Abstract
The preparation of bis(3-methylthio-1-azulenyl)phenylmethyl cations and 1,4-phenylenebis[bis(3,6-di-tert-butyl-1-azulenyl)methyl] dications was accomplished by the hydride abstraction of the corresponding hydride derivatives, which were synthesized by the acid-catalyzed condensation of 1-azulenyl methyl sulfide with benzaldehyde and terephthalaldehyde with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. The intramolecular charge transfer among the azulene ring and the methylium moieties of these cations and dications was investigated by UV–Vis spectroscopy and electrochemical analyses. The pKR+ values of the cations were examined for their thermodynamic stability spectrophotometrically. The voltammetry experiments of these cations revealed their reversible reduction waves on their cyclic voltammograms. Moreover, a notable spectral change of cations was observed by spectroelectrochemistry during electrochemical reduction conditions.
1. Introduction
Azulene and its derivatives have attracted interest in terms of their specific optical properties [1,2,3,4,5,6] and pharmacological activity [7,8,9,10]. The azulene system significantly stabilizes cationic as well as anionic states through the contribution of tropylium and cyclopentadienide substructures. Utilizing this characteristic property, the synthesis of redox-active chromophores composed by the azulene derivatives has been employed in our group due to the creation of stabilized electrochromic materials [11,12,13,14,15,16,17,18,19,20,21]. As a part of this research, a high thermodynamic stability and reversible redox behavior under the electrochemical conditions were observed in bis(1-azulenyl)phenylmethyl cations and dications connected by a 1,4-phenylene spacer, namely, 1+ and 22+ (Figure 1) [11,12,22]. However, in spectroelectrochemical measurements, 1+ and 22+ showed significant decomposition under electrochemical reduction conditions.
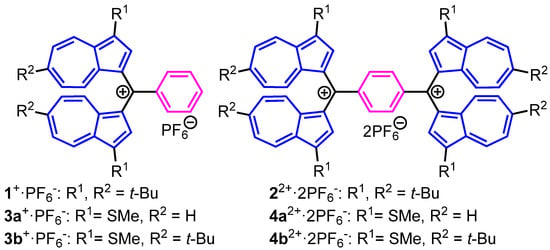
Figure 1.
Structures of bis(1-azulenyl)phenylmethylium hexafluorophosphates: 1+·PF6− and 3a,b+·PF6− and 1,4-phenylenebis[bis(1-azulenyl)methylium] bis(hexafluorophosphate)s; 22+·2PF6− and 4a,b2+·2PF6−.
Previously, we have described the effective synthetic procedure of several 1-azulenyl sulfides, as well as their unique reactivity and properties [23,24,25,26,27,28]. In these studies, we found that 1-azulenyl sulfides exhibit remarkable redox stability toward the electrochemical reactions. Considering these results, cations and dications incorporating the 1-methylthioazulene moieties should improve electrochemical stability in the electrochromic systems, i.e., having high reversibility in the redox behavior, in addition to a large thermodynamic stability in the ionic states.
We describe herein the preparation of bis(3-methylthio-1-azulenyl)phenylmethyl cations 3a,b+·PF6− and dications 4a,b2+·2PF6− connected by a 1,4-phenylene spacer. The thermodynamic stability of cations 3a,b+·PF6− and dications 4a,b2+·2PF6− were measured spectrophotometrically. Their redox behavior, as examined by cyclic voltammetry (CV) and differential pulse voltammetry (DPV), were also discussed. The spectroelectrochemistry of these cations was also examined, which showed notable spectral changes in the visible region in different redox states.
2. Results and Discussion
Synthesis: The strategy of reacting the aldehydes with azulene derivatives under acidic conditions to obtain the condensation product has been reported previously by several researchers, including our group [29,30,31,32,33,34,35]. The synthesis of bis(3-methylthio-1-azulenyl)methylbenzenes 6a,b was accomplished by the condensation of 5a,b [23] with benzaldehyde in acetic acid (AcOH), followed by the usual workup process in 93% and 100% yields, respectively (Figure 2). The higher reaction yields are considered to be due to the reaction control by the methyl sulfide group at the 1-position of the azulene ring, which suppresses the undesired oligomerization reactions occurring at both the 1- and 3-positions [36,37]. Compounds 7a,b were obtained in 87% and 95% yields by the reaction of 5a,b with terephthalaldehyde in a similar manner (Figure 2).
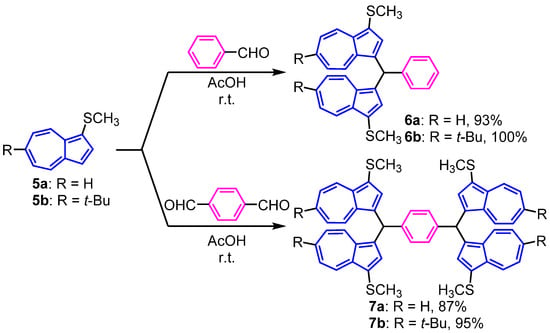
Figure 2.
Synthesis of 6a,b and 7a,b from 1-methylthioazulenes 5a,b.
The synthesis of 3a,b+·PF6− and 4a,b2+·2PF6− was established by the reaction of 6a,b, and 7a,b with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), as shown in Figure 3. The reactions of 6a and 6b with DDQ in dichloromethane (CH2Cl2), followed by the treatment with 60% HPF6 solution, produced 3a+·PF6− and 3b+·PF6− in 99% and 91% yield, respectively (Figure 3). Similar to the synthesis of 3a,b+·PF6−, the hydride abstraction of 7a,b with a two-fold amount of DDQ gave the dications 4a,b2+·2PF6− in 91% and 99% yields, after the addition of aq. HPF6 (Figure 3).
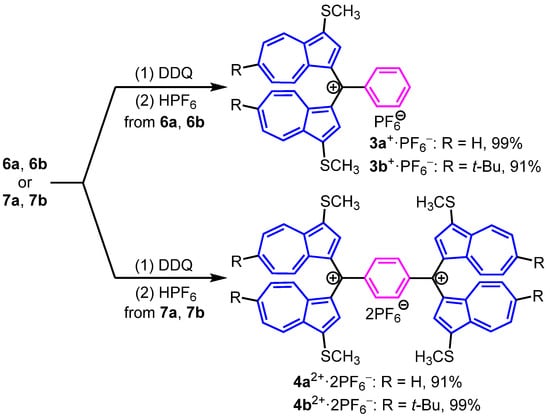
Figure 3.
Synthesis of cation 3a,b+·PF6− and dications 4a,b2+·2PF6−.
Spectroscopic properties: Cations 3a,b+·PF6− and 4a,b2+·2PF6− were fully characterized by spectroscopic data, as appears in the “Materials and Methods” section. High-resolution mass spectra (HRMS) revealed the correct molecular ion peaks. The chemical shifts in 1H NMR spectra of the azulene moiety for 3a,b+·PF6− and 4a,b2+·2PF6− revealed low-field shifts compared with those of 6a,b and 7a,b, attributing to the electron-withdrawing property of the methylium ion attached.
As expected by their cationic structures, UV–Vis spectra of 3a,b+·PF6− and 4a,b2+·2PF6− displayed the characteristic strong absorption band in the visible region (Table 1). For instance, the UV–Vis spectra of 3a,b+·PF6− and 4a,b2+·2PF6− in MeCN showed an absorption band at λmax = 740 nm (3a+·PF6−), 738 nm (3b+·PF6−), 773 nm (4a2+·2PF6−), and 766 nm (4b2+·2PF6−), which spread into the near-infrared region. The absorption maxima of 4a,b2+·2PF6− showed a modest red shift compared with those of 3a,b+·PF6−, implying the expansion of the π-electron system through the 1,4-phenylene spacer. The molar absorption coefficients of 4a,b2+·2PF6− are approximately twice as large as those of 3a,b+·PF6−, owing to the two bis(1-azulenyl)methylium units substituted. The strong and broad absorption bands of these cations can be ascribed to intramolecular charge transfer (ICT) across the two azulene rings, as illustrated by the resonance structure (Figure 4).

Table 1.
The longest absorption maxima [nm] and their coefficients (log ε) of 3a,b+·PF6− and 4a,b2+·2PF6− in several solvents, and those of 1+·PF6− and 22+·2PF6− in acetonitrile (MeCN) as a reference.
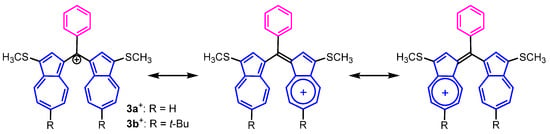
Figure 4.
Plausible resonance structures of 3a,b+·PF6−.
Solvatochromism is one of the characteristic features of dipolar molecules [38,39,40]. The solvent dependence of the UV–Vis spectra in the visible region of 3a,b+·PF6− and 4a,b2+·2PF6− implied the ICT character of the absorption band. A strong absorption band of 3a+·PF6− at λmax = 755 nm in CH2Cl2 showed a hypsochromic shift to λmax = 732 nm in the less-polar solvent, i.e., in 10% CH2Cl2/hexane-mixed solvent (Figure 5). Similar solvent effects observed in CH2Cl2 and 10% CH2Cl2/hexane in 3a+·PF6− can also be observed in 3b+·PF6− and 4a,b2+·2PF6−.
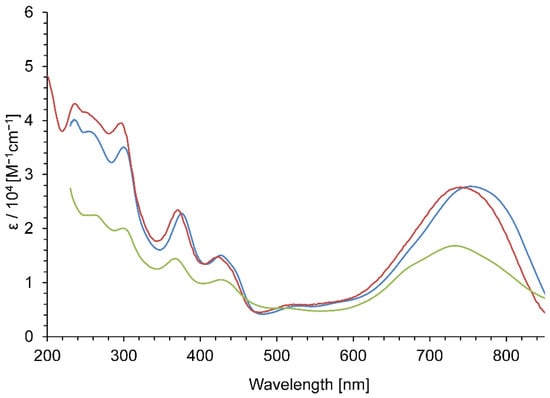
Figure 5.
UV–Vis spectra of 3a+·PF6− in CH2Cl2 (blue line), in MeCN (red line), and in 10% CH2Cl2/hexane (light green line).
The pKR+ values of 3a,b+·PF6− and 4a,b2+·2PF6− were measured spectrophotometrically to investigate the thermodynamic stability of the cations (Table 2). The pKR+ value of 3b+·PF6− (pKR+ = 11.6 ± 0.1) was higher than that of 3a+·PF6− (pKR+ = 9.9 ± 0.1). These outcomes could be ascribed to the stabilization by the hyperconjugation of the tert-butyl group at the seven-membered moiety of the azulene ring [23]. The 4a2+·2PF6− (pKR+ = 9.8 ± 0.1) and 4b2+·2PF6− (pKR+ = 11.7 ± 0.1) with two cation units were neutralized simultaneously. Thus, 4a2+·2PF6− and 4b2+·2PF6− exhibit high stability, as similar to those of 3a+·PF6− and 3b+·PF6−. The pKR+ values of 3a+·PF6− and 3b+·PF6− are lower than that of 1+·PF6− (pKR+ = 12.4). These outcomes indicate that the stabilization by the methylthio group is less effective than the tert-butyl group, which is most likely attributed to the ineffective electron-donating ability of the sulfur atom.
Table 2.
pKR+ Values a of 3a,b+·PF6−, 4a,b2+·2PF6−, 1+·PF6−, and 22+·2PF6−.
The redox behaviors of 3a,b+·PF6− and 4a,b2+·2PF6− were investigated by CV and DPV, in order to clarify their electrochemical properties (Table 3). Cation 3a+·PF6− revealed a reversible reduction wave at E1red = −0.60 V on the CV, indicating the generation of a neutral radical species (Figure 6). The half-wave potential of E1red = –0.68 V on the CV was observed in the electrochemical reduction of 3b+·PF6−. The E1red value of 3b+·PF6− showed a slight cathodic shift compared with that of 3a+·PF6−, indicating the stabilization of the carbocation by the tert-butyl groups, as expected from the results of the pKR+ measurements.
Table 3.
Redox Potentials a of 3a,b+·PF6−, 4a,b2+·2PF6−, 1+·PF6−, and 22+·2PF6− in benzonitrile.
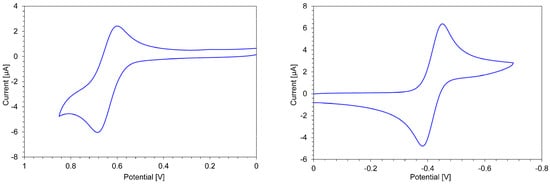
Figure 6.
Cyclic voltammogram for the oxidation (left) and reduction (right) of 4b+·PF6− in benzonitrile (1 mM) containing Et4NClO4 (0.1 M) as a supporting electrolyte.
Although the pKR+ values are almost equal each other, the E1red of 4a2+·2PF6− (E1red = –0.35 V) displayed a more anodic shift than that of 3a+·PF6− (E1red = –0.60 V). The anodic shift of 4a2+·2PF6− may be ascribed to the electrochemical instability resulting from the electrostatic repulsion between the cations through the benzene ring connected. The reversible reduction waves of 4a,b2+·2PF6− may be attributable to the one-step reduction of the two cation units forming the closed-shell quinoidal structures 8a and 8b, as shown in Figure 7 [41].
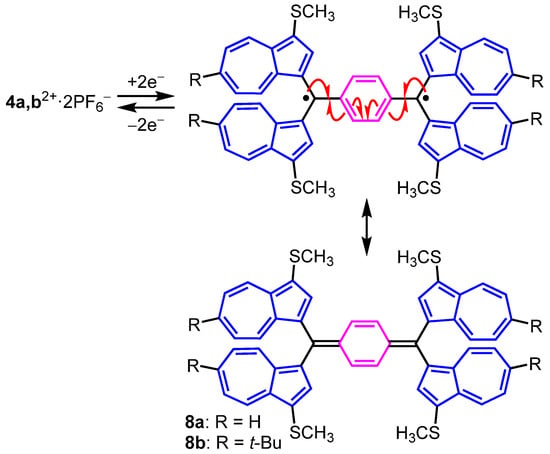
Figure 7.
Plausible redox behavior of 4a,b2+·2PF6− and generated quinoidal species 8a,b.
Electrochromism is a phenomenon that displays a significant color change in different redox states and is observed in molecules that show reversible redox behavior. Since high redox stability is required for the application to electrochromic materials, durability toward the redox cycle is an important factor to construct such materials [42,43,44]. The concept of violene–cyanine hybrids was proposed by Hünig et al. as a guideline for the creation of electrochromic materials [45,46,47,48,49]. The hybrid consists of a violene substructure involving delocalized, closed-shell polymethine (e.g., cyanine) dyes as terminal groups. Thus, in a redox system with a hybrid structure, the color of the cyanine dye could be observed by overall two-electron transfer (Figure 8). In contrast to the violene-type redox system, hybrid structures should improve the stability of electrochromic systems because both colored and discolored species comprise a closed shell structure.
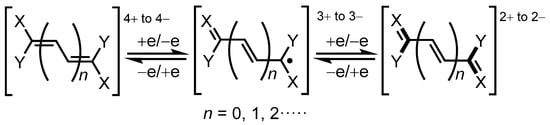
Figure 8.
The general structure of violene–cyanine hybrids proposed by Hünig et al.; cyanine-type substructures produced by two-electron transfer are illustrated by bold lines.
Therefore, spectroelectrochemical measurements of 3a,b+·PF6− and 4a,b2+·2PF6− were investigated under an electrochemical reaction. In the electrochemical reduction, the longest absorption band of λmax = 740 nm for 3a+·PF6− gradually decreased, at which time the color of the solution changed from green to yellow. However, despite the good reversibility observed on the CV, the reverse oxidation of the yellow-colored solution did not reproduce the original spectrum of 3a+·PF6− (27% recovery, Figure 9). The less reversible color change might be explained by the generation of unstable neutral radical species under the measurement conditions. The absorption band at λmax = 738 nm of 3b+·PF6− was also reduced upon the electrochemical reduction, along with the green-colored solution turning to yellow (see Supplementary Materials). Similar to 3a+·PF6−-, the visible spectrum of 3b+·PF6− was incompletely recovered when the reduced solution was reversely oxidized (12% recovery).
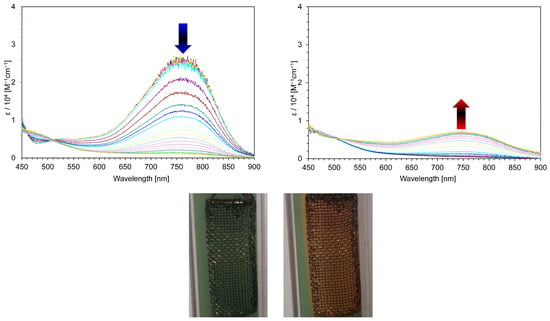
Figure 9.
Continuous change in visible spectra of 3a+·PF6− in benzonitrile containing Et4NClO4 (0.1 M): constant-current electrochemical reduction (50 μA) at 30 s intervals (left) and the reverse oxidation (right) of the reduced species (50 μA) at 30 s intervals. Photo: Color changes of 3a+·PF6− upon the electrochromic analysis in benzonitrile containing Et4NClO4 (0.1 M) (50 μA); before electrochemical reduction (left) and after electrochemical reduction (right).
The color change of 4a,b2+·2PF6− was also observed under the electrochemical reduction. In the absorption spectrum of 4a,b2+·2PF6−, an absorption band was developed at around λmax = 600 nm during the electrolytic reduction, accompanied by the disappearance of the absorption band centered at λmax = 773 nm. The original absorption spectrum of 4a2+·2PF6− was regenerated by the reverse oxidation of the reducing species (51% recovery) (see Supplementary Materials). This reversible spectral change was also confirmed by the electrochemical reduction of 4b2+·2PF6−, with a gradual development of the absorption band at around λmax = 610 nm, resulting in the green color of the solution turning blue. Reverse oxidation resulted in the disappearance of the newly generated absorption band, together with the regeneration of the original absorption of 4b2+·2PF6− (64% recovery, Figure 10). The higher reversibility of 4a,b2+·2PF6−, compared to that of 3a,b+·PF6−, could be explained by the generation of closed-shell species, i.e., quinoidal species 8a,b, by electrochemical reduction.
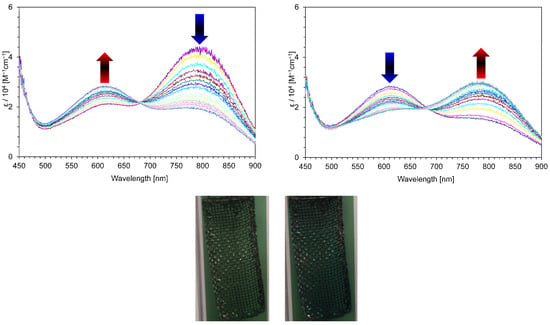
Figure 10.
Continuous change in visible spectra of 4b2+·2PF6− in benzonitrile containing Et4NClO4 (0.1 M): constant-current electrochemical reduction (50 μA) at 30 s intervals (top) and the reverse oxidation (bottom) of the reduced species (50 μA) at 30 s intervals. Photo: Color changes of 4a2+·2PF6− upon the electrochromic analysis in benzonitrile containing Et4NClO4 (0.1 M) (50 μA); before electrochemical reduction (left) and after electrochemical reduction (right).
Dications 4a,b2+·2PF6− contain two bis(3-methylthio-1-azulenyl)methylium units as the end groups, which are regarded as cyanine-type substructures to form a delocalized closed-shell system in the violene–cyanine hybrid structure. Thus, the electrochemical reduction of 4a,b2+·2PF6− could generate the other closed-shell systems 8a,b by two-electron transfer. As seen from the electrochemical behavior of 4a,b2+·2PF6−, the radical cationic species are not important in the redox systems, so the color change in the absorption spectra is mainly due to these two closed-shell species throughout. Since the dications 4a,b2+·2PF6− displayed distinct spectral changes at the different redox states, the electrochromic behavior of these dications should serve as the violene–cyanine hybrid, in which the four end groups X and Y in Figure 8 are azulene rings to form a quinoidal substructure in their reduced form.
3. Materials and Methods
Melting points were measured with a Yanagimoto MPS3 micro melting apparatus. High-resolution mass spectra were obtained with a Bruker APEX II instrument. IR spectra were measured with a JASCO FT/IR-4100 spectrophotometer (JASCO Corporation, Tokyo, Japan). The UV–Vis spectra were recorded with a Shimadzu UV-2550 spectrophotometer (Shimadzu Corporation, Kyoto, Japan). 1H and 13C NMR spectra were recorded in CDCl3 with a Bruker Avance 400 at 400 MHz and 100 MHz (Bruker BioSpin, Rheinstetten, Germany), respectively.
Bis(3-methylthio-1-azulenyl)phenylmethane (6a): a mixture of 5a (273 mg, 1.57 mmol) and benzaldehyde (55 mg, 0.52 mmol) in acetic acid (5 mL) was stirred at room temperature for 24 h. The reaction mixture was diluted with CH2Cl2. The organic layer was washed with a 5% NaHCO3 solution and water, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with CH2Cl2 to afford 6a (212 mg, 93%) as greenish-blue crystals. M.p. 186.0–188.0 °C (CH2Cl2); IR (KBr disk): νmax = 3021 (w), 2915 (w), 1574 (s), 1493 (m), 1451 (w), 1399 (s), 1381 (m), 1333 (w), 1248 (w), 1129 (w), 1075 (w), 1030 (w), 945 (w), 914 (w), 878 (w), 862 (w), 737 (s), 706 (m), 573 (w), 561 (w) cm−1; UV–Vis (CH2Cl2): λmax (log ε) = 240 (4.60), 296 sh (4.75), 299 (4.76), 366 sh (4.02), 374 (4.06), 619 (2.67) nm; 1H NMR (400 MHz, CDCl3): δ = 8.44 (d, 2H, J = 9.6 Hz, H-4), 8.04 (d, 2H, J = 9.6 Hz, H-8), 7.36 (s, 2H, H-2), 7.32 (t, 2H, J = 9.6 Hz, H-6), 7.14–7.02 (m, 5H, Ph), 6.98 (t, 2H, J = 9.6 Hz, H-5), 6.80 (d, 2H, J = 9.6 Hz, H-7), 6.54 (s, 1H, CH), 2.22 ppm (s, 6H, SCH3); 13C NMR (100 MHz, CDCl3): δ = 145.37, 141.33, 140.10, 138.90, 137.16, 135.93, 134.48, 132.67, 129.27, 128.92, 126.74, 123.45, 123.42, 120.85, 43.14, 20.99 ppm; HRMS (ESI): Calculated for C29H24S2 + Na+ [M + Na]+ 459.1212; found: 459.1211.
Bis(6-tert-butyl-3-methylthio-1-azulenyl)phenylmethane (6b): the procedure used for the preparation of 6a was adopted here. Reaction of 5b (1.00 g, 4.34 mmol) and benzaldehyde (185 mg, 1.74 mmol) in acetic acid (15 mL) at room temperature for 24 h, followed by column chromatography on silica gel with CH2Cl2 to afford 6b (660 mg, 100%) as a blue solid. M.p. 152.0–155.0 °C (CH2Cl2); IR (KBr disk): νmax = 2965 (m), 2869 (w), 1580 (s), 1493 (w), 1408 (w), 1397 (m), 1364 (w), 1306 (w), 1254 (w), 1067 (w), 963 (w), 839 (w), 716 (w), 698 (w), 677 (w), 581 (w) cm−1; UV–Vis (CH2Cl2): λmax (log ε) = 241 (4.60), 305 (4.91), 360 (4.07), 376 (4.08), 600 (2.87) nm; 1H NMR (400 MHz, CDCl3): δ = 8.51 (d, 2H, J = 10.4 Hz, H-8), 8.12 (d, 2H, J = 10.4 Hz, H-4), 7.38 (s, 2H, H-2), 7.34 (dd, 2H, J = 10.4, 1.6 Hz, H-7), 7.17 (dd, 2H, J = 10.4, 1.6 Hz, H-5), 7.3–7.1 (m, 5H, Ph), 6.60 (s, 1H, CH), 2.35 (s, 6H, SCH3), 1.40 ppm (s, 18H, tBu); 13C NMR (100 MHz, CDCl3): δ = 162.56, 145.55, 140.69, 138.87, 135.89, 134.70, 133.50, 132.34, 129.24, 128.75, 126.48, 121.59, 121.45, 119.87, 42.86, 38.98, 32.22, 21.16 ppm; HRMS (ESI): Calculated for C37H40S2 + Na+ [M + Na]+ 571.2464; found: 571.2462.
1,4-Bis[bis(3-methylthio-1-azulenyl)methyl]benzene (7a): the procedure used for the preparation of 6a was adopted here. Reaction of 5a (527 mg, 3.03 mmol) and terephthalaldehyde (67 mg, 0.50 mmol) in acetic acid (15 mL) at 50 °C for 24 h, followed by column chromatography on silica gel with CH2Cl2 to afford 7a (347 mg, 87%) as dark green crystals. M.p. 183.5–187.0 °C (CH2Cl2); IR (KBr disk): νmax = 3019 (w), 2919 (w), 1572 (s), 1504 (m), 1489 (w), 1449 (w), 1418 (w), 1395 (m), 1377 (m), 1333 (w), 1312 (w), 1217 (w), 1022 (w), 961 (w), 943 (w), 772 (w), 749 (m), 729 (s), 559 (w) cm−1; UV–Vis (CH2Cl2): λmax (log ε) = 242 (4.89), 291 (5.04), 299 (5.03), 321 sh (4.61), 363 sh (4.31), 374 (4.34), 614 (3.08), 647 sh (3.07) nm; 1H NMR (400 MHz, CDCl3): δ = 8.55 (d, 4H, J = 9.6 Hz, H-8), 8.16 (d, 4H, J = 9.6 Hz, H-4), 7.54 (t, 4H, J = 9.6 Hz, H-6), 7.45 (s, 4H, H-2), 7.15 (t, 4H, J = 9.6 Hz, H-7), 7.05 (s, 4H, Ph), 7.00 (t, 4H, J = 9.6 Hz, H-5), 6.62 (s, 2H, CH), 2.36 (s, 12H, SCH3) ppm; Low solubility hampered the measurement of 13C NMR. HRMS (ESI): Calculated for C52H42S4 + Na+ [M + Na]+ 817.2062; found: 817.2056.
1,4-Bis[bis(6-tert-butyl-3-methylthio-1-azulenyl)methyl]benzene (7b): the procedure used for the preparation of 6a was adopted here. Reaction of 5b (1.98 g, 8.59 mmol) and terephthalaldehyde (196 mg, 1.46 mmol) in acetic acid (20 mL) and CH2Cl2 (2 mL) at room temperature for 24 h, followed by column chromatography on silica gel with CH2Cl2 to afford 7b (1.42 g, 95%) as green crystals. M.p. > 300 °C (CH2Cl2); IR (KBr disk): νmax = 2963 (m), 2917 (w), 2869 (w), 1578 (s), 1549 (w), 1505 (w), 1408 (w), 1362 (w), 1337 (w), 1308 (w), 1252 (w), 1067 (w), 1021 (w), 965 (w), 835 (m), 822 (w), 675 (w), 588 (w), 450 (w) cm−1; UV–Vis (CH2Cl2): λmax (log ε) = 241 (4.89), 296 sh (5.15), 305 (5.18), 360 (4.36), 376 (4.36), 601 (3.15) nm; 1H NMR (400 MHz, CDCl3): δ = 8.48 (d, 4H, J = 10.0 Hz, H-4), 8.13 (d, 4H, J = 10.0 Hz, H-8), 7.39 (s, 4H, H-2), 7.33 (dd, 4H, J = 10.0, 1.2 Hz, H-5), 7.17 (dd, 4H, J = 10.0, 1.2 Hz, H-7), 7.06 (s, 4H, Ph), 6.58 (s, 2H, CH), 2.35 (s, 12H, SCH3), 1.39 (s, 36H, tBu) ppm; 13C NMR (100 MHz, CDCl3): δ = 145.37, 141.33, 140.10, 138.90, 137.16, 135.93, 134.48, 132.67, 129.27, 128.92, 126.74, 123.45, 123.42, 120.85, 43.14, 20.99, 15.78 ppm; HRMS (ESI): Calculated for C68H74S4 + Na+ [M + Na]+ 1041.4566; found: 1041.4560.
General Procedure for the Preparation of Hexafluorophosphates 3a,b+∙PF6− and 4a,b2+∙2PF6−: DDQ was added to a solution of 6a, 6b, 7a, and 7b in CH2Cl2 and the solution was stirred at room temperature for 30 min. 60% HPF6 solution was added to the mixture and stirred for an additional 15 min. Water was added to the mixture and the resulting suspension was collected by filtration. The filtrate was also extracted with CH2Cl2, washed with water, dried with MgSO4, and concentrated under reduced pressure. The residue was crystallized from CH2Cl2 and Et2O. The precipitated crystals were collected by filtration and washed with Et2O to give the corresponding cations 3a+, 3b+, 4b2+, and 4b2+ as hexafluorophosphates.
Bis(3-methylthio-1-azulenyl)phenylmethylium Hexafluorophosphate (3a+∙PF6−): the general procedure was followed using DDQ (65 mg, 0.29 mmol), 6a (104 mg, 0.24 mmol), and 60% HPF6 (5 mL) in CH2Cl2 (25 mL). Recrystallization from CH2Cl2/ether gave 3a+∙PF6− (131 mg, 99%) as dark green crystals. M.p. > 300 °C (decomp.); IR (KBr disk): νmax = 2923 (w), 1592 (w), 1570 (w), 1538 (w), 1470 (s), 1437 (m), 1408 (s), 1325 (s), 1310 (s), 1277 (s), 1227 (m), 1200 (w), 1090 (w), 999 (w), 968 (w), 922 (w), 878 (m), 839 (s), 739 (w), 693 (w), 596 (w), 558 (m), 486 (w), 453 (w), 434 (w) cm−1; UV–Vis (CH3CN): λmax (log ε) = 235 (4.68), 254 sh (4.62), 295 (4.60), 370 (4.37), 422 (4.17), 518 sh (3.78), 740 (4.44) nm; UV–Vis (CH2Cl2): λmax (log ε) = 237 (4.60), 256 sh (4.58), 300 (4.54), 376 (4.36), 428 (4.18), 521 sh (3.75), 755 (4.44) nm; UV–Vis (Hexane): λmax (log ε) = 266 sh (4.54), 300 (4.49), 367 (4.35), 427 (4.21), 671 sh (4.30), 733 (4.42) nm; 1H NMR (400 MHz, CD3CN): δ = 8.80 (dd, 2H, J = 10.0, 1.2 Hz, H-4), 8.16 (t, 2H, J = 10.0 Hz, H-6), 8.05 (ddd, 2H, J = 10.0, 0.8, 0.8 Hz, H-5), 7.93 (dd, 2H, J = 10.0, 0.8 Hz, H-8), 7.84 (s, 2H, H-2), 7.83 (tt, 1H, J = 8.0, 1.2 Hz, p-Ph), 7.64 (ddd, 2H, J = 8.0, 1.2, 1.2 Hz, m-Ph), 7.57 (ddd, 2H, J = 10.0, 0.8, 0.8 Hz, H-7), 7.50 (dd, 2H, J = 8.0, 1.2 Hz, o-Ph), 2.61 ppm (s, 6H, SCH3) ppm; 13C NMR (100 MHz, CD3CN): δ = 160.30, 151.35, 150.45, 145.75, 143.72, 143.13, 141.49, 140.26, 136.56, 136.46, 136.44, 136.38, 134.71, 134.53, 130.52, 17.69 ppm; HRMS (ESI): Calculated for C29H23S2+ [M − PF6]+ 435.1241; found: 435.1241.
Bis(6-tert-butyl-3-methylthio-1-azulenyl)phenylmethylium Hexafluorophosphate (3b+∙PF6−): the general procedure was followed by using DDQ (272 mg, 1.20 mmol), 6b (549 mg, 1.00 mmol), and 60% HPF6 (10 mL) in CH2Cl2 (50 mL). Recrystallization from CH2Cl2/ether gave 3b+∙PF6− (630 mg, 91%) as dark green crystals. M.p. 168.0–175.0 °C (decomp.); IR (KBr disk): νmax = 2963 (w), 2872 (w), 1572 (w), 1497 (w), 1470 (s), 1441 (m), 1416 (s), 1370 (w), 1347 (m), 1323 (s), 1294 (s), 1244 (s), 1192 (m), 1111 (m), 1075 (m), 870 (w), 839 (s), 733 (w), 704 (w), 558 (m) cm−1; UV–Vis (CH3CN): λmax (log ε) = 258 (4.57), 304 (4.55), 370 (4.31), 426 (4.13), 515 sh (3.74), 740 (4.40) nm; UV–Vis (CH2Cl2): λmax (log ε) = 261 (4.55), 306 (4.54), 376 (4.29), 429 (4.11), 515 sh (3.73), 738 (4.40) nm; UV-Vis (Hexane): λmax (log ε) = 262 (4.55), 306 (4.52), 362 (4.29), 424 (4.02), 672 sh (4.30), 723 (4.38) nm; 1H NMR (400 MHz, CD3CN): δ = 8.69 (d, 2H, J = 10.4 Hz, H-4), 8.23 (dd, 2H, J = 10.4, 0.8 Hz, H-5), 7.80 (t, 1H, J = 7.6 Hz, p-Ph), 7.78 (d, 2H, J = 10.8 Hz, H-8), 7.71 (dd, 2H, J = 10.4, 0.8 Hz, H-7), 7.70 (s, 2H, H-2), 7.63 (t, 2H, J = 7.6 Hz, m-Ph), 7.50 (d, 2H, J = 7.6 Hz, o-Ph), 2.58 (s, 6H, SCH3), 1.40 (s, 18H, 6-tBu) ppm; 13C NMR (100 MHz, CD3CN): δ = 171.37, 159.23, 150.07, 148.90, 143.10, 142.78, 140.46, 138.96, 136.23, 135.64, 134.95, 134.46, 134.34, 134.18, 130.42, 40.78, 32.08, 17.86 ppm; HRMS (ESI): Calculated for C37H39S2+ [M − PF6]+ 547.2488; found: 547.2488.
1,4-Phenylenebis[bis(3-methylthio-1-azulenyl)methylium] Bis(hexafluorophosphate) (4a2+∙2PF6−): the general procedure was followed by using DDQ (36 mg, 0.16 mmol), 7a (52 mg, 0.065 mmol), and 60% HPF6 (4 mL) in CH2Cl2 (30 mL). Recrystallization from CH2Cl2/ether gave 4a2+∙2PF6− (65 mg, 91%) as dark green crystals. M.p. > 300 °C (decomp.); IR (KBr disk): νmax = 2960 (w), 1472 (m), 1437 (w), 1408 (m), 1325 (s), 1308 (s), 1277 (s), 1229 (m), 1086 (w), 968 (w), 926 (w), 878 (w), 837 (s), 760 (w), 739 (w), 691 (w), 558 (m), 509 (w), 455 (w) cm−1; UV-Vis (CH3CN): λmax (log ε) = 236 (4.82), 255 (4.82), 298 (4.76), 376 (4.46), 423 (4.49), 607 (4.29), 773 (4.55) nm; UV–Vis (CH2Cl2): λmax (log ε) = 260 (4.83), 302 (4.77), 382 (4.51), 428 (4.50), 612 sh (4.31), 785 (4.55) nm; UV–Vis (Hexane): λmax (log ε) = 259 (4.60), 306 (4.52), 384 (4.39), 442 (4.32), 634 sh (4.19) nm; 1H NMR (400 MHz, CD3CN): δ = 8.80 (d, 4H, J = 9.6 Hz, H-4), 8.22 (t, 4H, J = 9.6 Hz, H-6), 8.11 (d, 4H, J = 9.6 Hz, H-8), 8.09 (t, 4H, J = 9.6 Hz, H-5), 7.99 (s, 4H, H-2), 7.70 (s, 4H, Ph), 7.66 (t, 4H, J = 9.6 Hz, H-7), 2.67 (s, 12H, SCH3) ppm; 13C NMR (100 MHz, CD3CN): δ = 156.92, 151.52, 150.56, 146.92, 146.03, 143.09, 141.73, 140.37, 137.33, 137.02, 136.92, 136.72, 134.98, 17.58 ppm; HRMS (ESI): Calculated for C52H40S42+ [M − 2PF6]2+ 396.1001; found: 396.1001.
1,4-Phenylenebis[bis(6-tert-butyl-3-methylthio-1-azulenyl)methylium] Bis(hexafluorophosphate) (4b2+∙2PF6−): the general procedure was followed by using DDQ (60 mg, 0.26 mmol), 7b (109 mg, 0.11 mmol), and 60% HPF6 (5 mL) in CH2Cl2 (25 mL). Recrystallization from CH2Cl2/ether gave 4b2+∙2PF6− (128 mg, 99%) as dark green crystals. M.p. 252.0–257.0 °C (CH2Cl2/ether); IR (KBr disk): νmax = 2965 (w), 2872 (w), 1574 (m), 1497 (m), 1472 (s), 1443 (s), 1416 (s), 1370 (m), 1320 (s), 1296 (s), 1246 (s), 1194 (m), 1111 (m), 1076 (m), 1044 (w), 972 (w), 924 (w), 839 (s), 737 (w), 700 (w), 558 (m), 507 (w), 469 (w) cm−1; UV-Vis (CH3CN): λmax (log ε) = 236 (4.85), 264 (4.88), 303 (4.89), 386 sh (4.55), 416 (4.57), 604 (4.33), 766 (4.62) nm; UV–Vis (CH2Cl2): λmax (log ε) = 266 (4.79), 307 (4.80), 385 (4.50), 424 (4.50), 612 sh (4.31), 777 (4.54) nm; UV–Vis (Hexane): λmax (log ε) = 265 (4.65), 306 (4.67), 424 (4.41), 766 (4.44) nm; 1H NMR (400 MHz, CD3CN): δ = 8.73 (d, 4H, J = 10.8 Hz, H-4), 8.28 (dd, 4H, J = 10.8, 2.0 Hz, H-5), 7.92 (s, 4H, H-2), 7.88 (d, 4H, J = 10.8 Hz, H-8), 7.76 (dd, 4H, J = 10.8, 2.0 Hz, H-7), 7.75 (s, 4H, Ph), 2.66 (s, 12H, SCH3), 1.42 (s, 36H, t-Bu) ppm; 13C NMR (100 MHz, CD3CN): δ = 171.67, 155.99, 150.23, 148.98, 146.87, 142.16, 140.56, 139.08, 136.48, 136.36, 135.31, 134.95, 134.60, 40.88, 32.09, 17.82; HRMS (ESI): Calculated for C63H72S42+ [M − 2PF6]2+ 508.2253; found: 508.2253.
4. Conclusions
In summary, we have prepared cations 3a,b+·PF6− and dications 4a,b2+·2PF6− and clarified their properties. Although the pKR+ values of 3a,b+·PF6− and 4a,b2+·2PF6− indicated a lower thermodynamic stability compared with 1+·PF6− and dications 22+·2PF6−, these cations still exhibited high pKR+ values, indicating high thermodynamic stability. These observations indicate the less effective stabilization of the methylthio moiety compared with that of the tert-butyl groups at the same position. The CV experiments showed that 3a,b+·PF6− and 4a,b2+·2PF6− exhibited a reversible reduction wave. Furthermore, a noticeable color change was observed during the electrochemical reduction of 3a,b+·PF6− and 4a,b2+·2PF6−. In particular, 4a,b2+·2PF6− displayed a remarkable color change resulting from the formation of the quinoidal structures 8a,b. These facts indicate that 4a,b2+·2PF6− served as the violene–cyanine hybrid in terms of a one-step two-electron reduction to generate a quinoidal form.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/org3040034/s1, Supplementary File S1: charts of 1H NMR, 13C NMR and COSY spectra, Supplementary File S2: UV–Vis spectra, Supplementary File S3: continuous change in the visible spectra and their photos, and Supplementary File S4: cyclic voltammograms of 3a,b+·PF6− and 4a,b2+·2PF6−.
Author Contributions
Conceptualization, T.S.; methodology, T.S. and N.S.; formal analysis, T.S., N.S., R.S. and S.I.; investigation, T.S., N.S., R.S. and S.I.; resources, T.S., N.S., R.S. and S.I.; writing–original draft preparation, T.S.; writing–review and editing, T.S., N.S., R.S. and S.I.; supervision, T.S.; funding acquisition, T.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Number 21K05037.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zeller, K.-P. Azulene in Methoden. Org. Chem. 1952, V Pt 2c, 127–418. [Google Scholar]
- Ito, S.; Morita, N. Creation of Stabilized Electrochromic Materials by Taking Advantage of Azulene Skeletons. Eur. J. Org. Chem. 2009, 2009, 4567–4579. [Google Scholar] [CrossRef]
- Ito, S.; Shoji, T.; Morita, N. Recent Advances in the Development of Methods for the Preparation of Functionalized Azulenes for Electrochromic Applications. Synlett 2011, 16, 2279–2298. [Google Scholar] [CrossRef]
- Shoji, T.; Ito, S. The Preparation and Properties of Heteroarylazulenes and Hetero-Fused Azulenes. Adv. Heterocycl. Chem. 2018, 126, 1–54. [Google Scholar]
- Shoji, T.; Okujima, T.; Ito, S. Development of Heterocycle-Substituted and Fused Azulenes in the Last Decade (2010–2020). Int. J. Mol. Sci. 2020, 21, 7087. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Ito, S.; Yasunami, M. Synthesis of Azulene Derivatives from 2H-Cyclohepta[b]furan-2-ones as StartingMaterials: Their Reactivity and Properties. Int. J. Mol. Sci. 2021, 22, 10686. [Google Scholar] [CrossRef]
- Tomiyama, T.; Wakabayashi, S.; Kosakai, K.; Yokota, M. Azulene derivatives: New non-prostanoid thromboxane A2 receptor antagonists. J. Med. Chem. 1990, 33, 2323–2326. [Google Scholar] [CrossRef]
- Tomiyama, T.; Yokota, M.; Wakabayashi, S.; Kosakai, K.; Yanagisawa, T. Design, synthesis, and pharmacology of 3-substituted sodium azulene-1-sulfontes and related compounds: Non-prostanoid thromboxane A2 receptor antagonists. J. Med. Chem. 1993, 36, 791–800. [Google Scholar] [CrossRef]
- Nakamura, H.; Sekido, M.; Yamamoto, Y. Synthesis of Carboranes Containing an Azulene Framework and in vitro Evaluation as Boron Carriers. J. Med. Chem. 1997, 40, 2825–2830. [Google Scholar] [CrossRef]
- Rekka, E.; Chrysselis, M.; Siskou, I.; Kourounakis, A. Synthesis of New Azulene Derivatives and Study of Their Effect on Lipid Peroxidation and Lipoxygenase Activity. Chem. Pharm. Bull. 2002, 50, 904–907. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, S.; Okujima, T.; Morita, N.; Asao, T. Synthesis, Properties, and Redox Behavior of Di(1-azulenyl)(2- and 3-thienyl)methyl Cations and Dications Composed of Two Di(1-azulenyl)methylium Units Connected with 2,5-Thiophenediyl and 2,5-Thienothiophenediyl Spacers. J. Org. Chem. 2001, 66, 2470–2479. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Nomura, A.; Morita, N.; Kabuto, C.; Kobayashi, H.; Maejima, S.; Fujimori, K.; Yasunami, M. Synthesis and Two-Electron Redox Behavior of Diazuleno[2,1-a:1,2-c]naphthalenes. J. Org. Chem. 2002, 67, 7295–7302. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Inabe, H.; Morita, N.; Ohta, K.; Kitamura, T.; Imafuku, K. Synthesis of Poly(6-azulenylethynyl)benzene Derivatives as a Multielectron Redox System with Liquid Crystalline Behavior. J. Am. Chem. Soc. 2003, 125, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Kubo, T.; Morita, N.; Ikoma, T.; Tero-Kubota, S.; Kawakami, J.; Tajiri, A. Azulene-Substituted Aromatic Amines. Synthesis and Amphoteric Redox Behavior of N,N-Di(6-azulenyl)-p-toluidine and N,N,N‘,N‘-Tetra(6-azulenyl)-p-phenylenediamine and Their Derivatives. J. Org. Chem. 2005, 70, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Akimoto, K.; Kawakami, J.; Tajiri, A.; Shoji, T.; Satake, H.; Morita, N. Synthesis, Stabilities, and Redox Behavior of Mono-, Di-, and Tetracations Composed of Di(1-azulenyl)methylium Units Connected to a Benzene Ring by Phenyl- and 2-Thienylacetylene Spacers. A Concept of a Cyanine−Cyanine Hybrid as a Stabilized Electrochromic System. J. Org. Chem. 2007, 72, 162–172. [Google Scholar]
- Shoji, T.; Ito, S.; Toyota, K.; Yasunami, M.; Morita, N. Synthesis, Properties, and Redox Behavior of Mono-, Bis-, and Tris[1,1,4,4,-tetracyano-2-(1-azulenyl)-3-butadienyl] Chromophores Binding with Benzene and Thiophene Cores. Chem. Eur. J. 2008, 14, 8398–8408. [Google Scholar] [CrossRef]
- Shoji, T.; Ito, S.; Toyota, K.; Iwamoto, T.; Yasunami, M.; Morita, N. Reactions between 1-Ethynylazulenes and 7,7,8,8-Tetracyanoquinodimethane (TCNQ): Preparation, Properties, and Redox Behavior of Novel Azulene-Substituted Redox-Active Chromophores. Eur. J. Org. Chem. 2009, 2009, 4316–4324. [Google Scholar] [CrossRef]
- Shoji, T.; Ito, S.; Okujima, T.; Morita, N. Synthesis of 2-Azulenyl-1,1,4,4-tetracyano-3-ferrocenyl-1,3-butadienes by [2+2] Cycloaddition of (Ferrocenylethynyl)azulenes with Tetracyanoethylene. Chem. Eur. J. 2013, 19, 5721–5730. [Google Scholar] [CrossRef]
- Shoji, T.; Maruyama, M.; Shimomura, E.; Maruyama, A.; Ito, S.; Okujima, T.; Toyota, K.; Morita, N. Synthesis, Properties, and Redox Behavior of Tetracyanobutadiene and Dicyanoquinodimethane Chromophores Bearing Two Azulenyl Substituents. J. Org. Chem. 2013, 78, 12513–12524. [Google Scholar] [CrossRef]
- Shoji, T.; Maruyama, M.; Maruyama, A.; Ito, S.; Okujima, T.; Toyota, K. Synthesis of 1,3-bis(tetracyano-2-azulenyl-3-butadienyl)azulenes by the [2+2] cycloaddition-retroelectrocyclization of 1,3-bis(azulenylethynyl)azulenes with tetracyanoethylene. Chem. Eur. J. 2014, 20, 11903–11912. [Google Scholar] [CrossRef]
- Shoji, T.; Ito, S. Azulene-Based Donor–Acceptor Systems: Synthesis, Optical, and Electrochemical Properties. Chem. Eur. J. 2017, 23, 16696–16709. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Morita, N.; Asao, T. Synthesis, Properties, and Redox Behaviors of Di- and Trications Composed of Di(1-azulenyl)methylium Units Connected by p- and m-Phenylene and 1,3,5-Benzenetriyl Spacers. Bull. Chem. Soc. Jpn. 2000, 73, 1865–1874. [Google Scholar] [CrossRef]
- Shoji, T.; Higashi, J.; Ito, S.; Toyota, K.; Yasunami, M.; Fujimori, K.; Asao, T.; Morita, N. Synthesis and Redox Behavior of 1-Azulenyl Sulfides and Efficient Synthesis of 1,1’-Biazulenes. Eur. J. Org. Chem. 2008, 2008, 1242–1252. [Google Scholar] [CrossRef]
- Shoji, T.; Maruyama, A.; Maruyama, M.; Ito, S.; Okujima, T.; Higashi, J.; Toyota, K.; Morita, N. Synthesis and Properties of 6-Methoxy- and 6-Dimethylamino-1-methylthio- and 1,3-Bis(methylthio)azulenes and Triflic Anhydride-Mediated Synthesis of Their Biaryl Derivatives. Synthesis and Properties of 6-Methoxy- and 6-Dimethylamino-1-methylthio- and 1,3-Bis(methylthio)azulenes and Triflic Anhydride-Mediated Synthesis of Their Biaryl Derivatives. Bull. Chem. Soc. Jpn. 2014, 87, 141–154. [Google Scholar]
- Shoji, T.; Ito, S.; Toyota, K.; Yasunami, M.; Morita, N. The novel transition metal free synthesis of 1,1’-biazulene. Tetrahedron Lett. 2007, 48, 4999–5002. [Google Scholar] [CrossRef]
- Higashi, J.; Shoji, T.; Ito, S.; Toyota, K.; Yasunami, M.; Morita, N. Heteroarylation of 1-Azulenyl Methyl Sulfide: Two-Step Synthetic Strategy for 1-Methylthio-3-(heteroaryl)azulenes Using the Triflate of N-Containing Heterocycles. Eur. J. Org. Chem. 2008, 2008, 5823–5831. [Google Scholar] [CrossRef]
- Shoji, T.; Ito, S.; Toyota, K.; Iwamoto, T.; Yasunami, M.; Morita, N. Synthesis and Redox Behavior of 1,3-Bis(methylthio-) and 1,3-Bis(phenylthio)azulenes Bearing 2- and 3-Thienyl Substituents by Palladium-Catalyzed Cross-Coupling Reaction of 2- and 6-Haloazulenes with Thienylmagnesium Ate Complexes. Eur. J. Org. Chem. 2009, 2009, 4307–4315. [Google Scholar] [CrossRef]
- Shoji, T.; Higashi, H.; Ito, S.; Morita, N. Synthesis, Properties, and Redox Behavior of Ferrocene-Substituted Bis(3-methylthio-1-azulenyl)methylium Ions. Eur. J. Inorg. Chem. 2010, 2010, 4886–4891. [Google Scholar] [CrossRef]
- Kirby, E.C.; Reid, D.H. Conjugated cyclic hydrocarbons and their heterocyclic analogues. Part II. The condensation of azulenes with homocyclic and heterocyclic aromatic aldehydes in the presence of perchloric acid. J. Chem. Soc. 1960, 494–501. [Google Scholar] [CrossRef]
- Kirby, E.C.; Reid, D.H. Conjugated cyclic hydrocarbons and their heterocyclic analogues. Part V. 1-1′-Azulenylmethyleneazulenium salts and 1-ethoxymethyl-eneazulenium salts. J. Chem. Soc. 1961, 1724–1730. [Google Scholar] [CrossRef]
- Kirby, E.C.; Reid, D.H. Conjugated cyclic hydrocarbons and their heterocyclic analogues. Part VI. The condensation of azulenes with aliphatic aldehydes in the presence of perchloric acid. J. Chem. Soc. 1961, 3579–3593. [Google Scholar] [CrossRef]
- Takekuma, S.-i.; Takekuma, H.; Matsubara, Y.; Hirai, A.; Yamamoto, H.; Nozoe, T. Reactions of Guaiazulene with Aldehyde Reagents in Acetic Acid—An Efficient Preparation of 3,3’-Methylenediguaiazulene Derivatives. Nippon Kagaku Kaishi 1996, 1996, 419–423. [Google Scholar] [CrossRef][Green Version]
- Takekuma, S.-i.; Takekuma, H.; Hanaoka, Y.; Yamamoto, H. Reactions of Azulene with Heteroaromatic Carbaldehydes in Acetic Acid. Nippon Kagaku Kaishi 1996, 1996, 659–662. [Google Scholar] [CrossRef][Green Version]
- Takekuma, S.-i.; Takekuma, H.; Hatanaka, Y.; Kawaguchi, J.; Yamamoto, H. Reactions of Guaiazulene with Phthalaldehyde, Isophthalaldehyde and Terephthalaldehyde in Acetic Acid. Nippon Kagaku Kaishi 1998, 1998, 275–279. [Google Scholar] [CrossRef]
- Takekuma, S.-i.; Sasaki, M.; Takekuma, H.; Yamamoto, H. Preparation and Characteristic Properties of 1,4-Bis(3-guaiazulenylmethylium)benzene Bishexafluorophosphate. Chem. Lett. 1999, 28, 999–1000. [Google Scholar] [CrossRef]
- Ito, S.; Morita, N.; Asao, T. Azulene analogues of triphenylmethyl cation; extremely stable hydrocarbon carbocations. Tetrahedron Lett. 1991, 32, 773–776. [Google Scholar] [CrossRef]
- Cowper, P.; Pockett, A.; Kociok-Köhn, G.; Cameron, P.J.; Lewis, S.E. Azulene-Thiophene-Cyanoacrylic acid dyes with donor-π-acceptor structures. Synthesis, characterisation and evaluation in dye-sensitized solar cells. Tetrahedron 2018, 74, 2775–2786. [Google Scholar] [CrossRef]
- Suppan, P.; Ghoneim, N. Solvatochromism, The Royal Society of Chemistry; Londres: Cambridge, UK, 1997. [Google Scholar]
- Suppan, P. Invited review solvatochromic shifts: The influence of the medium on the energy of electronic states. J. Photochem. Photobiol. A 1990, 50, 293–330. [Google Scholar] [CrossRef]
- Christian, R. Solvent and Solvent Effects in Organic Chemistry; Wiley-VCH: New York, NY, USA, 2004. [Google Scholar]
- Takekuma, S.-I.; Sasaki, M.; Takekuma, H.; Yamamoto, H. The isolation of quinoid compound was accomplished by Takekuma. An Efficient Preparation and Characteristic Properties of Tetramethyl 3,3′,3″,3‴-(p–Quinodimethane-7,7,8,8-tetrayltetraazulene-1,1′,1″,1‴-tetracarboxylate. Nippon Kagaku Kaishi 2000, 2000, 107–114. [Google Scholar] [CrossRef]
- Komatsu, K.; Ohta, K.; Fujimoto, T.; Yamamoto, I. Chromic materials. Part 1.-Liquid-crystalline behaviour and electrochromism in bis(octakis-n-alkylphthalocyaninato)lutetium(III) complexes. J. Mater. Chem. 1994, 4, 533–536. [Google Scholar] [CrossRef]
- Rosseinsky, D.R.; Monk, P.M.S. Studies of tetra-(bipyridilium) salts as possible polyelectrochromic materials. J. Appl. Electrochem. 1994, 24, 1213–1221. [Google Scholar] [CrossRef]
- Monk, P.M.S.; Mortimer, R.J.; Rosseinsky, D.R. Electrochromism and Electrochromic Devices; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Hünig, S.; Kemmer, M.; Wenner, H.; Perepichka, I.F.; Bäuerle, P.; Emge, A.; Gescheidt, G. Violene/Cyanine Hybrids: A General Structure for Electrochromic Systems. Chem. Eur. J. 1999, 5, 1969–1973. [Google Scholar] [CrossRef]
- Hünig, S.; Kemmer, M.; Wenner, H.; Barbosa, F.; Gescheidt, G.; Perepichka, I.F.; Bäuerle, P.; Emge, A.; Peters, K. Violene/Cyanine Hybrids as Electrochromics Part 2: Tetrakis(4-dimethylaminophenyl)ethene and Its Derivatives. Chem. Eur. J. 2000, 6, 2618–2632. [Google Scholar] [CrossRef] [PubMed]
- Hunig, S.; Perepichka, I.F.; Kemmer, M.; Wenner, H.; Bauerle, P.; Emge, A. Violene/cyanine hybrids as electrochromic systems. Part 3: Heterocyclic onium end groups. Tetrahedron 2000, 56, 4203–4211. [Google Scholar] [CrossRef]
- Hünig, S.; Briehn, C.A.; Bäuerle, P.; Emge, A. Electrochromics by Intramolecular Redox Switching of Single Bonds. Chem. Eur. J. 2001, 7, 2745–2757. [Google Scholar] [CrossRef]
- Hünig, S.; Langels, A.; Schmittel, M.; Wenner, H.; Perepichka, I.F.; Peters, K. Violene/Cyanine Hybrids as Electrochromic Systems: A New Variation of the General Structure. Eur. J. Org. Chem. 2001, 2001, 1393–1399. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).