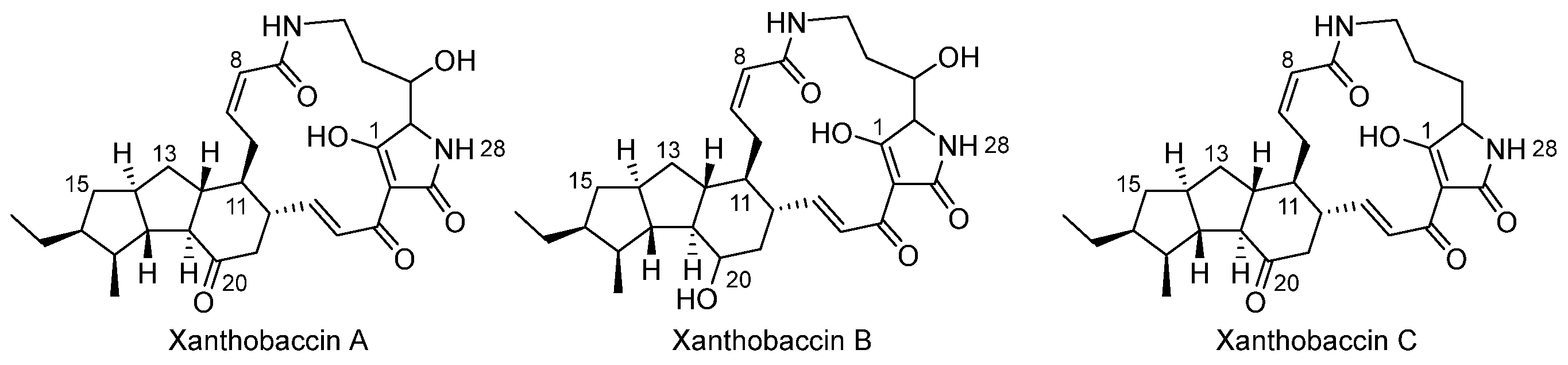
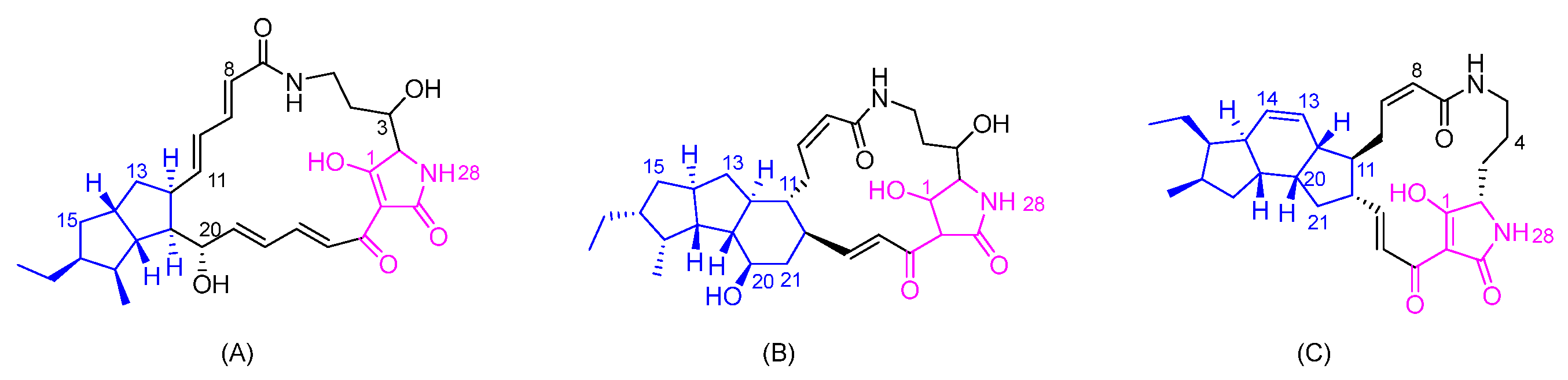
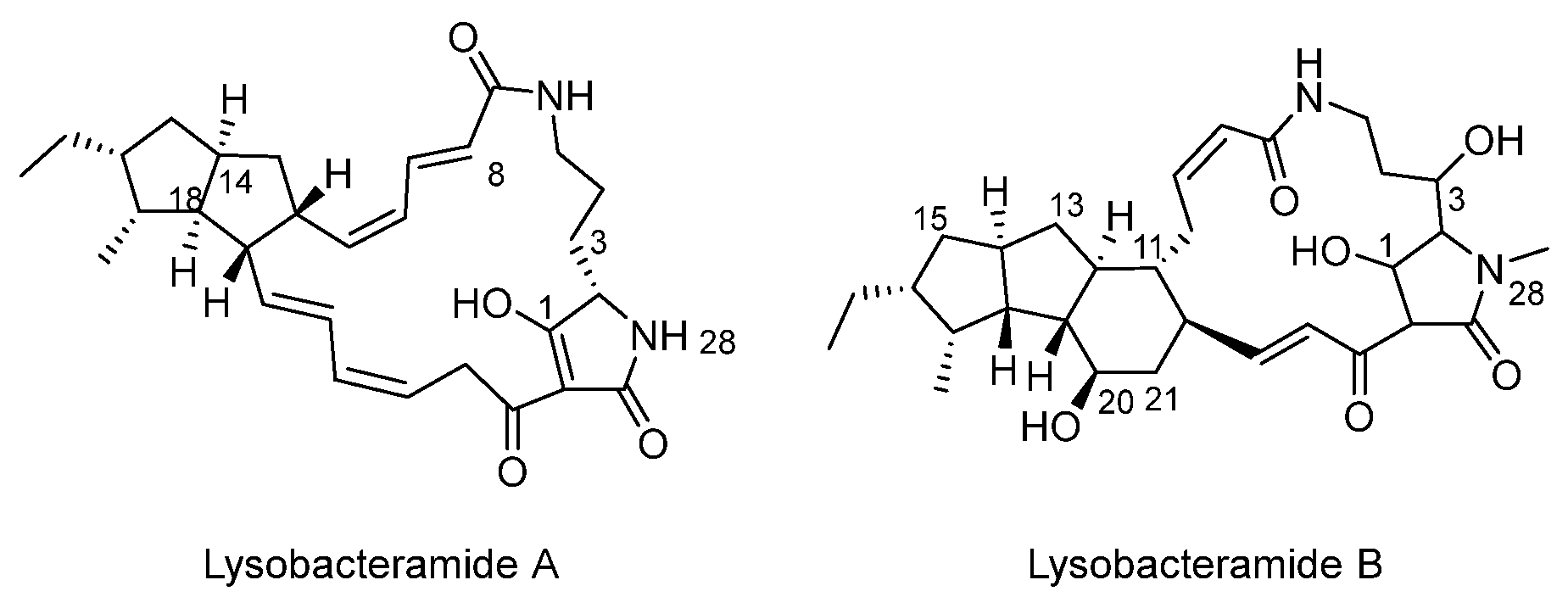
3.1. Basic Structure
3.2. Biosynthetic Pathway
3.3. Past Research to Support Anticancer Potential
3.3.1. PoTeMs Significantly Inhibit Tumor Cell Proliferation
3.3.2. Polycyclic Tetramate Macrolactam with Potential Anticancer Properties from S. zhaozhouensis Subspecies
4. Anticancer Mechanistic Pathways of PoTeMs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Demain, A.L. Importance of microbial natural products and the need to revitalize their discovery. J. Ind. Microbiol. Biotechnol. 2014, 41, 185–201. [Google Scholar] [CrossRef] [PubMed]
- David, B.; Wolfender, J.-L.; Dias, D.A. The pharmaceutical industry and natural products: Historical status and new trends. Phytochem. Rev. 2015, 14, 299–315. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, H.; Qian, L.; Liu, Y.; Zhu, D.; Xu, Z.; Chang, W.; Xu, J.; Wang, L.; Sun, B.; et al. Targeting GDP-Dissociation Inhibitor Beta (GDI2) with a Benzo[a]quinolizidine Library to Induce Paraptosis for Cancer Therapy. JACS Au 2023, 3, 2749–2762. [Google Scholar] [CrossRef]
- Huang, M.; Lu, J.J.; Ding, J. Natural Products in Cancer Therapy: Past, Present and Future. Nat. Prod. Bioprospect. 2021, 11, 5–13. [Google Scholar] [CrossRef]
- Zheng, H.; Dong, Y.; Li, L.; Sun, B.; Liu, L.; Yuan, H.; Lou, H. Novel Benzo[a]quinolizidine Analogs Induce Cancer Cell Death through Paraptosis and Apoptosis. J. Med. Chem. 2016, 59, 5063–5076. [Google Scholar] [CrossRef]
- Wang, H.; He, Y.; Jian, M.; Fu, X.; Cheng, Y.; He, Y.; Fang, J.; Li, L.; Zhang, D. Breaking the Bottleneck in Anticancer Drug Development: Efficient Utilization of Synthetic Biology. Molecules 2022, 27, 7480. [Google Scholar] [CrossRef]
- Surade, S.; Blundell, T.L. Structural biology and drug discovery of difficult targets: The limits of ligandability. Chem. Biol. 2012, 19, 42–50. [Google Scholar] [CrossRef]
- Mallinson, J.; Collins, I. Macrocycles in new drug discovery. Future Med. Chem. 2012, 4, 1409–1438. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, W.; Jin, H.; Zhang, Q.; Chen, Y.; Jiang, X.; Zhang, G.; Zhang, L.; Zhang, W.; She, Z.; et al. Genome Mining of Marine-Derived Streptomyces sp. SCSIO 40010 Leads to Cytotoxic New Polycyclic Tetramate Macrolactams. Mar. Drugs 2019, 17, 663. [Google Scholar] [CrossRef] [PubMed]
- Blodgett, J.A.; Oh, D.C.; Cao, S.; Currie, C.R.; Kolter, R.; Clardy, J. Common biosynthetic origins for polycyclic tetramate macrolactams from phylogenetically diverse bacteria. Proc. Natl. Acad. Sci. USA 2010, 107, 11692–11697. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Huang, H.; Liang, J.; Wang, M.; Lu, L.; Shao, Z.; Cobb, R.E.; Zhao, H. Activation and characterization of a cryptic polycyclic tetramate macrolactam biosynthetic gene cluster. Nat. Commun. 2013, 4, 2894. [Google Scholar] [CrossRef]
- Medema, M.H.; de Rond, T.; Moore, B.S. Mining genomes to illuminate the specialized chemistry of life. Nat. Rev. Genet. 2021, 22, 553–571. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Tu, J.; Zhang, H.; Wei, X.; Ju, J.; Li, Q. Genome Mining and Metabolic Profiling Uncover Polycyclic Tetramate Macrolactams from Streptomyces koyangensis SCSIO 5802. Mar. Drugs 2021, 19, 440. [Google Scholar] [CrossRef]
- Mo, X.; Li, Q.; Ju, J. Naturally occurring tetramic acid products: Isolation, structure elucidation and biological activity. RSC Adv. 2014, 4, 50566–50593. [Google Scholar] [CrossRef]
- Lowery, C.A.; Park, J.; Gloeckner, C.; Meijler, M.M.; Mueller, R.S.; Boshoff, H.I.; Ulrich, R.L.; Barry, C.E., 3rd; Bartlett, D.H.; Kravchenko, V.V.; et al. Defining the mode of action of tetramic acid antibacterials derived from Pseudomonas aeruginosa quorum sensing signals. J. Am. Chem. Soc. 2009, 131, 14473–14479. [Google Scholar] [CrossRef]
- Bodenschatz, K.; Stoeckl, J.; Winterer, M.; Schobert, R. A synthetic approach to 5/5/6-polycyclic tetramate macrolactams of the discodermide type. Tetrahedron 2022, 104, 132113. [Google Scholar] [CrossRef]
- Xu, L.; Wu, P.; Wright, S.J.; Du, L.; Wei, X. Bioactive Polycyclic Tetramate Macrolactams from Lysobacter enzymogenes and Their Absolute Configurations by Theoretical ECD Calculations. J. Nat. Prod. 2015, 78, 1841–1847. [Google Scholar] [CrossRef]
- Saha, S.; Zhang, W.; Zhang, G.; Zhu, Y.; Chen, Y.; Liu, W.; Yuan, C.; Zhang, Q.; Zhang, H.; Zhang, L.; et al. Activation and characterization of a cryptic gene cluster reveals a cyclization cascade for polycyclic tetramate macrolactams. Chem. Sci. 2017, 8, 1607–1612. [Google Scholar] [CrossRef]
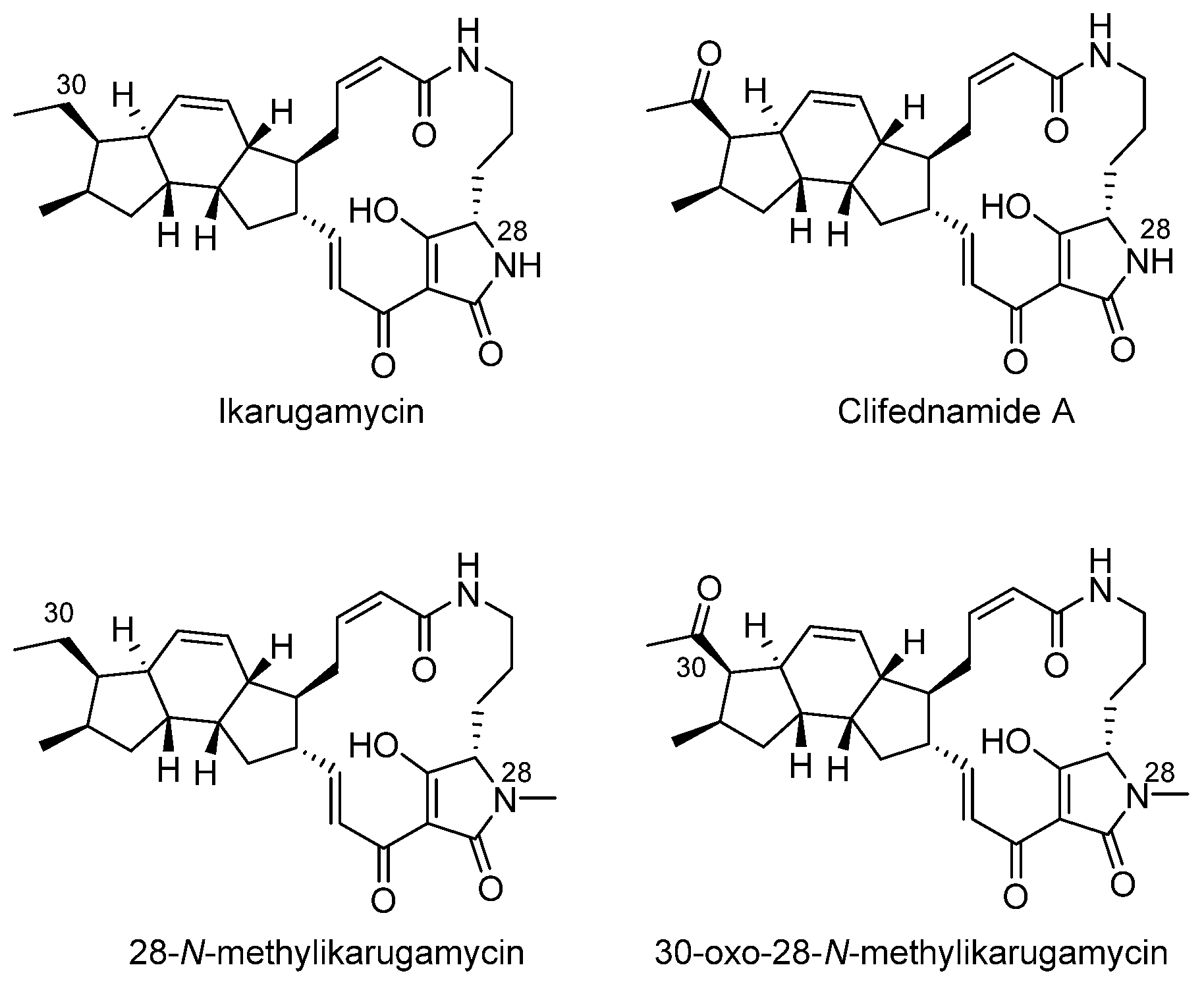
- Jin, H.; Zhang, W.; Zhang, G.; Zhang, L.; Liu, W.; Zhang, C. Engineered Biosynthesis of 5/5/6 Type Polycyclic Tetramate Macrolactams in an Ikarugamycin (5/6/5 Type)-Producing Chassis. Org. Lett. 2020, 22, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Liu, Y.; Jiao, Y.; Li, S.; Shen, Y.; Du, L. Biosynthesis of the Polycyclic System in the Antifungal HSAF and Analogues from Lysobacter enzymogenes. Angew. Chem. (Int. Ed. Engl.) 2018, 57, 6221–6225. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Shen, Y.; Li, Y.; Du, L. OX4 Is an NADPH-Dependent Dehydrogenase Catalyzing an Extended Michael Addition Reaction to Form the Six-Membered Ring in the Antifungal HSAF. Biochemistry 2019, 58, 5245–5248. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.P.; Day, A.; Tsingos, M.; Ding, E.; Zeng, E.; Stumpf, S.D.; Qi, Y.; Robinson, A.; Greif, J.; Blodgett, J.A.V. Critical analysis of polycyclic tetramate macrolactam biosynthetic gene cluster phylogeny and functional diversity. Appl. Environ. Microbiol. 2024, 90, e0060024. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, H.; Song, Y.; Zhu, D.; Shen, Y.; Li, Y. Combinatorial Biosynthesis of Oxidized Combamides Using Cytochrome P450 Enzymes from Different Polycyclic Tetramate Macrolactam Pathways. ACS Synth. Biol. 2021, 10, 2434–2439. [Google Scholar] [CrossRef]
- Jiang, P.; Jin, H.; Zhang, G.; Zhang, W.; Liu, W.; Zhu, Y.; Zhang, C.; Zhang, L. A Mechanistic Understanding of the Distinct Regio- and Chemoselectivity of Multifunctional P450s by Structural Comparison of IkaD and CftA Complexed with Common Substrates. Angew. Chem. (Int. Ed. Engl.) 2023, 62, e202310728. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr.; Rosenberg, S.A. Two hundred years of cancer research. N. Engl. J. Med. 2012, 366, 2207–2214. [Google Scholar] [CrossRef]
- Sullivan, R.; Alatise, O.I.; Anderson, B.O.; Audisio, R.; Autier, P.; Aggarwal, A.; Balch, C.; Brennan, M.F.; Dare, A.; D’Cruz, A.; et al. Global cancer surgery: Delivering safe, affordable, and timely cancer surgery. Lancet Oncol. 2015, 16, 1193–1224. [Google Scholar] [CrossRef] [PubMed]
- Craver, L.F. Recent advances in treatment of inoperable cancer. Bull. N. Y. Acad. Med. 1952, 28, 385–407. [Google Scholar]
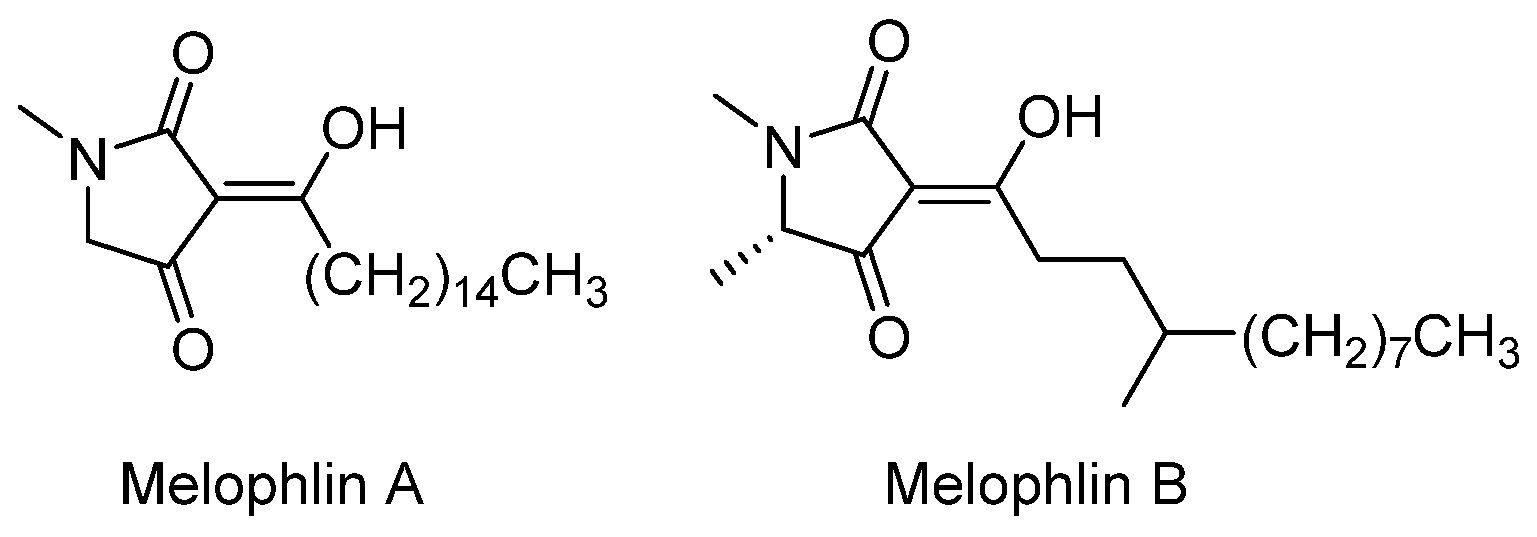
- Aoki, S.; Higuchi, K.; Ye, Y.; Satari, R.; Kobayashi, M. Melophlins A and B, novel tetramic acids reversing the phenotype of ras-transformed cells, from the marine sponge Melophlus sarassinorum. Tetrahedron 2000, 56, 1833–1836. [Google Scholar] [CrossRef]
- Birnie, G.D. The HL60 cell line: A model system for studying human myeloid cell differentiation. Br. J. Cancer. Suppl. 1988, 9, 41–45. [Google Scholar] [PubMed]
- Li, X.; Liu, Q.; Zou, H.; Luo, J.; Jiao, Y.; Wang, H.; Du, L.; Shen, Y.; Li, Y. Discovery and biosynthesis of pseudoamides reveal enzymatic cyclization of the polyene precursor to 5–5 bicyclic tetramate macrolactams. ACS Catal. 2023, 13, 4760–4767. [Google Scholar] [CrossRef]
- Chen, Y.; Jin, H.; Xiong, W.; Fang, Z.; Sun, W.; Zhu, Y.; Zhang, L.; Zhang, Y.; Zhang, W.; Zhang, C. Discovery of Aburatubolactams Reveals Biosynthetic Logic for Distinct 5/5-Type Polycyclic Tetramate Macrolactams. Org. Lett. 2024, 26, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Dhaneesha, M.; Hasin, O.; Sivakumar, K.C.; Ravinesh, R.; Naman, C.B.; Carmeli, S.; Sajeevan, T.P. DNA Binding and Molecular Dynamic Studies of Polycyclic Tetramate Macrolactams (PTM) with Potential Anticancer Activity Isolated from a Sponge-Associated Streptomyces zhaozhouensis subsp. mycale subsp. nov. Mar. Biotechnol. 2019, 21, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Khoo, J.C.; Miller, E.; McLoughlin, P.; Steinberg, D. Enhanced macrophage uptake of low density lipoprotein after self-aggregation. Arteriosclerosis 1988, 8, 348–358. [Google Scholar] [CrossRef]
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam Cells in Atherosclerosis: Novel Insights into Its Origins, Consequences, and Molecular Mechanisms. Front. Cardiovasc. Med. 2022, 9, 845942. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and its impact on cardiovascular health: Focus on atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef]
- Hasumi, K.; Shinohara, C.; Naganuma, S.; Endo, A. Inhibition of the uptake of oxidized low-density lipoprotein in macrophage J774 by the antibiotic ikarugamycin. Eur. J. Biochem. 1992, 205, 841–846. [Google Scholar] [CrossRef]
- Elkin, S.R.; Oswald, N.W.; Reed, D.K.; Mettlen, M.; MacMillan, J.B.; Schmid, S.L. Ikarugamycin: A Natural Product Inhibitor of Clathrin-Mediated Endocytosis. Traffic 2016, 17, 1139–1149. [Google Scholar] [CrossRef]
- Motley, A.; Bright, N.A.; Seaman, M.N.; Robinson, M.S. Clathrin-mediated endocytosis in AP-2-depleted cells. J. Cell Biol. 2003, 162, 909–918. [Google Scholar] [CrossRef]
- Luo, T.; Fredericksen, B.L.; Hasumi, K.; Endo, A.; Garcia, J.V. Human immunodeficiency virus type 1 Nef-induced CD4 cell surface downregulation is inhibited by ikarugamycin. J. Virol. 2001, 75, 2488–2492. [Google Scholar] [CrossRef] [PubMed]
- Popescu, R.; Heiss, E.H.; Ferk, F.; Peschel, A.; Knasmueller, S.; Dirsch, V.M.; Krupitza, G.; Kopp, B. Ikarugamycin induces DNA damage, intracellular calcium increase, p38 MAP kinase activation and apoptosis in HL-60 human promyelocytic leukemia cells. Mutat. Res. 2011, 709–710, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Bertasso, M.; Holzenkampfer, M.; Zeeck, A.; Stackebrandt, E.; Beil, W.; Fiedler, H.-P. Ripromycin and other polycyclic macrolactams from Streptomyces sp. Tu 6239: Taxonomy, fermentation, isolation and biological properties. J. Antibiot. 2020, 56, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.H.; Dong, F.Y.; Da, L.T.; Yang, X.M.; Wang, X.X.; Weng, J.Y.; Feng, L.; Zhu, L.L.; Zhang, Y.L.; Zhang, Z.G.; et al. Ikarugamycin inhibits pancreatic cancer cell glycolysis by targeting hexokinase 2. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 3943–3955. [Google Scholar] [CrossRef]
- Minamidate, A.; Onizawa, M.; Saito, C.; Hikichi, R.; Mochimaru, T.; Murakami, M.; Sakuma, C.; Asakawa, T.; Hiraoka, Y.; Oshima, S.; et al. A potent endocytosis inhibitor Ikarugamycin up-regulates TNF production. Biochem. Biophys. Rep. 2021, 27, 101065. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montavon, A.; Marchán-Rivadeneira, M.R.; Han, Y. Polycyclic Tetramate Macrolactams and Their Potential as Anticancer Agents. Organics 2024, 5, 361-377. https://doi.org/10.3390/org5040019
Montavon A, Marchán-Rivadeneira MR, Han Y. Polycyclic Tetramate Macrolactams and Their Potential as Anticancer Agents. Organics. 2024; 5(4):361-377. https://doi.org/10.3390/org5040019
Chicago/Turabian StyleMontavon, Alexandria, M. Raquel Marchán-Rivadeneira, and Yong Han. 2024. "Polycyclic Tetramate Macrolactams and Their Potential as Anticancer Agents" Organics 5, no. 4: 361-377. https://doi.org/10.3390/org5040019
APA StyleMontavon, A., Marchán-Rivadeneira, M. R., & Han, Y. (2024). Polycyclic Tetramate Macrolactams and Their Potential as Anticancer Agents. Organics, 5(4), 361-377. https://doi.org/10.3390/org5040019