
Neighboring Nitrogen Atom-Induced Reactions of Azidoacetyl Hydrazides, including Unexpected Nitrogen-Nitrogen Bond Cleavage of the Hydrazide


Abstract
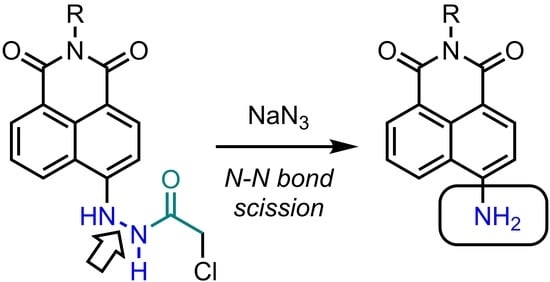
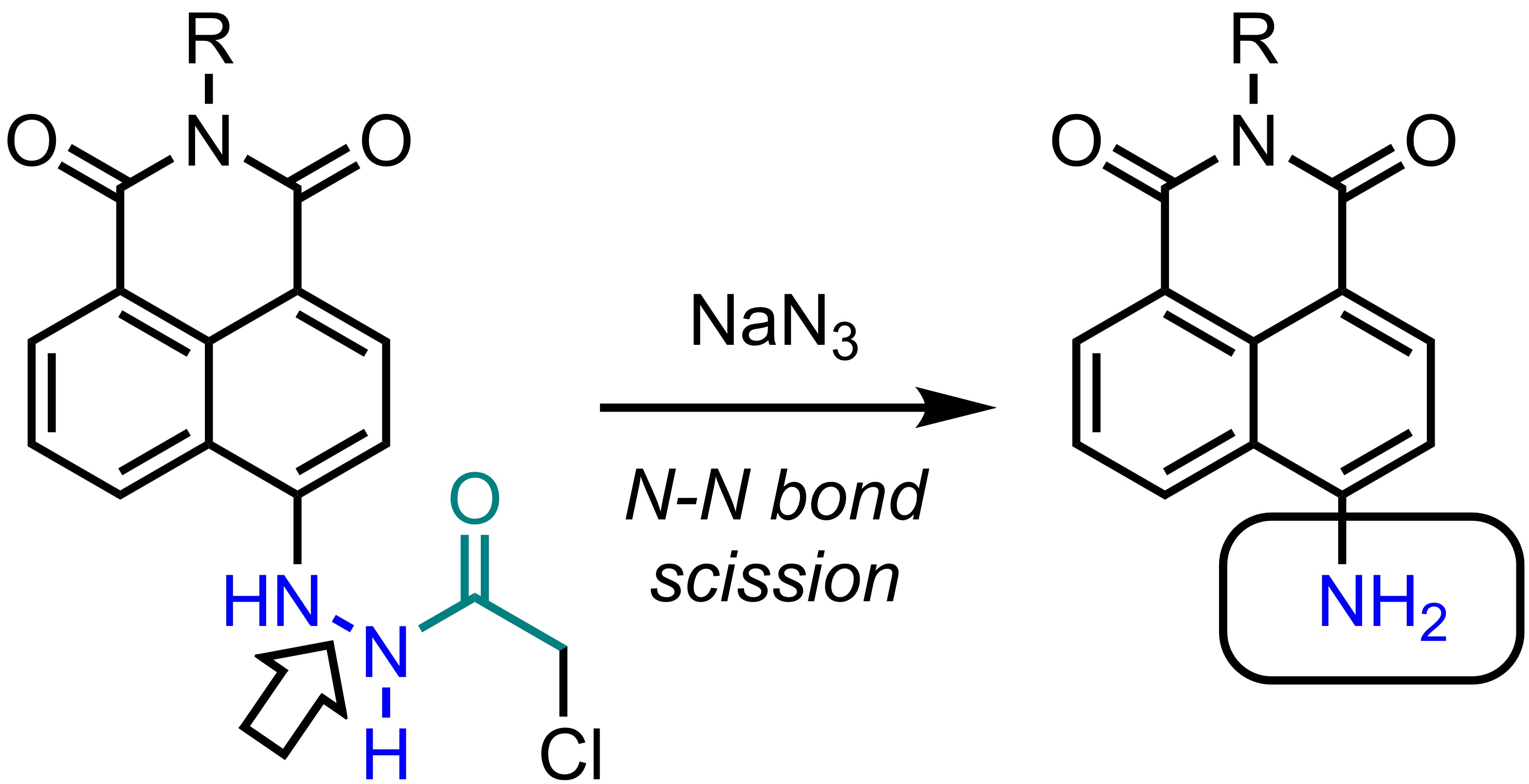{kind=link}
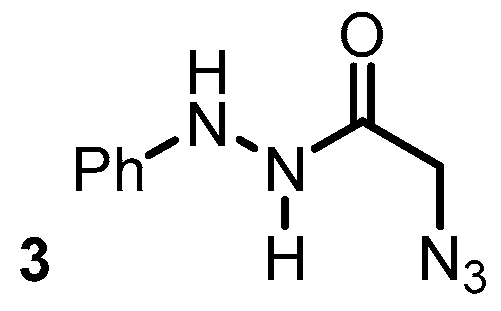
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
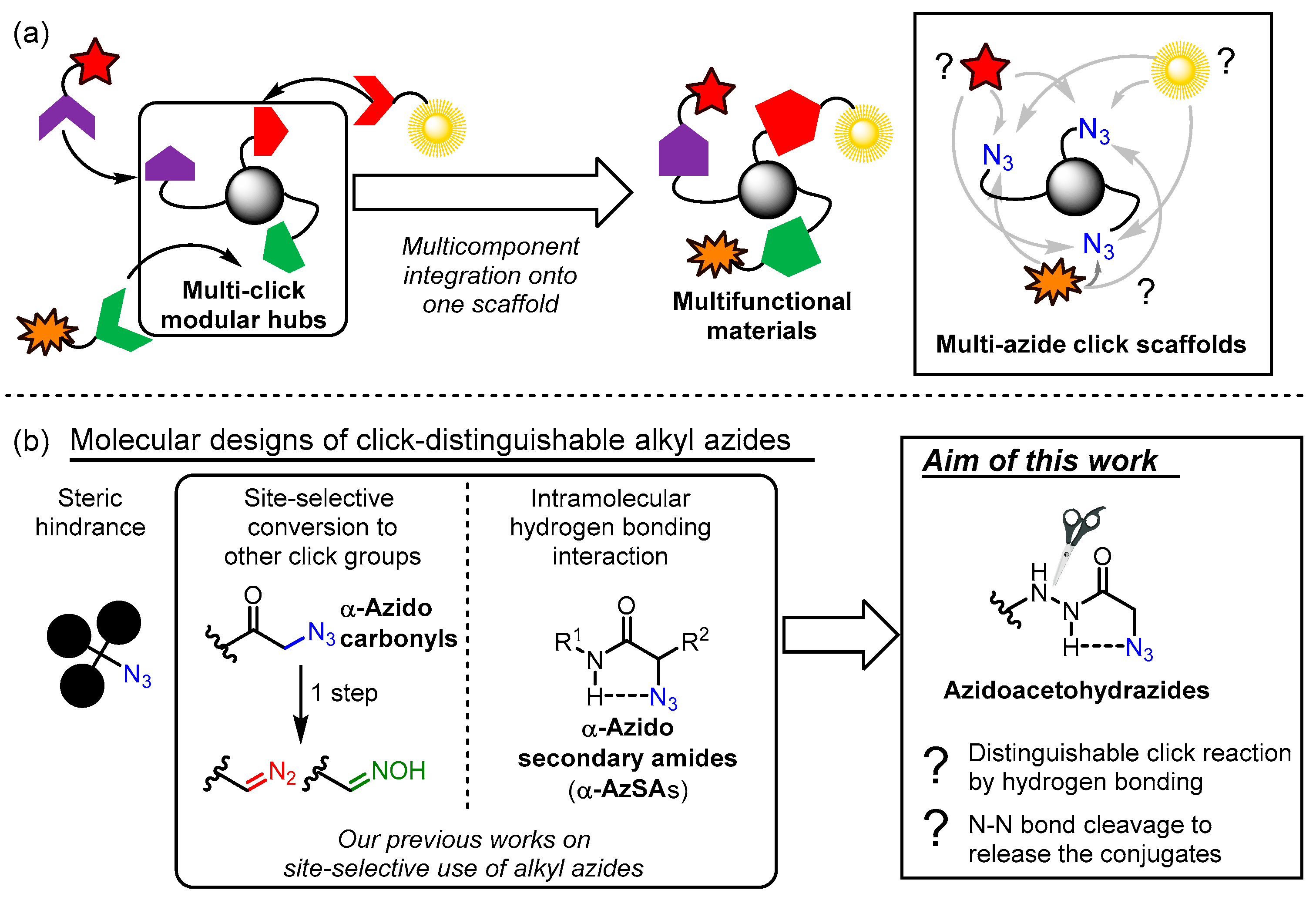
1. Introduction
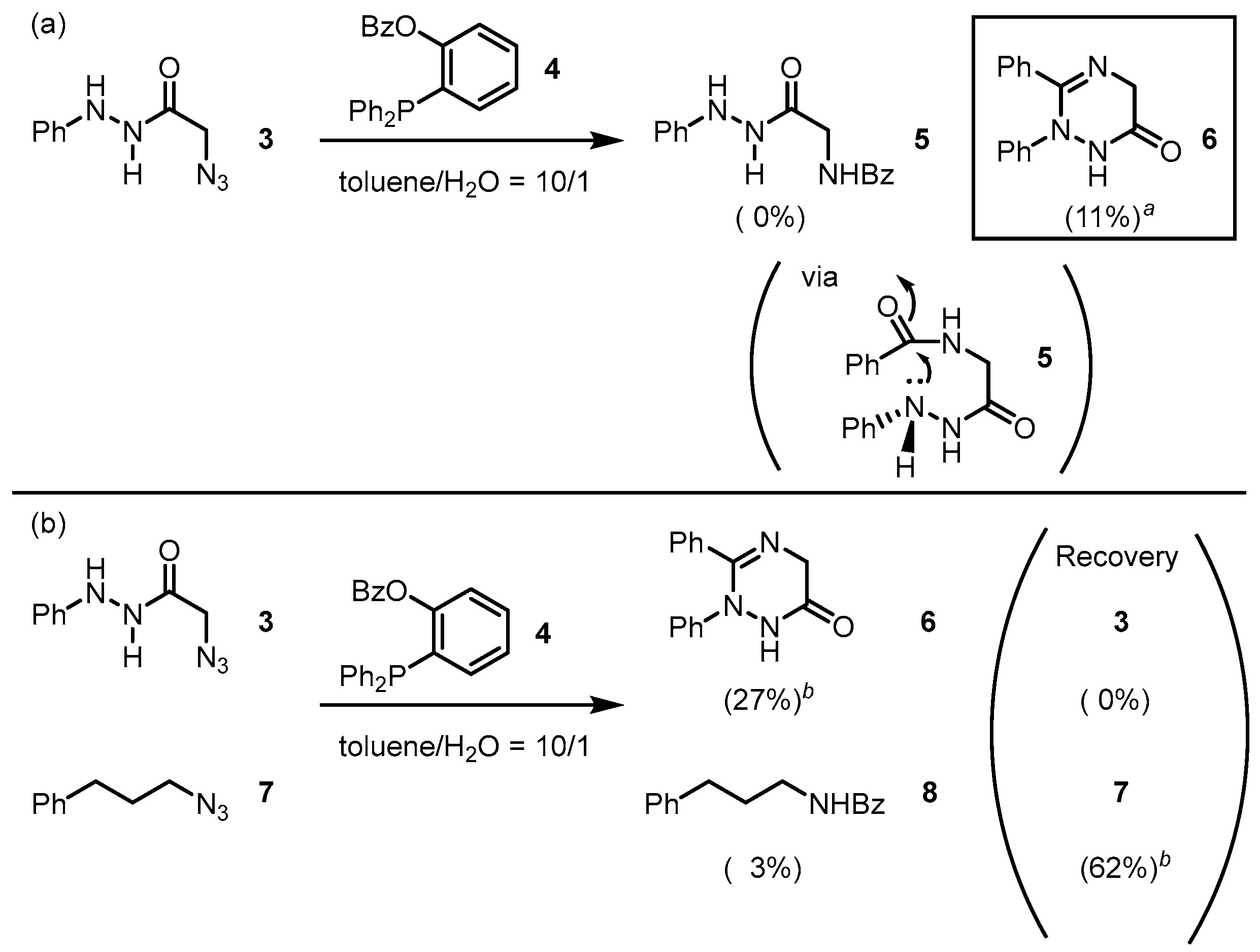
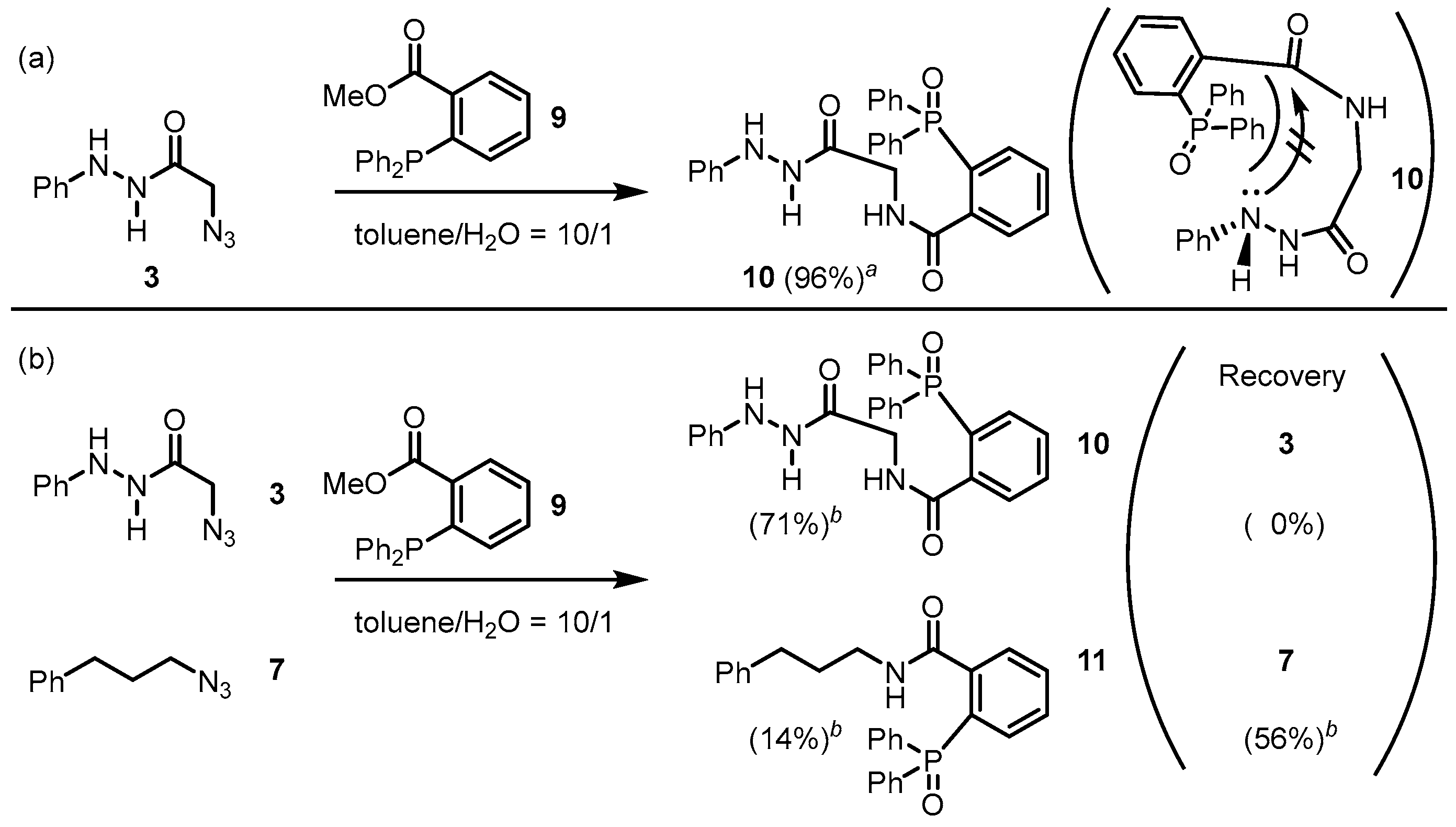
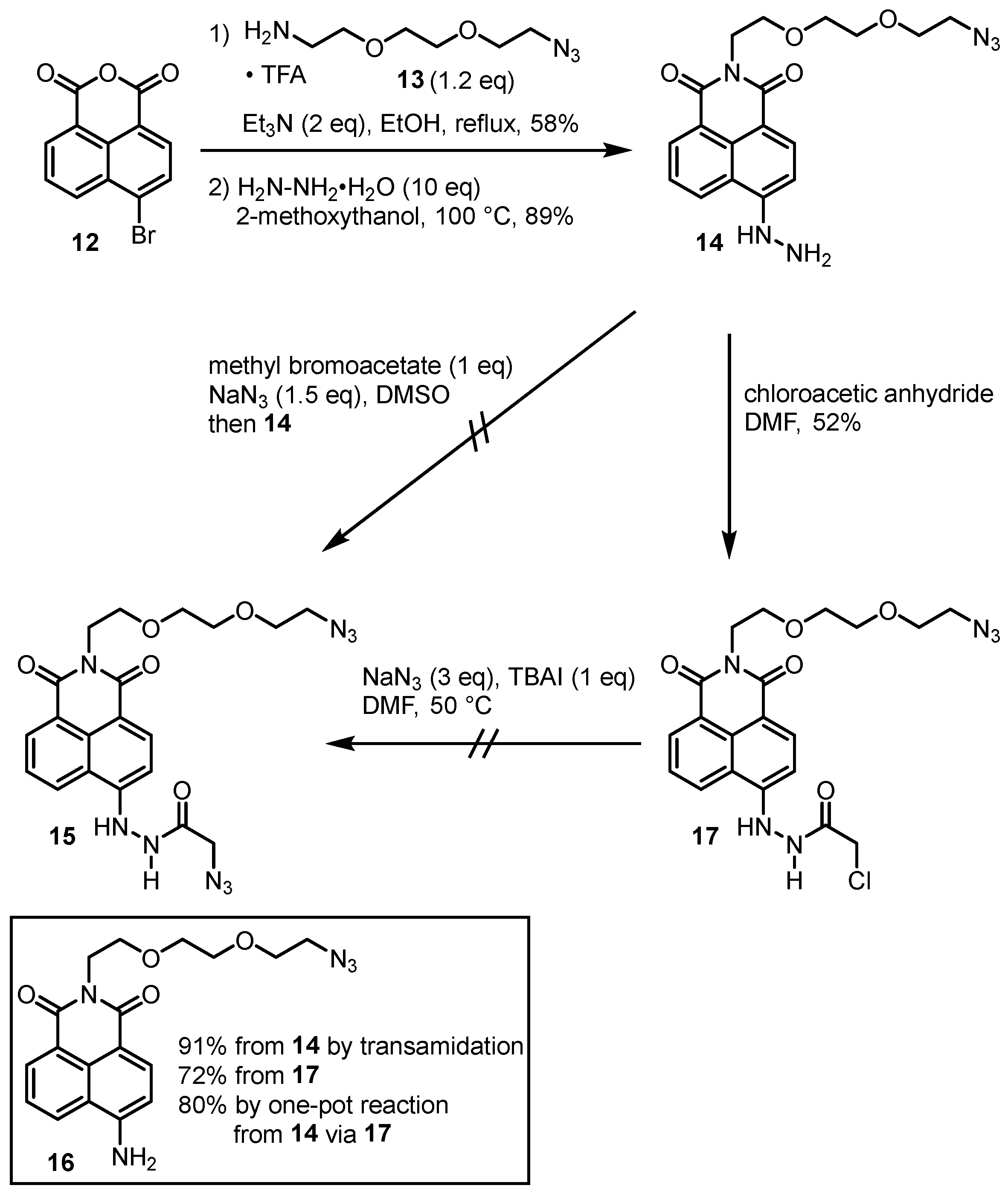
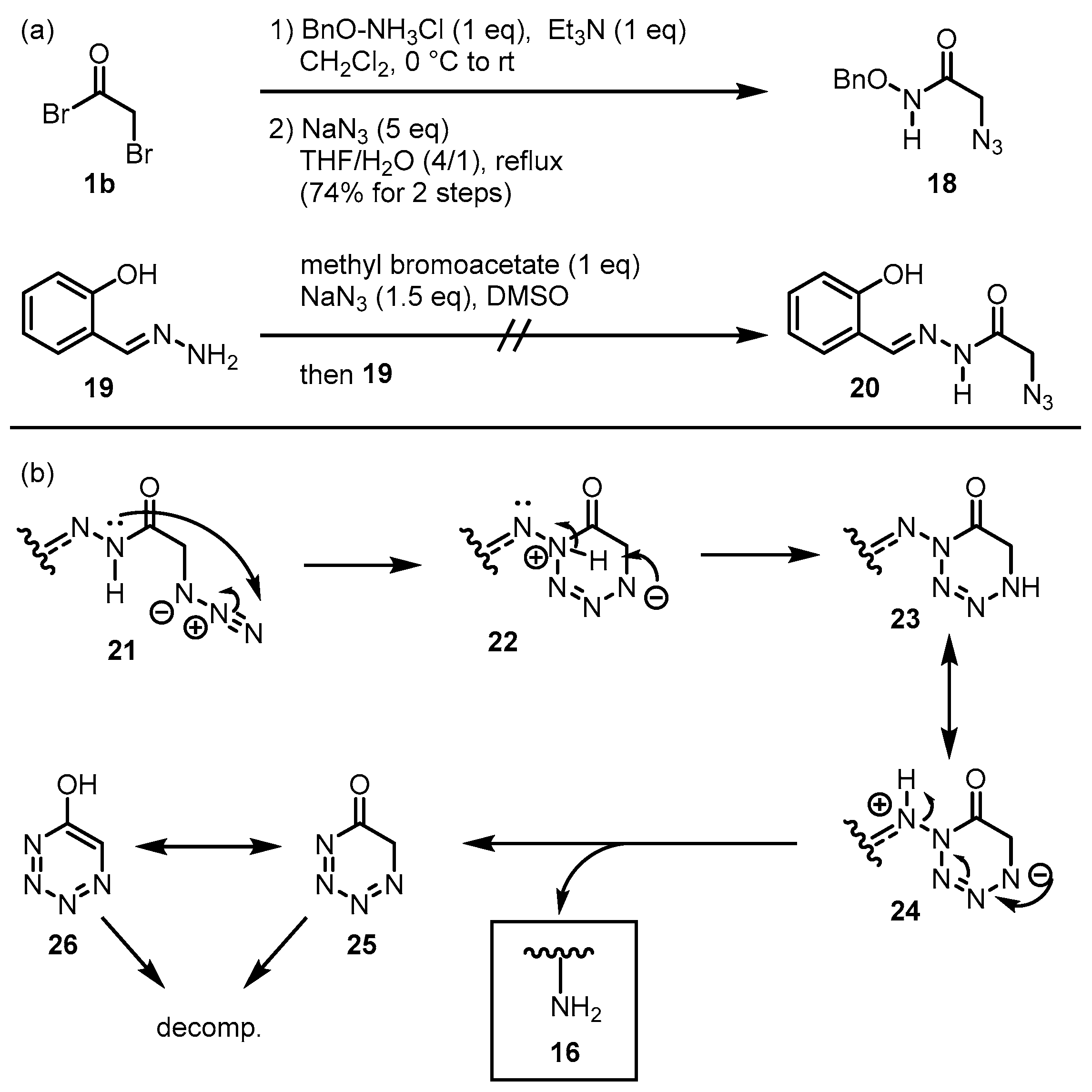
2. Results
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. General Information including Important Notices
5.2. Synthesis of Substrates
5.3. Competitive Staudinger and Traceless Staudinger Ligation Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Cu-Catalyzed Azide-Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Lahann, J. (Ed.) Click Chemistry for Biotechnology and Materials Science; John Wiley & Sons: West Sussex, UK, 2009. [Google Scholar]
- Schock, M.; Bräse, S. Reactive & Efficient: Organic Azides as Cross-Linkers in Material Sciences. Molecules 2020, 25, 1009. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S. Sequential conjugation methods based on triazole formations and related reactions using azides. Org. Biomol. Chem. 2020, 18, 1550–1562. [Google Scholar] [CrossRef]
- Sato, D.; Wu, Z.; Fujita, H.; Lindsey, J.S. Design, synthesis, and utility of defined molecular scaffolds. Organics 2021, 2, 161–273. [Google Scholar] [CrossRef]
- Bräse, S.; Banert, K. (Eds.) Organic Azides: Synthesis and Applications; John Wiley & Sons: West Sussex, UK, 2010. [Google Scholar]
- Tanimoto, H.; Kakiuchi, K. Recent Applications and Developments of Organic Azides in Total Synthesis of Natural Products. Nat. Prod. Commun. 2013, 8, 1021–1034. [Google Scholar] [CrossRef]
- Huang, G.; Yan, G. Recent Advances in Reactions of Azides. Adv. Synth. Catal. 2017, 359, 1600–1619. [Google Scholar] [CrossRef]
- Sala, R.; Loro, C.; Foschi, F.; Broggini, G. Transition Metal Catalyzed Azidation Reactions. Catalysts 2020, 10, 1173. [Google Scholar] [CrossRef]
- Yokoi, T.; Tanimoto, H.; Ueda, T.; Morimoto, T.; Kakiuchi, K. Site-selective conversion of azido groups at carbonyl α-positions to diazo groups in diazido and triazido compounds. J. Org. Chem. 2018, 83, 12103–12121. [Google Scholar] [CrossRef]
- Yokoi, T.; Ueda, T.; Tanimoto, H.; Morimoto, T.; Kakiuchi, K. Site-selective conversion of azido groups at carbonyl α-positions into oxime groups leading triazide to triple click conjugation scaffold. Chem. Commun. 2019, 55, 1891–1894. [Google Scholar] [CrossRef]
- Maegawa, K.; Tanimoto, H.; Onishi, S.; Tomohiro, T.; Morimoto, T.; Kakiuchi, K. Taming the reactivity of alkyl azides by intramolecular hydrogen bonding: Site-selective conjugation of unhindered diazides. Org. Chem. Front. 2021, 8, 5793–5803. [Google Scholar] [CrossRef]
- Kenny, P.W. Hydrogen-Bond Donors in Drug Design. J. Med. Chem. 2022, 65, 14261–14275. [Google Scholar] [CrossRef] [PubMed]
- Soloviev, D.O.; Hanna, F.E.; Misuraca, M.C.; Hunter, C.A. H-Bond Cooperativity: Polarisation Effects on Secondary Amides. Chem. Sci. 2022, 13, 11863. [Google Scholar] [CrossRef] [PubMed]
- Saxon, E.; Bertozzi, C.R. Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef]
- Saxon, E.; Luchansky, S.J.; Hang, H.C.; Yu, C.; Lee, S.C.; Bertozzi, C.R. Investigating Cellular Metabolism of Synthetic Azidosugars with the Staudinger Ligation. J. Am. Chem. Soc. 2002, 124, 14893–14902. [Google Scholar] [CrossRef]
- Liu, S.; Edgar, K.J. Staudinger Reactions for Selective Functionalization of Polysaccharides: A Review. Biomacromolecules 2015, 16, 2556–2571. [Google Scholar] [CrossRef]
- Bednarek, C.; Wehl, I.; Jung, N.; Schepers, U.; Bräse, S. The Staudinger Ligation. Chem. Rev. 2020, 120, 4301–4354. [Google Scholar] [CrossRef]
- Heiss, T.K.; Dorn, R.S.; Prescher, J.A. Bioorthogonal Reactions of Triarylphosphines and Related Analogues. Chem. Rev. 2021, 121, 6802–6849. [Google Scholar] [CrossRef]
- Zhang, H.; Tanimoto, H.; Morimoto, T.; Nishiyama, Y.; Kakiuchi, K. Regioselective Rapid Synthesis of Fully Substituted 1,2,3-Triazoles Mediated by Propargyl Cations. Org. Lett. 2013, 15, 5222–5225. [Google Scholar] [CrossRef]
- Zhang, H.; Tanimoto, H.; Morimoto, T.; Nishiyama, Y.; Kakiuchi, K. Acid-mediated synthesis of fully substituted 1, 2, 3-triazoles: Multicomponent coupling reactions, mechanistic study, synthesis of serine hydrolase inhibitor and its derivatives. Tetrahedron 2014, 70, 9828–9835. [Google Scholar] [CrossRef]
- Fernández, R.; Ferrete, A.; Llera, J.M.; Magriz, A.; Martín-Zamora, E.; Díez, E.; Lassaletta, J.M. A Practical Oxidative Method for the Cleavage of Hydrazide N-N Bonds. Chem. Eur. J. 2004, 10, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zheng, N. Photoinduced Cleavage of N-N Bonds of Aromatic Hydrazines and Hydrazides by Visible Light. Synthesis 2011, 2011, 2223–2236. [Google Scholar] [CrossRef] [PubMed]
- Hotta, Y.; Kaneko, T.; Hayashi, R.; Yamamoto, A.; Morimoto, S.; Chiba, J.; Tomohiro, T. Photoinduced Electron Transfer-Regulated Protein Labeling with a Coumarin-Based Multifunctional Photocrosslinker. Chem. Asian J. 2019, 14, 398–402. [Google Scholar] [CrossRef]
- Saxon, E.; Armstrong, J.I.; Bertozzi, C.R. A “Traceless” Staudinger Ligation for the Chemoselective Synthesis of Amide Bonds. Org. Lett. 2000, 2, 2141–2143. [Google Scholar] [CrossRef] [PubMed]
- Soellner, M.B.; Nilsson, B.L.; Raines, R.T. Reaction Mechanism and Kinetics of the Traceless Staudinger Ligation. J. Am. Chem. Soc. 2006, 128, 8820–8828. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-P.A.; Tiana, C.-L.; Zheng, J.-S. The Recent Developments and Applications of the Traceless-Staudinger Reaction in Chemical Biology Study. RSC Adv. 2015, 5, 107192–107199. [Google Scholar] [CrossRef]
- Jemas, A.; Xie, Y.; Pigga, J.E.; Caplan, J.L.; am Ende, C.W.; Fox, J.M. Catalytic Activation of Bioorthogonal Chemistry with Light (CABL) Enables Rapid, Spatiotemporally Controlled Labeling and No-Wash, Subcellular 3D-Patterning in Live Cells Using Long Wavelength Light. J. Am. Chem. Soc. 2022, 144, 1647–1662. [Google Scholar] [CrossRef]
- Nigst, T.A.; Antipova, A.; Mayr, H. Nucleophilic Reactivities of Hydrazines and Amines: The Futile Search for the α-Effect in Hydrazine Reactivities. J. Org. Chem. 2012, 77, 8142–8155. [Google Scholar] [CrossRef]
- Cho, H.-J.; Um, I.-H. The α-Effect in SNAr Reaction of 1-Fluoro-2,4-dinitrobenzene with Hydrazine: Ground-State Destabilization versus Transition-State Stabilization. Bull. Korean Chem. Soc. 2014, 35, 2371–2374. [Google Scholar] [CrossRef]
- Betancourth, J.G.; Castaño, J.A.; Visbal, R.; Chaur, M.N. Versatility of the Amino Group in Hydrazone-based Molecular and Supramolecular Systems. Eur. J. Org. Chem. 2022, 2022, e202200228. [Google Scholar] [CrossRef]
- Nakajima, M.; Oda, Y.; Wada, T.; Minamikawa, R.; Shirokane, K.; Sato, T.; Chida, N. Chemoselective Reductive Nucleophilic Addition to Tertiary Amides, Secondary Amides, and N-Methoxyamides. Chem. Eur. J. 2014, 20, 17565–17571. [Google Scholar] [CrossRef] [PubMed]
- Bouzayani, N.; Kraïem, J.; Marque, S.; Kacem, Y.; Carlin-Sinclair, A.; Marrot, J.; Hassine, B.B. Green synthesis of new chiral 1-(arylamino)imidazo[2,1-a]isoindole-2,5-diones from the corresponding α-amino acid arylhydrazides in aqueous medium. Beilstein J. Org. Chem. 2018, 14, 2923–2930. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, K.; Li, P.; Yao, H.; Lin, A. [3 + 3] Cycloaddition of Azides with in Situ Formed Azaoxyallyl Cations To Synthesize 1,2,3,4-Tetrazines. Org. Lett. 2018, 20, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Motiwala, H.F.; Armaly, A.M.; Cacioppo, J.G.; Coombs, T.C.; Koehn, K.R.K.; Norwood, V.M., IV; Aubé, J. HFIP in Organic Synthesis. Chem. Rev. 2022, 122, 12544–12747. [Google Scholar] [CrossRef]
- Colomer, I.; Chamberlain, A.E.R.; Haughey, M.B.; Donohoe, T.J. Hexafluoroisopropanol as a highly versatile solvent. Nat. Rev. Chem. 2017, 1, 0088. [Google Scholar] [CrossRef]
- Aubé, J.; Fehl, C.; Liu, R.; McLeod, M.C.; Motiwala, H.F. Hofmann, Curtius, Schmidt, Lossen, and Related Reactions. In Comprehensive Organic Synthesis, 2nd ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2014; Volume 6, pp. 598–635. [Google Scholar]
- Ardiansah, B.; Tanimoto, H.; Tomohiro, T.; Morimoto, T.; Kakiuchi, K. Sulfonium Ion-Promoted Traceless Schmidt Reaction of Alkyl Azides. Chem. Commun. 2021, 57, 8738–8741. [Google Scholar] [CrossRef] [PubMed]
- Conrow, R.E.; Dean, W.D. Diazidomethane Explosion. Org. Proc. Res. Dev. 2008, 12, 1285–1286. [Google Scholar] [CrossRef]
- Treitler, D.S.; Leung, S. How Dangerous Is Too Dangerous? A Perspective on Azide Chemistry. J. Org. Chem. 2022, 87, 11293–11295. [Google Scholar] [CrossRef]
- Farias, R.R.; Mascarenhas, A.J.S.; Santos, T.J.; Victor, M.M. Are diazides really dangerous compounds under ordinary conditions? Tetrahedron Lett. 2020, 61, 152574. [Google Scholar] [CrossRef]
- Shimokawa, K.; Yamada, K.; Ohno, O.; Oba, Y.; Uemura, D. Design, synthesis, and biological evaluation of biotin-labeled (−)-ternatin, a potent fat-accumulation inhibitor against 3T3-L1 adipocytes. Bioorg. Med. Chem. Lett. 2009, 19, 92–95. [Google Scholar] [CrossRef]
- Barz, M.; Duro-Castano, A.; Vicent, M.J. A versatile post-polymerization modification method for polyglutamic acid: Synthesis of orthogonal reactive polyglutamates and their use in “click chemistry”. Polym. Chem. 2013, 4, 2989–2994. [Google Scholar] [CrossRef]
- Fray, M.; Mathiron, D.; Pilard, S.; Lesur, D.; Abidi, R.; Barhoumi-Slimi, T.; Cragg, P.J.; Benazza, M. Heteroglycoclusters through Unprecedented Orthogonal Chemistry Based on N-Alkylation of N-Acylhydrazone. Eur. J. Org. Chem. 2022, 2022, e202101537. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, J.; Li, J.; Yuan, S.; Li, D. An unprecedented cobalt-catalyzed selective aroylation of primary amines with aroyl peroxides. Tetrahedron Lett. 2020, 61, 152399. [Google Scholar] [CrossRef]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanimoto, H.; Adachi, R.; Otsuki, A.; Tomohiro, T. Neighboring Nitrogen Atom-Induced Reactions of Azidoacetyl Hydrazides, including Unexpected Nitrogen-Nitrogen Bond Cleavage of the Hydrazide. Organics 2022, 3, 520-533. https://doi.org/10.3390/org3040035
Tanimoto H, Adachi R, Otsuki A, Tomohiro T. Neighboring Nitrogen Atom-Induced Reactions of Azidoacetyl Hydrazides, including Unexpected Nitrogen-Nitrogen Bond Cleavage of the Hydrazide. Organics. 2022; 3(4):520-533. https://doi.org/10.3390/org3040035
Chicago/Turabian StyleTanimoto, Hiroki, Ryo Adachi, Aoi Otsuki, and Takenori Tomohiro. 2022. "Neighboring Nitrogen Atom-Induced Reactions of Azidoacetyl Hydrazides, including Unexpected Nitrogen-Nitrogen Bond Cleavage of the Hydrazide" Organics 3, no. 4: 520-533. https://doi.org/10.3390/org3040035
APA StyleTanimoto, H., Adachi, R., Otsuki, A., & Tomohiro, T. (2022). Neighboring Nitrogen Atom-Induced Reactions of Azidoacetyl Hydrazides, including Unexpected Nitrogen-Nitrogen Bond Cleavage of the Hydrazide. Organics, 3(4), 520-533. https://doi.org/10.3390/org3040035