1. Introduction
4-Formyl-1,2,3-triazoles, also known as 1,2,3-triazole-4-carbaldehydes, are widely present in the literature, mainly due to the synthetic versatility of the formyl group [
1,
2,
3,
4,
5,
6,
7]. In the field of medicinal chemistry [
7], triazole-4-carbaldehydes have been prepared as intermediates in the synthesis of anticancer [
8,
9,
10], antifungal [
11,
12], antituberculosis [
13,
14,
15], anti-inflammatory [
10,
16], antidiabetic [
17], and even bioimaging agents [
18]. Furthermore, imine derivatives have been synthesized as interesting ligands in coordination chemistry [
19,
20,
21]. 1-Substituted 4-formyl-1,2,3-triazoles are commonly prepared via a two-step synthesis involving the copper-catalyzed azide-alkyne cycloaddition reaction with azides and propargyl alcohol [
2,
9,
10,
11,
12,
13,
15,
16,
17,
18,
22] or acetal-protected propargyl aldehydes [
3,
5,
6,
8,
23,
24], followed by oxidation or hydrolysis, respectively. These methods are clever alternatives for the cycloaddition between azides and the low boiling propynal, which is inconvenient to handle. Commonly used oxidants include manganese dioxide [
11,
12,
18], 2-iodoxybenzoic acid (IBX) [
13,
15,
17], and hexavalent chromium compounds [
2,
9,
10,
16,
22]. Despite their wide application, a few drawbacks of the copper-catalyzed click reactions can be named. Firstly, the scope is limited to compounds that tolerate strong oxidative or acidic conditions. Secondly, the use of the copper catalyst and the need for different azido compounds for the synthesis of analogues can be seen as unfavorable features.
Throughout the past decade, methods have been reported that circumvent the isolation of different azides, strong oxidative or acidic conditions, or the use of a metal catalyst. Fletcher et al. showed that the substitution of alkyl bromides with sodium azide was feasible under ‘click’ conditions in a
tBuOH/water mixture [
24], and the alkyl azides were reacted in situ with propargylaldehyde diethyl acetal. Interestingly, the complete hydrolysis of the acetal function on the resulting triazoles was observed at 70 °C without the use of acid [
24,
25]. Yao et al. demonstrated the oxidative cycloaddition of benzyl azide to acrolein under copper catalysis in the presence of Hünig’s base and oxygen at 80 °C [
26]. A copper-free three-step method was exemplified by Li, Wang et al. who performed the regioselective cycloaddition of phenyl azide to methyl 3-methoxyacrylate, followed by a reduction and oxidation using DIBAL and PCC, respectively [
27]. Recently, Gao et al. disclosed a metal- and oxidant-free approach starting from
α-bromoacroleins and azides [
28]. This eliminative cycloaddition was carried out under mild conditions and had a wide substrate scope. However, the bromoacroleins are poorly available and need to be synthesized from acroleins and bromine at low temperature.
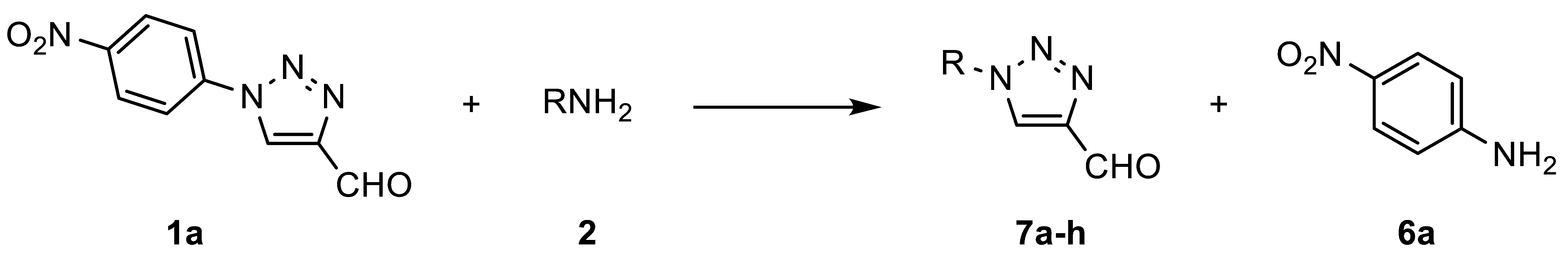
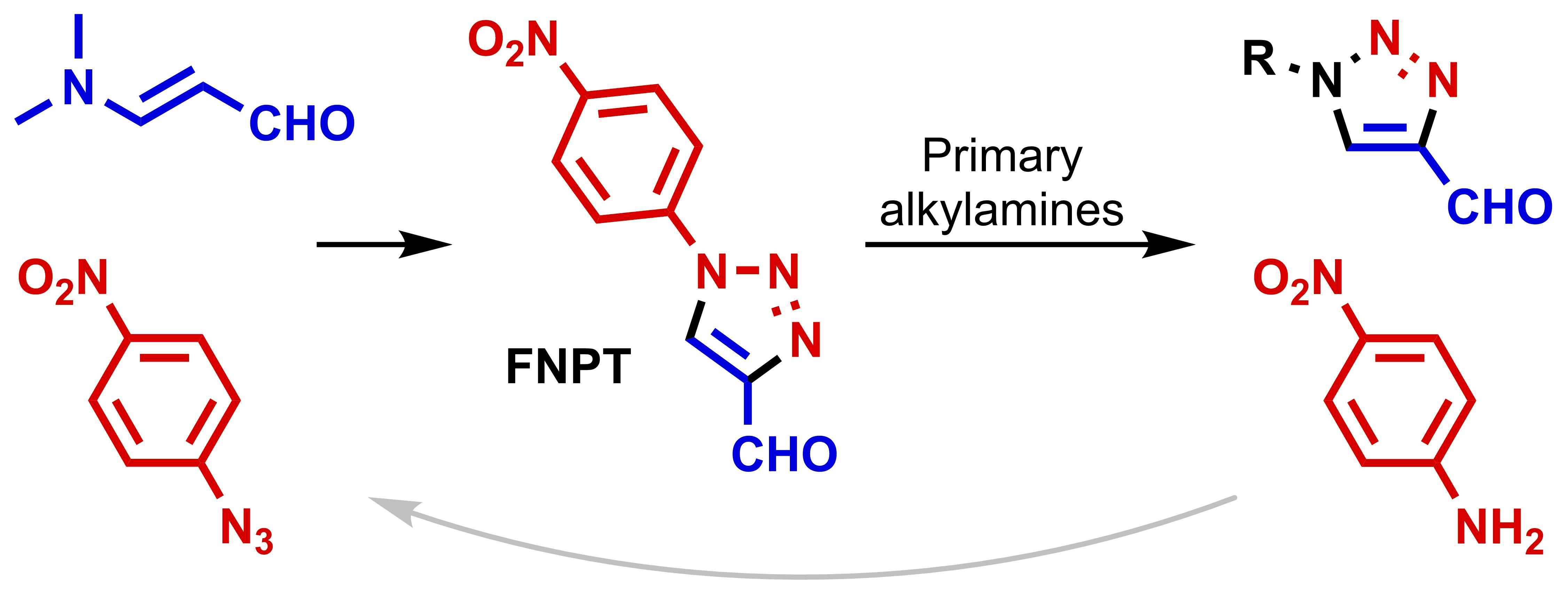
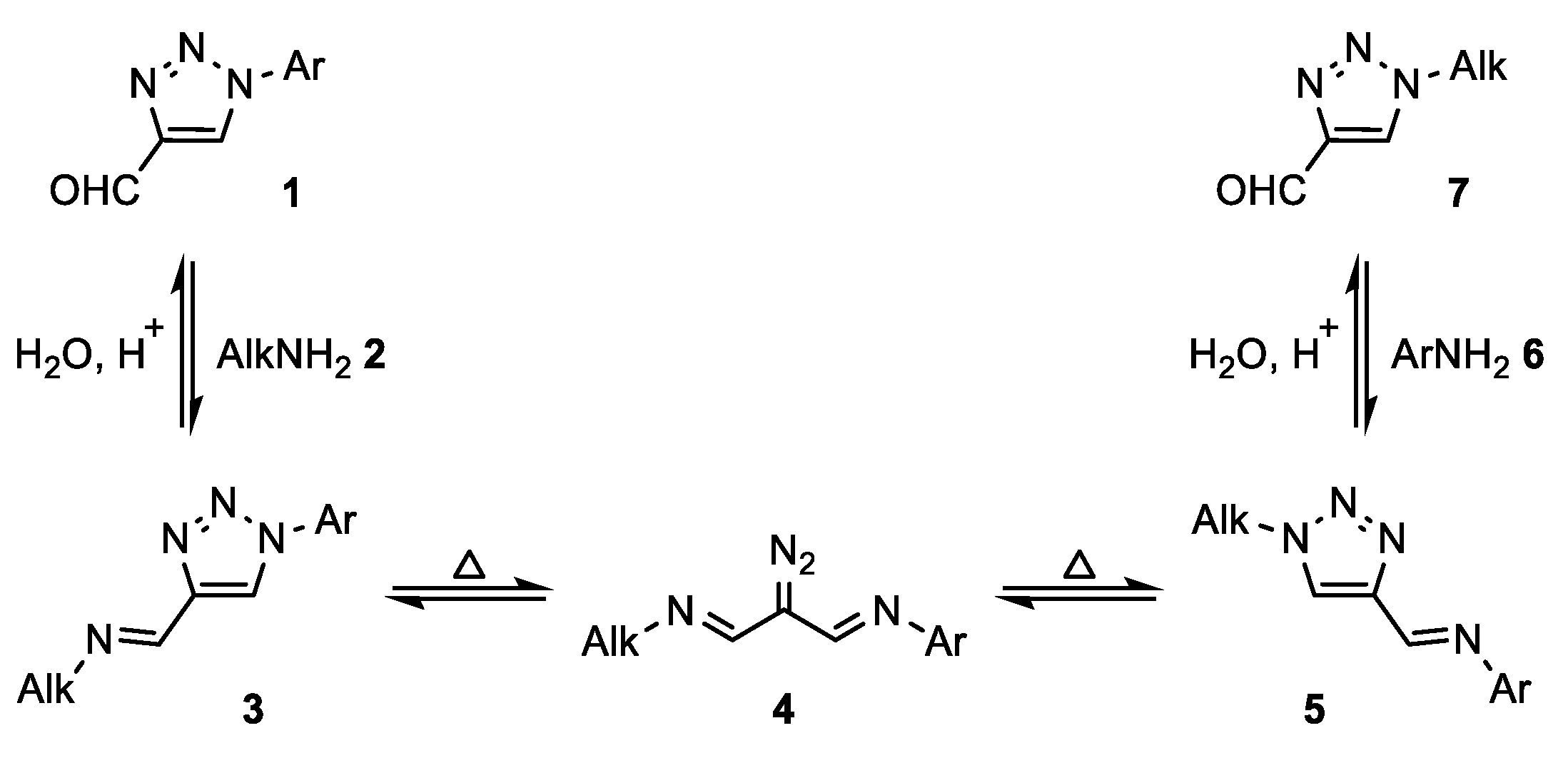
In 1989, L’abbé et al. studied the thermal rearrangement of 5-amino-4-iminomethyl-1-aryl-1,2,3-triazoles [
29]. Instead of Dimroth rearrangement towards 5-anilino-
NH-triazoles, the formation of 4-amidino-1,2,3-triazoles was observed as a result of the triazole-diazoimine equilibrium. This ring-degenerate rearrangement can be classified as a type of Cornforth rearrangement [
30] and was recently applied by our group for the synthesis of bitriazoles from α-ketotriazoles [
31]. L’abbé conceived the triazole–diazoimine equilibrium as a tool for the replacement of aryl by alkyl substituents in 1-substituted 4-formyl-1,2,3-triazoles
1 (
Scheme 1) [
32,
33]. The synthesis of 1-alkyl-1,2,3-triazoles
7 was carried out in three separate steps, namely imine formation, Cornforth rearrangement, and hydrolysis, and was introduced as a useful pathway to avoid the use of low-boiling explosive alkyl azides [
20,
21]. In addition, this method is particularly interesting when the primary amine is more available than its corresponding azide.
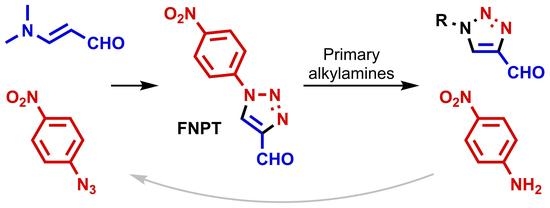
As described by L’abbé, the equilibrium of the Cornforth rearrangement is shifted toward the triazole with the most electron-deficient substituent on the imine nitrogen. Fletcher et al., who further studied this rearrangement in 2018, observed the same trend [
34]. The condensation of different 1-aryl-4-formyl-1,2,3-triazoles
1 and anilines gave dynamic mixtures with up to six different triazole compounds. In contrast, reactions with aniline and 4-formyl-1-(4-nitrophenyl)triazole
1a (FNPT;
Scheme 2) only gave rearranged products. As a consequence, FNPT
1a was put forward as a preferred reagent for the synthesis of 1-subsituted 4-formyltriazoles and can be seen as a safe alternative for diazomalonaldehyde [
14,
35]. Onoda et al. applied this strategy for the preparation of 1-alkyl-4-formyltriazoles
7 that could be used for the
N-terminal modification of proteins [
1]. Interestingly, a procedure was developed starting from FNPT
1a and primary amines that allowed for protein modification in excellent conversions without isolating the targeted aldehydes
7. Following our ongoing interest in the synthesis of 1,2,3-triazoles [
36,
37,
38], we recognized the potential of this approach for the metal-free synthesis of 1-alkyl-4-formyl-1,2,3-triazoles
7. However, to the best of our knowledge, a metal-free and straightforward synthesis of FNPT
1a on a preparative scale was still lacking.
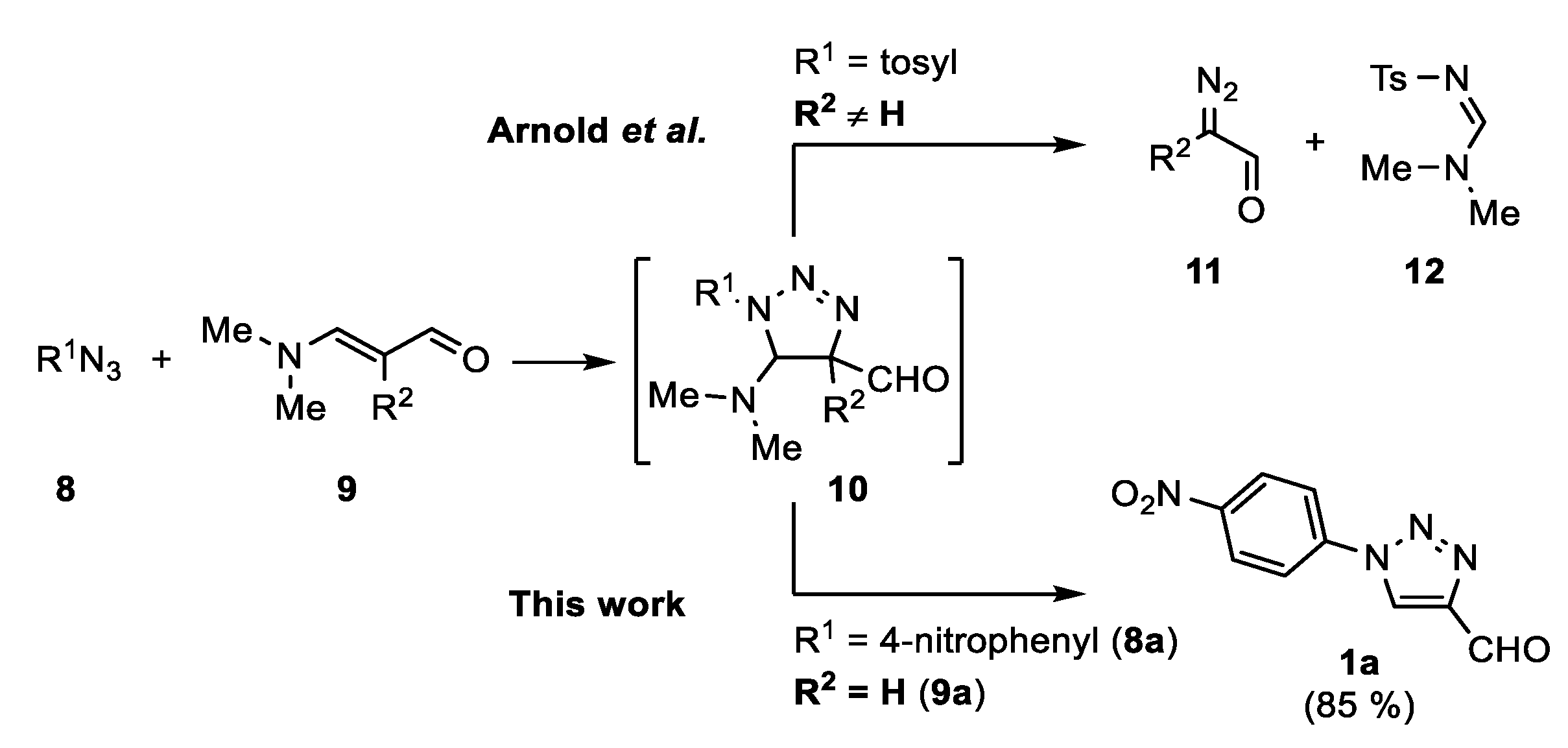
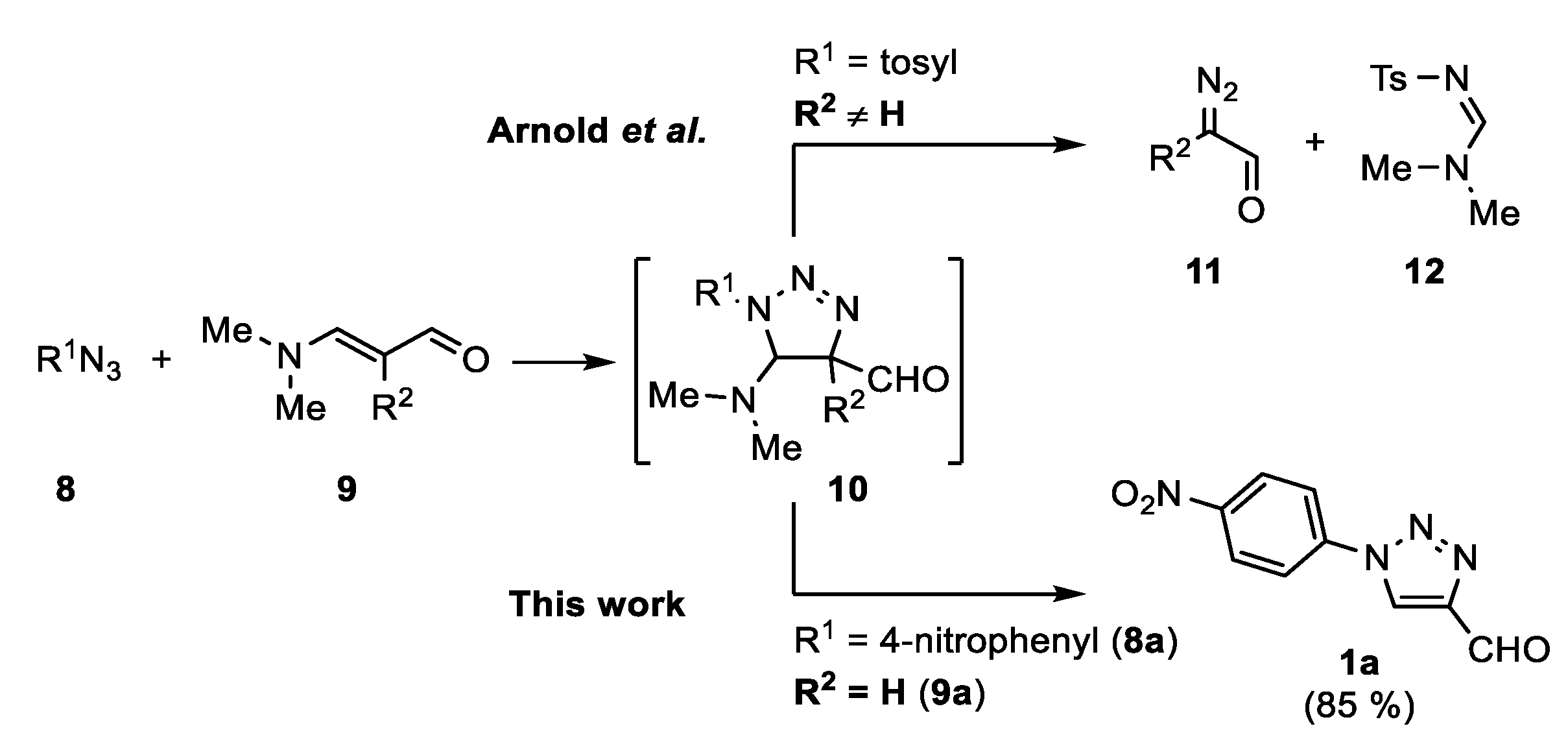
In 1970, Arnold et al. reported the synthesis of
α-diazoaldehydes
11 from 2-alkyl-3-dimethylaminoacroleins
9 (R
2 ≠ H) and tosyl azide (
Scheme 2) [
39]. This transformation involves a retro 1,3-dipolar cycloaddition of triazoline
10. When using 2-unsubstituted 3-dimethylaminoacroleins
9 (R
2 = H), however, only a trace amount of diazoacetalde hyde
11 was obtained. To the best of our knowledge, the reaction of aminoacroleins with azides was not further studied. As enamines are among the best known starting materials for the synthesis of 1,2,3-triazoles [
36,
37,
40,
41], it is rather surprising that the commercially available and rather inexpensive 3-dimethylaminoacrolein
9a somehow remained unnoticed as a possible reagent for the synthesis of 4-formyl-1,2,3-triazoles.
2. Results and Discussions
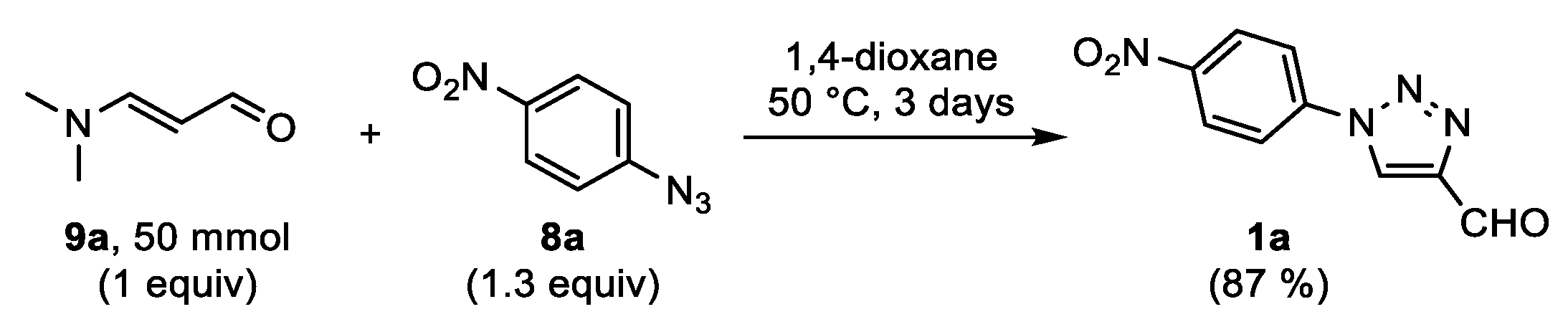
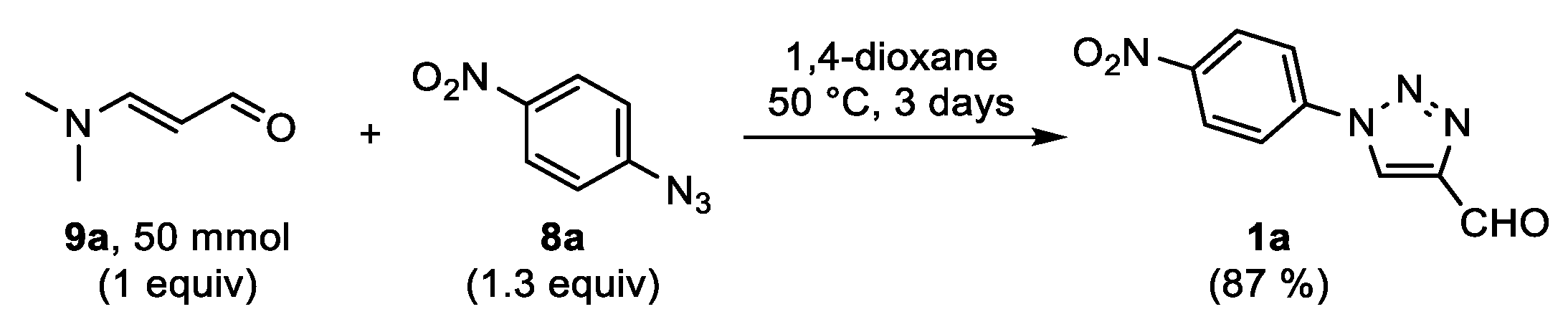
The first aim of this work was to develop a protocol for the metal-free synthesis of FNPT
1a on a preparative scale (
Scheme 3). The reaction between 3-dimethylaminoacrolein
9a and 4-nitrophenyl azide
8a was first evaluated on a submillimolar scale. To our delight, high yields above 80% of the desired 4-formyltriazole
1a were obtained from reactions in aprotic polar solvents (DMF, DMSO, 1,4-dioxane, MeCN) at 50 °C. Three days were needed in order to reach the desired conversion of the reagents in an equimolar ratio. In isopropanol, the cycloaddition was significantly slower, and protic solvents were therefore not further studied. DMSO and DMF were selected as the optimal solvents, affording reproducible yields of 86–88%. Our aim was to avoid chromatographic purification. Therefore, in initial experiments, the reaction mixtures were diluted with ethyl acetate, washed with water, and concentrated. The sufficiently pure product was obtained after precipitation from pentane. Unfortunately, this protocol failed for reactions on a gram scale in several ways. The yield decreased significantly, and a large amount of ethyl acetate was required in order to keep the product in solution. More importantly, the isolated product was less pure, as observed via
1H NMR analysis. Therefore, the conditions were re-evaluated on gram scale. An excess of the water soluble acrolein
9a (1.3 equiv) was used in order to attain full conversion. When using 1,4-dioxane as the reaction solvent, precipitation of the product by pouring the mixture in aqueous HCl could afford the crude product in circa 90% yield. Nevertheless, the desired purity was still not acquired. The reaction was also carried out under a constant nitrogen flow to remove the released dimethylamine. However, this had no influence on the reaction outcome. Additionally, attempts to grow crystals of the product failed. Eventually, it was found that the use of 1.3 equivalents of 4-nitrophenyl azide
8a gave a cleaner reaction mixture. The precipitation of the product
1a by adding equal portions of aqueous HCl and diethyl ether, filtration, and washing of the solid with water and diethyl ether gave a pure solid in an acceptable yield of 85% on gram scale. It should be noted that when washing the isolated solid with pentane, the resulting powder was statically charged and therefore difficult to handle. Finally, the procedure was repeated starting from 50 mmol of 3-dimethylaminoacrolein
9a, and the pure triazole
1a was isolated in a slightly lower but acceptable yield of 79% after filtration. It was assumed that a part of the product was taken up in the water layer as the hydrate, which was substantiated by the
1H NMR analysis of the acquired solid before the evaporation of the residual solvents under vacuo (see
Supplementary Materials, Figure S1). Nevertheless, we were pleased to see that further chromatographic purification of the ether phase of the filtrate gave another 0.94 g (9%) of the triazole
1a and allowed for the complete recovery of the excess of 4-nitrophenyl azide.
Although the cycloaddition of 4-nitrophenyl azide 8a to 3-dimethylaminoacrolein 9a was successful, the scope of azides was not further explored in this work. A preliminary experiment with phenyl azide showed that the cycloaddition was slow, even at 80 °C. Furthermore, increasing the temperature resulted in a crude product with an inferior purity profile compared to reactions at 50 °C. Based on this observation, it is expected that azides with electron-deficient aryl groups will be the most suitable substrates for the synthesis of 1-aryl-4-formyl-1,2,3-triazoles from 3-dimethylaminoacrolein.
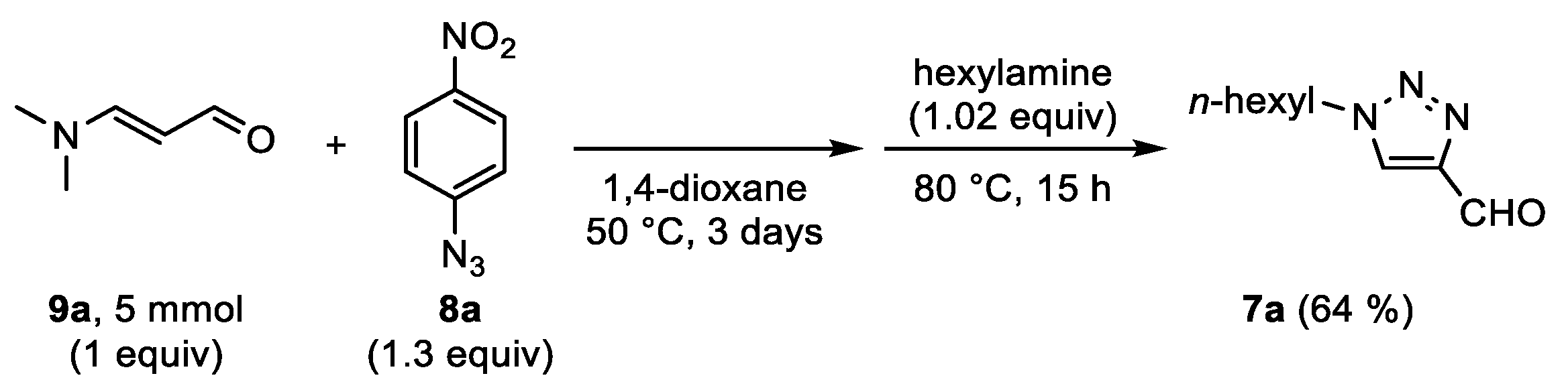
With a metal-free protocol for the multigram scale preparation of FNPT
1a in hand, we wanted to further explore the previously reported reaction with primary amines
2 [
1,
20,
32,
33,
34], i.e., imine formation, Cornforth rearrangement, and hydrolysis, in a single step (
Table 1). The study was initiated with hexylamine. Cornforth rearrangement of the in situ generated 4-(hexylimino)methyl-1,2,3-triazole occurred efficiently at 80 °C in solvents such as isopropanol (
iPrOH), 1,4-dioxane, and water. Full conversion toward the rearranged products was also achieved at 50 °C, although longer reaction times were required, i.e., in the order of 24 h in
iPrOH and 48 h in water. It is important to note that when water was used, a fine suspension of triazole
1a and the solubility of the amine in water were crucial for the reaction to run smoothly. Reactions with hexylamine were analyzed by means of
1H NMR after concentrating the crude mixtures under vacuo. For each solvent, a mixture of rearranged 4-iminomethyltriazoles and the desired 1-alkyl-4-formyltriazole
7a was obtained, with the latter as the major product. Since Onoda et al. observed the complete hydrolysis of rearranged iminomethyltriazoles after 30 min at 99 °C in DMSO-
d6 [
1], one could assume that the target aldehyde may partly recondense with the liberated 4-nitroaniline
6a when the mixtures were concentrated. Unfortunately, the complete removal of nitroaniline by means of acidic extractions with ethyl acetate or diethylether was unsuccessful and led to significant losses of product. Interestingly, L’abbé et al. reported that such imines hydrolyze during chromatographic work-up using silica [
42]. For those reasons, reactions mixtures with
iPrOH or dioxane were concentrated and immediately purified via column chromatography. To our delight, the pure triazole
7a was isolated in a 94% yield for both reaction solvents.
Next, other amines were evaluated (
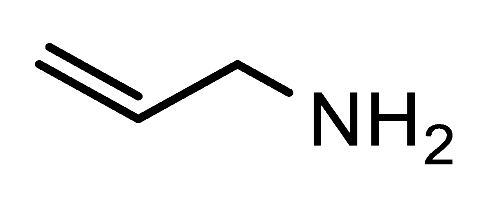
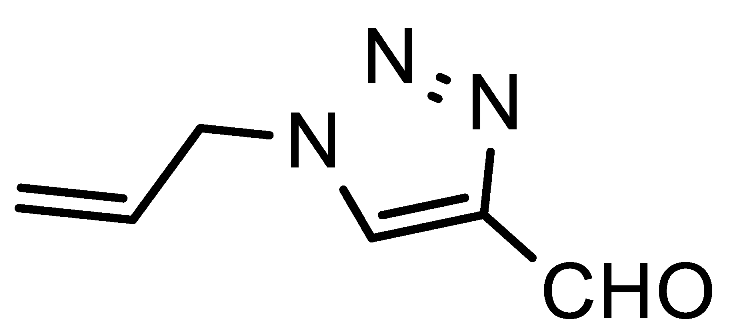
Table 1). Reactions with benzylamine, as well as the acid-sensitive allylamine and aminoacetaldehyde dimethyl acetal were successful and gave excellent yields of the respective formyltriazoles
7c,
7d, and
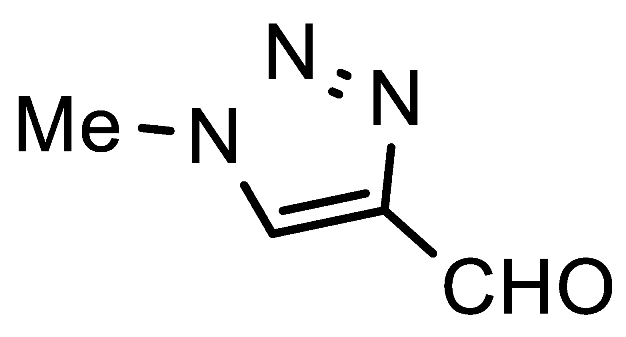
7f. 4-Formyl-1-methyl-1,2,3-triazole
7b was synthesized in 82% by using a commercial solution of methylamine in isopropanol. Due to its volatility under vacuum at elevated temperatures, sublimation in a Kugelrohr or cold finger equipment could be used to crystallize the product. The use of
tert-butyl glycinate resulted in lower but comparable yields of
7g in both
iPrOH (41%) and dioxane (46%). 1-Propargyl-4-formyltriazole
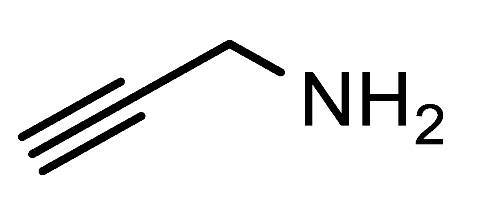
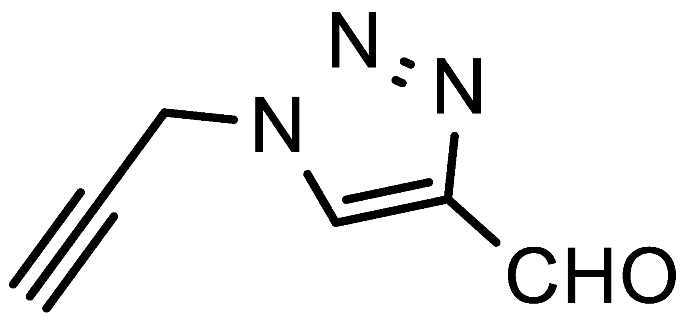
7e, which is currently not accessible by other means, was successfully prepared in a 65% yield from propargylamine in dioxane at 70 °C. Interestingly, tristriazole
7h could be synthesized in a 90% yield from tris(2-aminoethyl)amine by performing the reaction in a 1:3 mixture of isopropanol and water.
While suitable conditions were found for reactions with alkylamines, the reaction with aniline in 1,4-dioxane at 80 °C was slow. Moreover, hydrolysis of the resulting imine on silica failed, which hampered the isolation of 4-formyl-1-phenyl-1,2,3-triazole. We therefore believe that the protocol of Fletcher et al. [
34], among other synthetic methods, should be recommended for the preparation of 1-aryl-4-formyltriazoles.
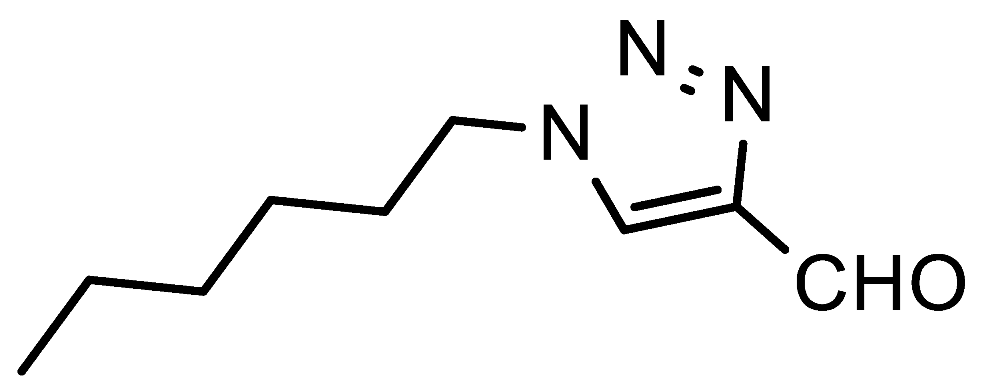
The reaction with hexylamine in isopropanol was then carried out on a multigram scale. Unfavorably, the hydrolysis of rearranged 4-iminomethyl-1,2,3-triazoles on silica was less efficient in this case, making the purification via column chromatography inconvenient. This issue could be largely resolved by removing a part of the nitroaniline
6a by means of an acidic extraction prior to the chromatographic purification. The product
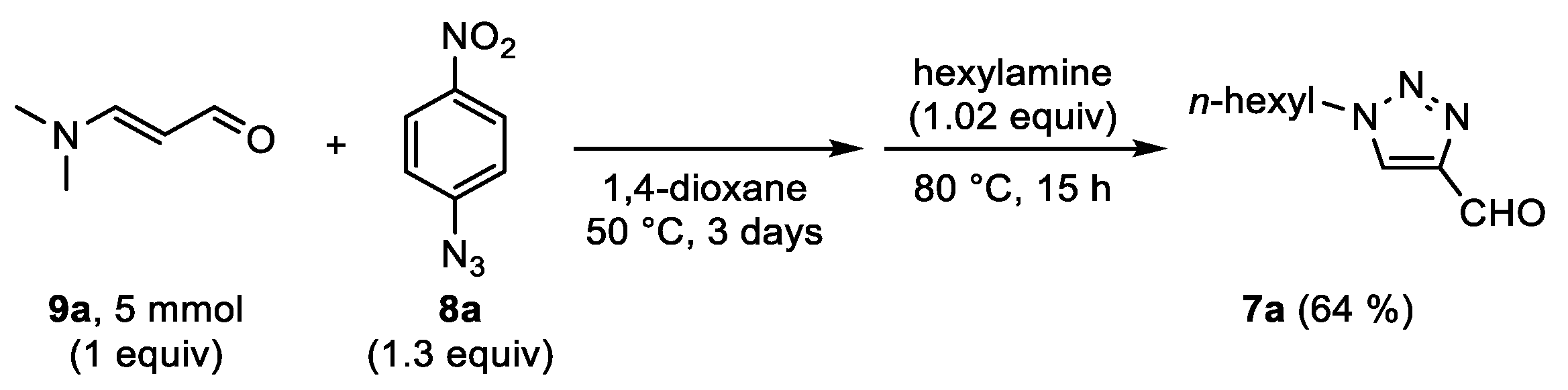
7a was eventually obtained in a yield of 73%. To further demonstrate the utility of this chemistry, 4-formyl-1-hexyl-1,2,3-triazole
7a was also successfully prepared via a two-step one-pot reaction, starting from 3-dimethylaminoacrolein
9a, 4-nitrophenyl azide
8a and hexylamine (
Scheme 4).
In a later stage of this work, we envisioned that the reactions in isopropanol or water could be part of a protocol for the direct regeneration of 4-nitrophenyl azide
8a. Our group earlier demonstrated the in situ diazotization of 4-nitroaniline
6a after a three-component triazole synthesis from ketones, primary amines, and 4-nitrophenyl azide [
43]. Although such a procedure would not be applicable to the acid-sensitive substrates, it could possibly enhance the hydrolysis of the rearranged iminomethyltriazoles, which would largely reduce the amount of solvent required for the chromatographic purification. After the reaction of FNPT
1a and hexylamine in isopropanol, 4-nitroaniline
6a was diazotized via the addition of isopropanol, aqueous HCl, and sodium nitrate. We were pleased to see that the imines were fully hydrolyzed under the diazotization conditions. In this way, the pure 4-formyl-1-hexyl-1,2,3-triazole
7a could be isolated in a 96% yield via a simple extraction on the scale of 0.5 mmol. For the gram scale synthesis, however, further purification of the crude product was required. While the formyltriazole
7a was isolated in a yield of 70%, nitrophenyl azide
8a was regenerated in 80% yield.
4. Materials and Methods
All chemicals were purchased from Acros Organics, Merck, Fluorochem, J&K Scientific, and TCI Europe, and used as received. 4-Nitrophenyl azide
8a was prepared on multigram scale according to a procedure previously reported in the literature [
45]. Thin layer chromatography (TLC) was performed using silica gel with fluorescent indicator 254 nm precoated on aluminum sheets (Merck). For column chromatography, 70–230 mesh silica 60 (Acros) was used as the stationary phase.
1H and
13C NMR spectra were recorded on a Bruker Avance 300, Bruker Avance III HD 400, or a Bruker Avance II
+ 600 spectrometer. Chemical shifts (δ) were reported in parts per million (ppm) referenced to tetramethylsilane (0.00 ppm) as an internal standard for samples in CDCl
3, or to the solvent signal for samples in DMSO-
d6 (2.50 ppm).
13C NMR spectra were referenced to the respective solvent signals (CDCl
3, 77.16 ppm; DMSO-
d6, 39.52 ppm). High-resolution mass spectra were acquired on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Milford, MA, USA). Samples were infused at 3 μL/min and spectra were obtained in positive ionization mode with a resolution of 15,000 (FWHM) using leucine enkephalin as lock mass. Melting points (not corrected) were determined using a Reichert Thermovar apparatus.
1-(4-Nitrophenyl)-1H-1,2,3-triazole-4-carbaldehyde(FNPT; 1a). To a screw-capped 40 mL reaction tube equipped with a magnetic stirring bar were added 4-nitrophenyl azide
8a (65 mmol, 10.7 g), 3-dimethylaminoacrolein
9a (50 mmol, 5 g), and 1,4-dioxane (25 mL). The mixture was stirred at 50 °C for 72 h, after which the solvent was reduced by half under reduced pressure. The reaction mixture was then diluted with 1 M HCl (aq., 50 mL) and Et
2O (50 mL), and the mixture was shaken in a separation funnel until a white suspension was obtained. The precipitate was filtered of and washed with water (3 × 50 mL) and Et
2O (4 x 50 mL). The solid was further dried in vacuo in order to obtain 8.58 g (79%) of the pure triazole
1a. The filtrate was transferred to a separation funnel and the organic layer was washed with 1 M HCl (aq., 120 mL), saturated NaHCO
3 (aq., 100 mL), and brine (100 mL). The ether phase was then dried over MgSO
4, filtered, and concentrated. The residue was purified via column chromatography using a petroleum-ether/DCM gradient (1:1) to recover the pure unreacted 4-nitrophenyl azide
8a (2.6 g), followed by a DCM/MeOH gradient (1:0–19:1) to collect another 0.94 g (9%) of triazole
1a. Off-white solid; yield: 9.52 g (87%); mp 173–174 °C;
1H NMR (300 MHz, DMSO-
d6) δ 10.12 (s, 1H), 9.75 (s, 1H), 8.45 (d,
J = 9.1 Hz, 2H), 8.28 (d,
J = 9.1 Hz, 2H);
13C NMR (75 MHz, DMSO) δ 184.9, 147.8, 147.3, 140.2, 126.8, 125.5, 121.4. NMR data was in accordance with the previously reported data for this compound [
1].
General procedurefor the synthesis of 1-alkyl-1H-1,2,3-triazole-4-carbaldehydes. 4-Formyl-1-(4-nitrophenyl)-1,2,3-triazole (FNPT) 1a (0.5 mmol), primary amine (0.55 mmol), water (45 µL), and iPrOH (or 1,4-dioxane) (1 mL) were added to a screw-capped reaction tube equipped with a magnetic stirring bar. The reaction mixture was stirred for 15 h at 80 °C, unless stated otherwise. Next, the reaction mixture was concentrated and directly purified via column chromatography.
1-Hexyl-1H-1,2,3-triazole-4-carbaldehyde (7a). Prepared according to the general procedure for the synthesis of 1-alkyl-1H-1,2,3-triazole-4-carbaldehydes; hexylamine (0.55 mmol, 56 mg), iPrOH as the reaction solvent. Purified via column chromatography using DCM as the eluent to remove 4-nitroaniline, followed by a DCM/EtOAc gradient (1:0—19:1). Clear yellowish oil that solidified over time; yield: 85 mg (94%); mp 31–33 °C; 1H NMR (400 MHz, CDCl3) δ 10.14 (s, 1H), 8.15 (s, 1H), 4.45 (t, J = 7.2 Hz, 2H), 2.09—1.85 (m, 2H), 1.45—1.18 (m, 6H), 0.99—0.78 (m, 3H); 13C NMR (151 MHz, CDCl3) δ 185.2, 147.8, 125.2, 50.8, 31.1, 30.1, 26.0, 22.4, 13.9.
Procedure for the recovery of 4-nitrophenyl azide: FNPT 1a (4.6 mmol, 1 g), hexylamine (5.98 mmol, 605 mg), water (22 mmol, 0.4 mL), and iPrOH (10 mL) were added to a screw-capped reaction tube equipped with a magnetic stirring bar. The reaction mixture was stirred for 25 h at 50 °C. Next, the mixture was allowed to cool down and 4 M HCl (aq., 7 mL) was added. At 0 °C, a solution of NaNO2 (5.5 mmol, 380 mg) in water (1 mL) was added dropwise while stirring, and the mixture was continued to stir for 30 min at 0 °C. The aqueous solution was extracted with cold EtOAc (20 mL, 0 °C) and separated. To the aqueous layer, a solution of NaN3 (6.9 mmol, 448 mg) in water (1.5 mL) was added dropwise at 0 °C and the mixture was subsequently stirred at room temperature for 3 h. 4-Nitrophenyl azide was extracted with Et2O (20 mL), and the organic layer was washed with 4 M HCl (aq., 29 mL), saturated NaHCO3 (aq., 20 mL), and brine (10 mL). The ether phase was then dried over MgSO4, filtered, and concentrated to afford 4-nitrophenyl azide 8a as a pure pale yellow solid; yield: 605 mg (80%). The EtOAc layer was also washed with saturated NaHCO3 (aq., 20 mL) and brine (10 mL), dried over MgSO4, filtered, and concentrated. Further purification via column chromatography using a DCM/EtOAc gradient (1:0—19:1) yielded the pure product 7a as a pale yellow semisolid; yield: 580 mg (70%).
One-pot procedure from 3-dimethylaminoacrolein: To a screw-capped 40 mL reaction tube equipped with a magnetic stirring bar were added 4-nitrophenyl azide 8a (6.5 mmol, 1.07 g), 3-dimethylaminoacrolein 9a (5 mmol, 0.5 g), and 1,4-dioxane (2.5 mL). The mixture was stirred at 50 °C for 70 h. Next, hexylamine (5.1 mmol, 516 mg), water (450 µL), and 1,4-dioxane (7.5 mL) were added and the mixture was stirred for 15 h at 80 °C. The reaction mixture was subsequently diluted with Et2O (100 mL) and washed with 3 M HCl (aq., 2 ϗ 100 mL), saturated NaHCO3 (aq., 50 mL), and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The crude product 7a was further purified via column chromatography using a petroleum–ether/DCM gradient (1:4—0:1) as the eluent to remove 4-nitroaniline, followed by a DCM/EtOAc gradient (1:0—4:1). Pale orange semisolid; yield: 581 mg (64%).
1-Methyl-1H-1,2,3-triazole-4-carbaldehyde (7b). FNPT
1a (2.5 mmol, 545 mg), a solution of methylamine in
iPrOH (2 M, 1.5 mL), and an additional amount of
iPrOH (3.5 mL) were added to a screw-capped reaction tube equipped with a magnetic stirring bar. The reaction mixture was stirred for 15 h at 80 °C. Next, the crude product was coated on silica and directly purified via column chromatography using DCM as the eluent to remove 4-nitroaniline, followed by a DCM/MTBE gradient (1:0—1:1). Further purification was possible by sublimation in a Kugelrohr. White crystalline solid; yield: 229 mg (82%); mp 112–114 °C;
1H NMR (300 MHz, CDCl
3) δ 10.14 (s, 1H), 8.15 (s, 1H), 4.21 (s, 3H);
13C NMR (75 MHz, CDCl
3) δ 185.1, 148.1, 126.3, 37.2;
1H NMR (300 MHz, DMSO-
d6) δ 10.01 (s, 1H), 8.81 (s, 1H), 4.13 (s, 3H);
13C NMR (151 MHz, DMSO) δ 185.0, 147.0, 128.8, 36.7. NMR data was in accordance with the previously reported data for this compound [
20].
1-Benzyl-1H-1,2,3-triazole-4-carbaldehyde (7c). Prepared according to the general procedure for the synthesis of 1-alkyl-1
H-1,2,3-triazole-4-carbaldehydes; benzylamine (0.55 mmol, 59 mg),
iPrOH as the reaction solvent. Purified via column chromatography using a petroleum-ether/DCM gradient (1:4—0:1) as the eluent to remove 4-nitroaniline, followed by a DCM/EtOAc gradient (19:1—1:9). Clear oil with yellowish hue that solidified over time to give an off-white solid; yield: 89 mg (95%); mp 82–86 °C;
1H NMR (400 MHz, CDCl
3) δ 10.12 (s, 1H), 8.02 (s, 1H), 7.44—7.28 (m, 5H), 5.60 (s, 2H);
13C NMR (101 MHz, CDCl
3) δ 185.2, 148.1, 133.5, 129.5, 129.4, 128.5, 125.3, 54.7. NMR data was in accordance with the previously reported data for this compound [
24].
1-Allyl-1H-1,2,3-triazole-4-carbaldehyde (7d). Prepared according to the general procedure for the synthesis of 1-alkyl-1
H-1,2,3-triazole-4-carbaldehydes; allylamine (0.55 mmol, 31 mg),
iPrOH as the reaction solvent. Purified via column chromatography using a petroleum-ether/DCM gradient (1:4—0:1) as the eluent to remove 4-nitroaniline, followed by a DCM/EtOAc gradient (19:1—1:9). Clear oil with greenish hue; yield: 64 mg (93%);
1H NMR (400 MHz, CDCl
3) δ 10.15 (s, 1H), 8.16 (s, 1H), 6.06 (ddt, J = 16.6, 10.1, 6.3 Hz, 1H), 5.50—5.34 (m, 2H), 5.08 (d, J = 6.3 Hz, 2H);
13C NMR (101 MHz, CDCl
3) δ 185.1, 148.0, 130.2, 125.2, 121.5, 53.1. NMR data was in accordance with the previously reported data for this compound [
24].
1-(Prop-2-yn-1-yl)-1H-1,2,3-triazole-4-carbaldehyde (7e). Prepared according to the general procedure for the synthesis of 1-alkyl-1H-1,2,3-triazole-4-carbaldehydes; propargylamine (0.55 mmol, 30 mg), 1,4-dioxane as the reaction solvent, 70 °C, 18h. Purified via column chromatography using DCM as the eluent to remove 4-nitroaniline, followed by a DCM/EtOAc gradient (9:1—4:1). Clear oil with yellowish hue; yield: 44 mg (65%); 1H NMR (400 MHz, CDCl3) δ 10.15 (s, 1H), 8.39 (s, 1H), 5.28 (d, J = 2.6 Hz, 2H), 2.69 (t, J = 2.6 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 184.9, 147.9, 125.3, 77.1, 74.1, 40.4; HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C6H5N3O: 136.0505; found: 136.0512.
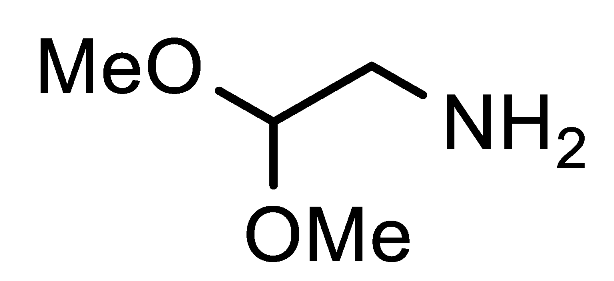
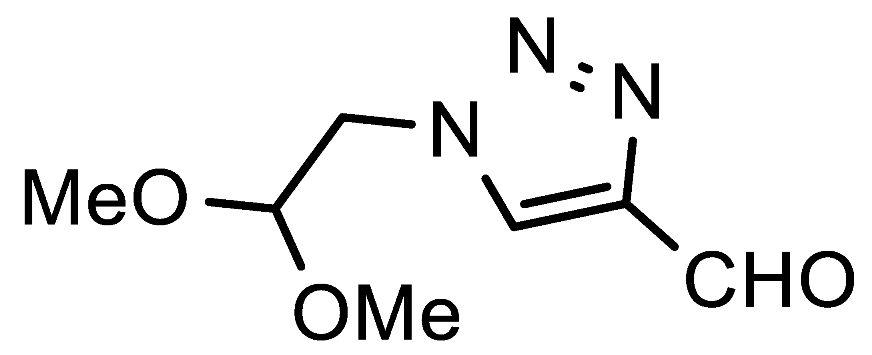
1-(2,2-Dimethoxyethyl)-1H-1,2,3-triazole-4-carbaldehyde (7f). Prepared according to the general procedure for the synthesis of 1-alkyl-1H-1,2,3-triazole-4-carbaldehydes; aminoacetaldehyde dimethyl acetal (0.55 mmol, 58 mg), iPrOH as the reaction solvent. Purified via column chromatography using DCM as the eluent to remove 4-nitroaniline, followed by a DCM/EtOAc gradient (1:0—4:1). Clear oil with yellowish hue; yield: 86 mg (93%); 1H NMR (300 MHz, CDCl3) δ 10.14 (s, 1H), 8.28 (s, 1H), 4.75—4.65 (m, 1H), 4.57 (d, J = 5.0 Hz, 2H), 3.44 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 184.9, 147.7, 126.8, 102.0, 55.1, 52.0.
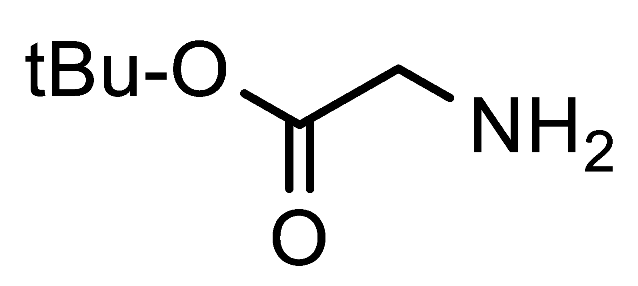
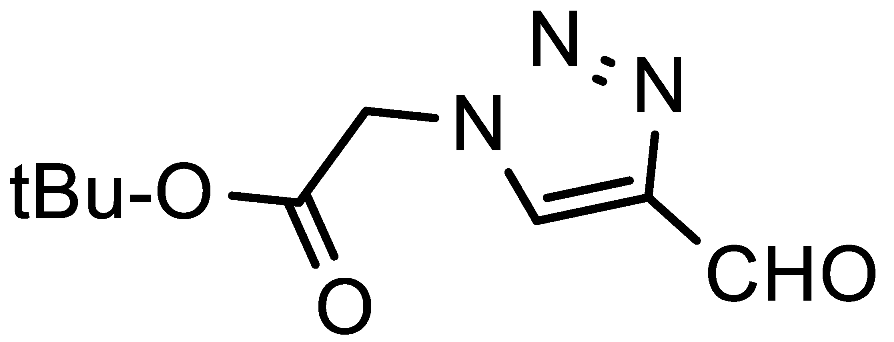
tert-Butyl 2-(4-formyl-1H-1,2,3-triazol-1-yl)acetate (7g). Prepared according to the general procedure for the synthesis of 1-alkyl-1
H-1,2,3-triazole-4-carbaldehydes;
tert-butyl glycinate (0.55 mmol, 72 mg), 1 mL
iPrOH or 1,4-dioxane as the reaction solvent. Purified via column chromatography using DCM as the eluent to remove 4-nitroaniline, followed by a DCM/EtOAc gradient (9:1—4:1). Off-white crystalline solid; yield: 43 mg (41%) and 47 mg (45%) for reactions in
iPrOH and dioxane, resp.; mp 93–95 °C;
1H NMR (400 MHz, CDCl
3) δ 10.15 (s, 1H), 8.30 (s, 1H), 5.17 (s, 2H), 1.50 (s, 9H);
13C NMR (101 MHz, CDCl
3) δ 184.9, 164.6, 148.0, 127.0, 84.6, 51.7, 28.0. NMR data was in accordance with the previously reported data for this compound [
28].
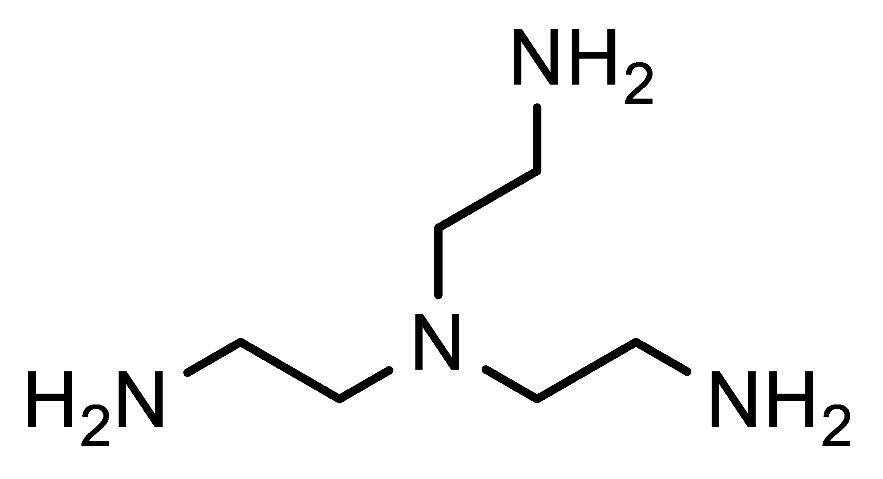
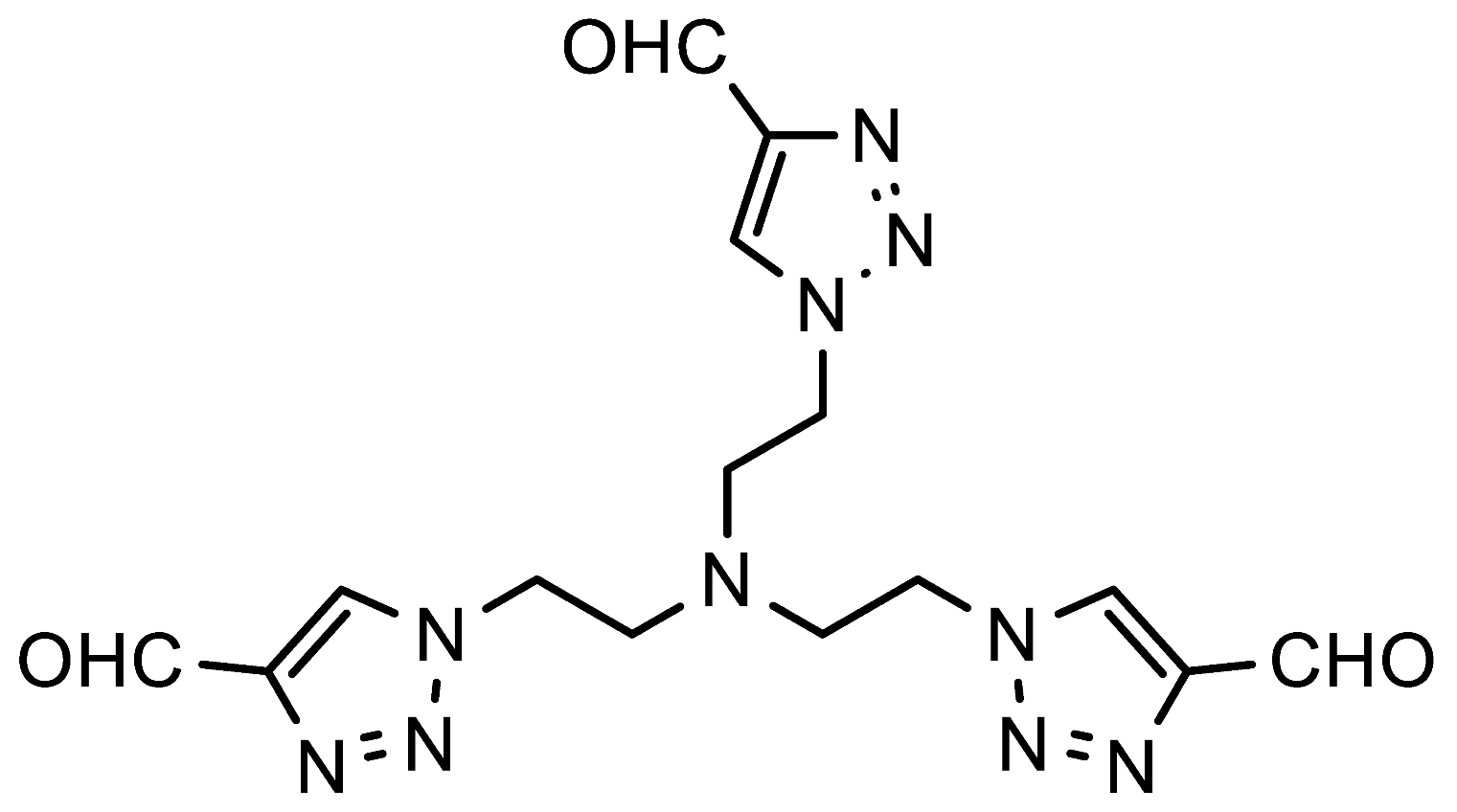
1,1′,1″-(Nitrilotris(ethane-2,1-diyl))tris(1H-1,2,3-triazole-4-carbaldehyde) (7h). FNPT 1a (0.83 mmol, 180 mg), tris(2-aminoethyl)amine (0.25 mmol, 36.5 mg), iPrOH (0.5 mL) and water (1.5 mL) were added in this sequence to a screw-capped reaction tube equipped with a magnetic stirring bar. The reaction mixture was stirred for 17 h at 80 °C. Next, the aqueous solution was extracted with DCM (2 × 10 mL) after which the combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude product was further purified via column chromatography using a DCM/EtOAc gradient (1:0—3:2) as the eluent to remove 4-nitroaniline and the starting triazole 1a, followed by a DCM/MeOH gradient (49:1—19:1). Off-white solid; yield: 87 mg (90%); mp 146–151 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 3H), 8.54 (s, 3H), 4.44 (t, J = 6.2 Hz, 6H), 3.06 (t, J = 6.3 Hz, 6H); 13C NMR (101 MHz, DMSO) δ 184.9, 146.7, 128.0, 52.0, 47.4; HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C15H18N10O3: 387.1636; found: 387.1633.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}