
Synthesis of Surrogate Blends Corresponding to Petroleum Middle Distillates, Oxidative and Extractive Desulfurization Using Imidazole Ionic Liquids


Abstract
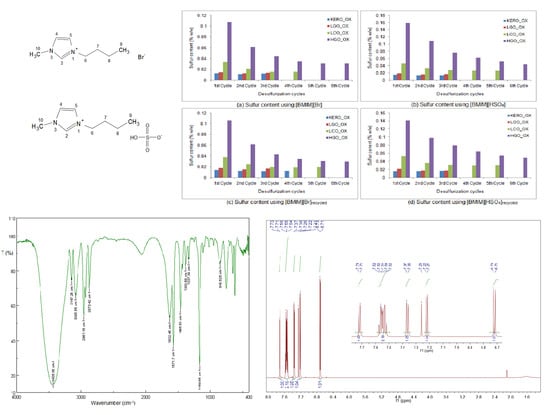
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Apparatus
2.3. Analysis
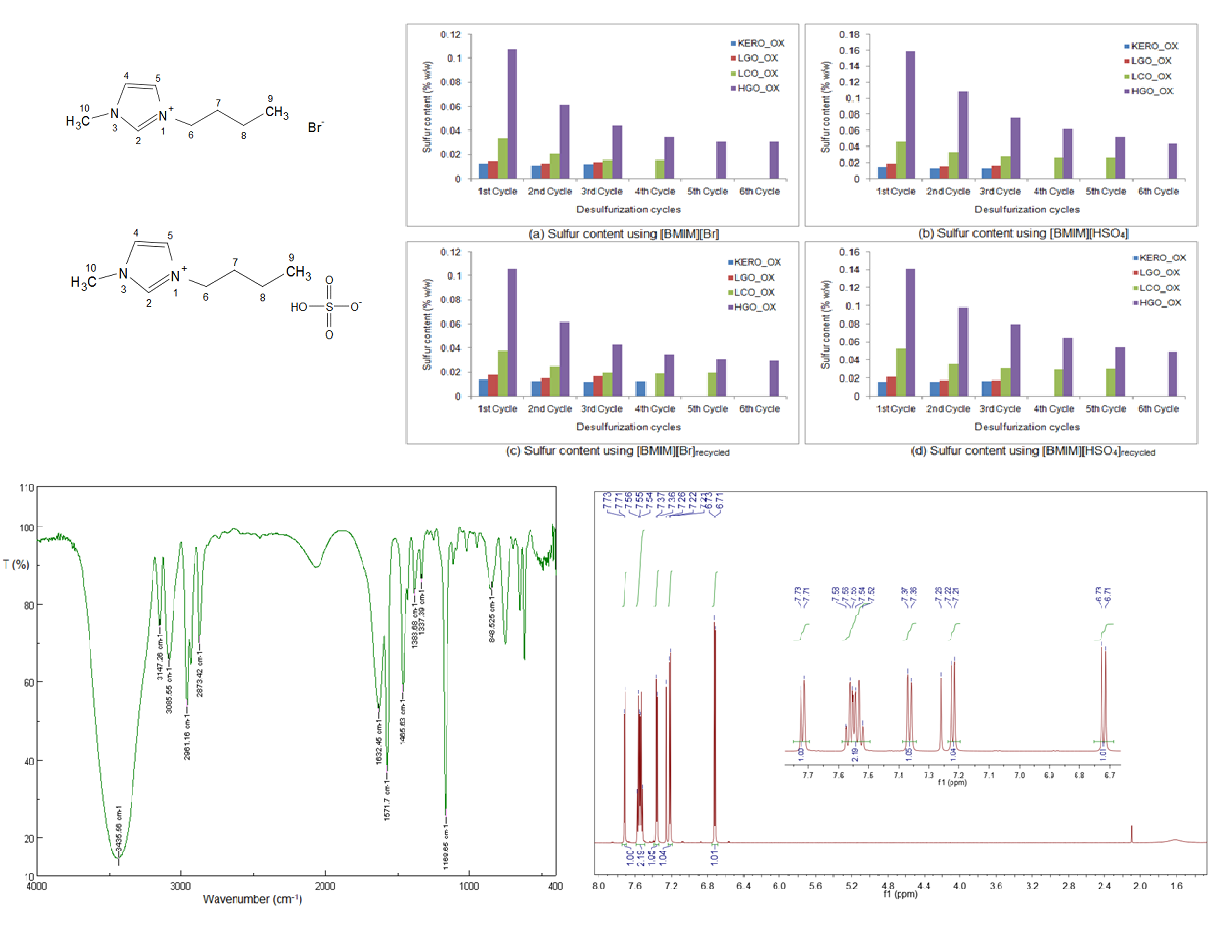
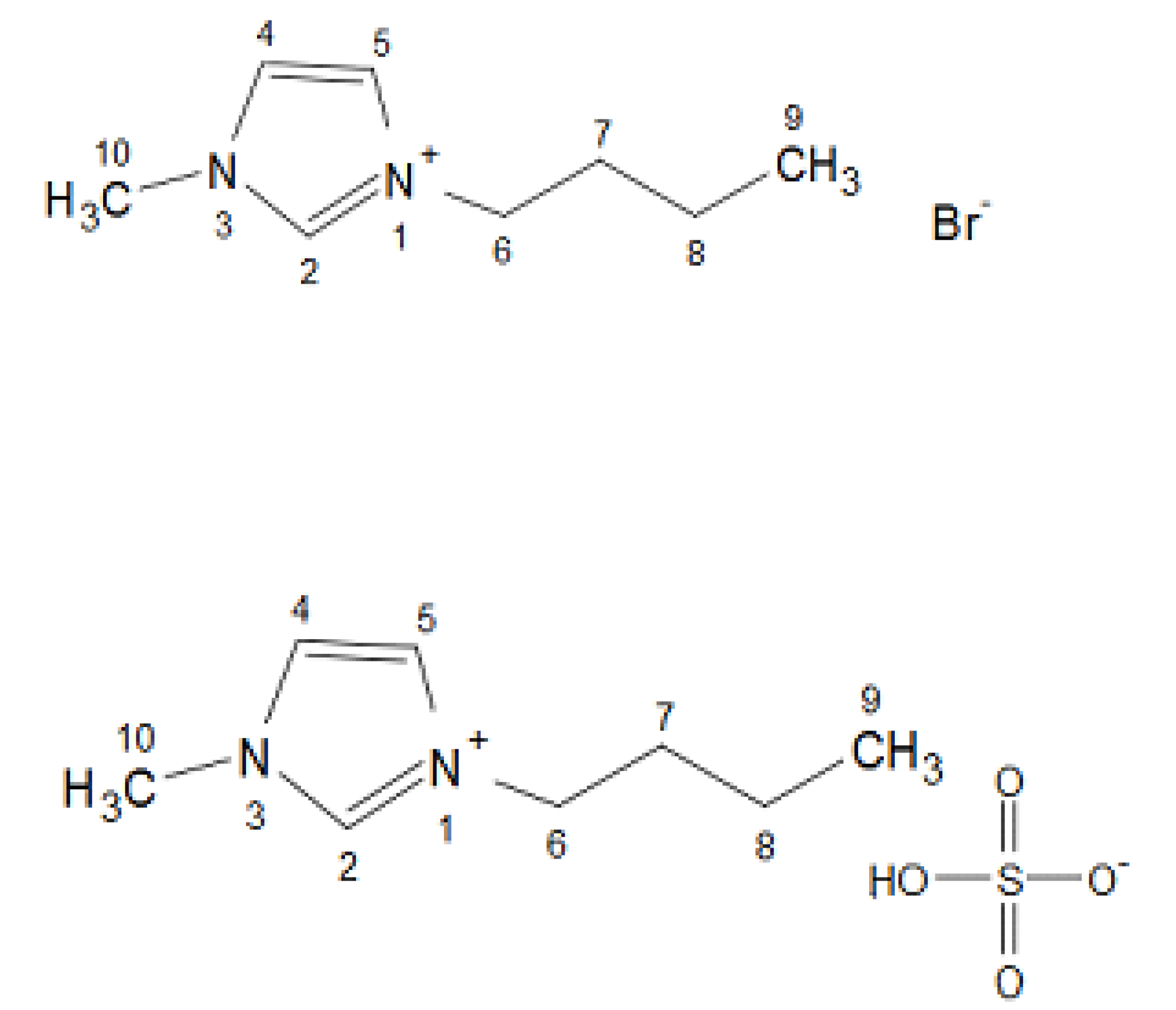
2.4. Synthesis of Ionic Liquids
2.4.1. Synthesis of [BMIM][Br]
2.4.2. Synthesis of [BMIM][HSO4]
2.5. Synthesis of the Surrogate Feeds
2.6. Oxidative Desulfurization
2.7. Extractive Desulfurization
2.7.1. Extraction with Solvents
2.7.2. Extraction with Ionic Liquids
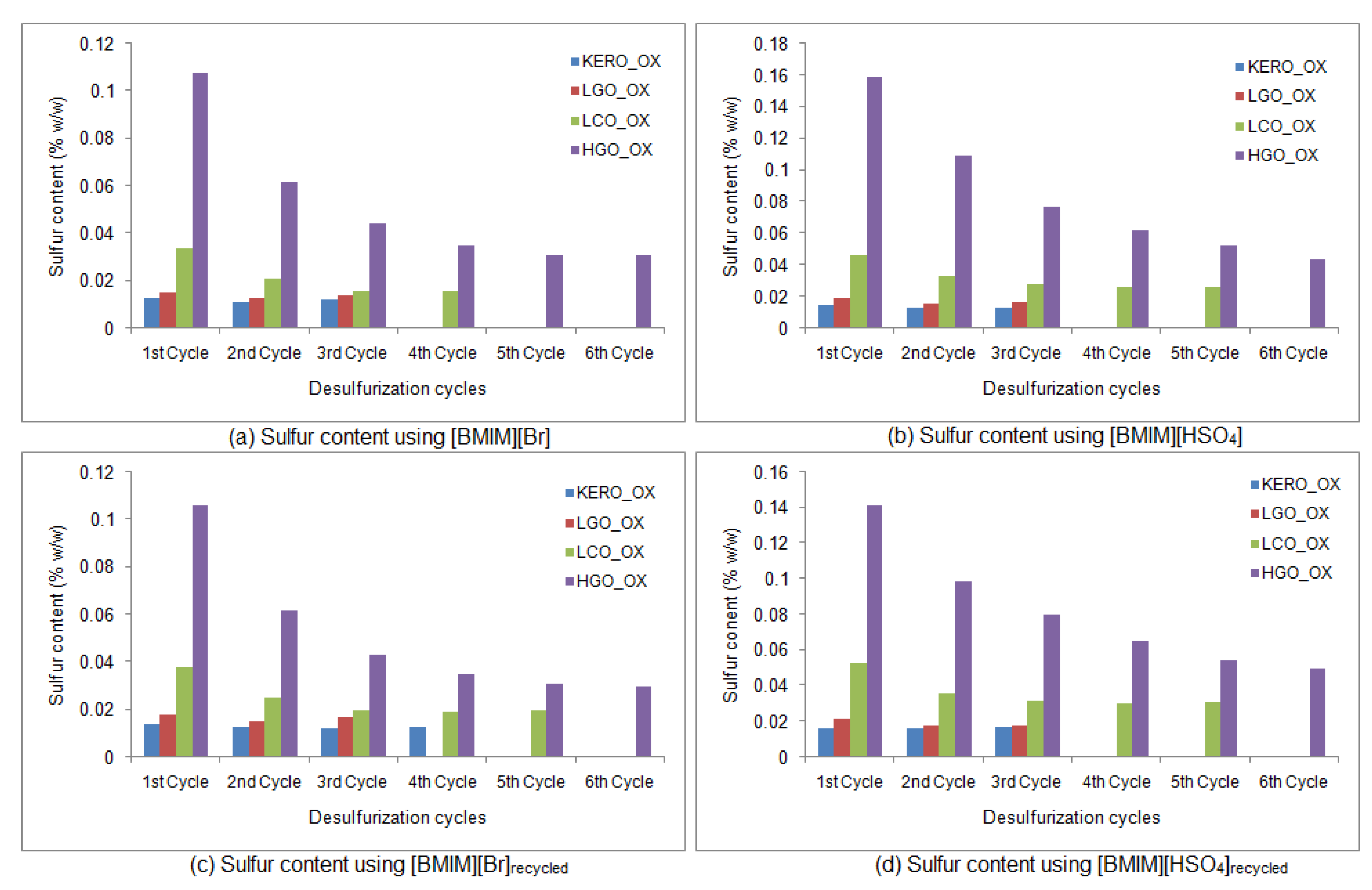
2.8. Regeneration of Ionic Liquids
3. Results and Discussion
3.1. Properties and Characterization of the Synthesized Ionic Liquids
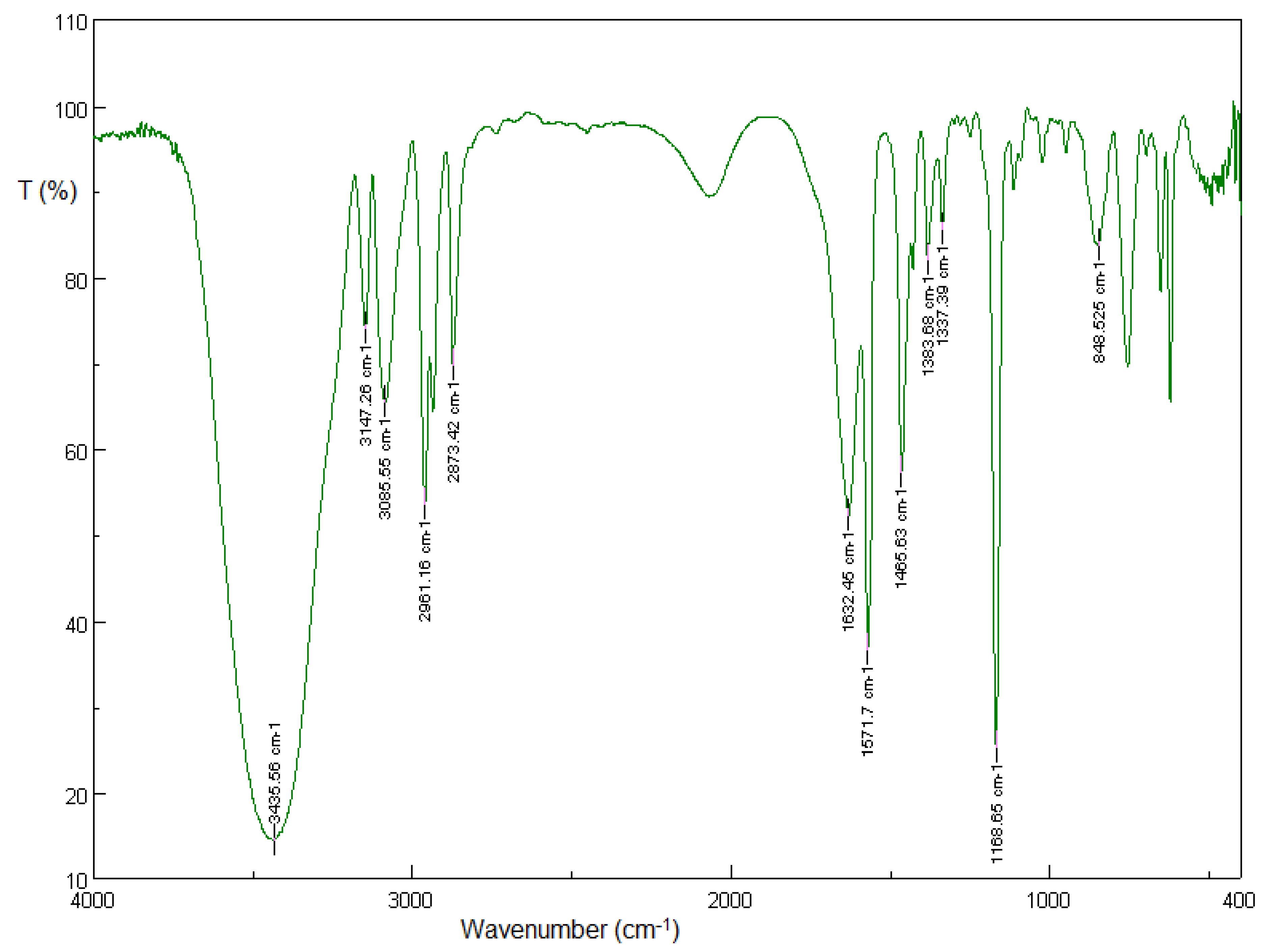
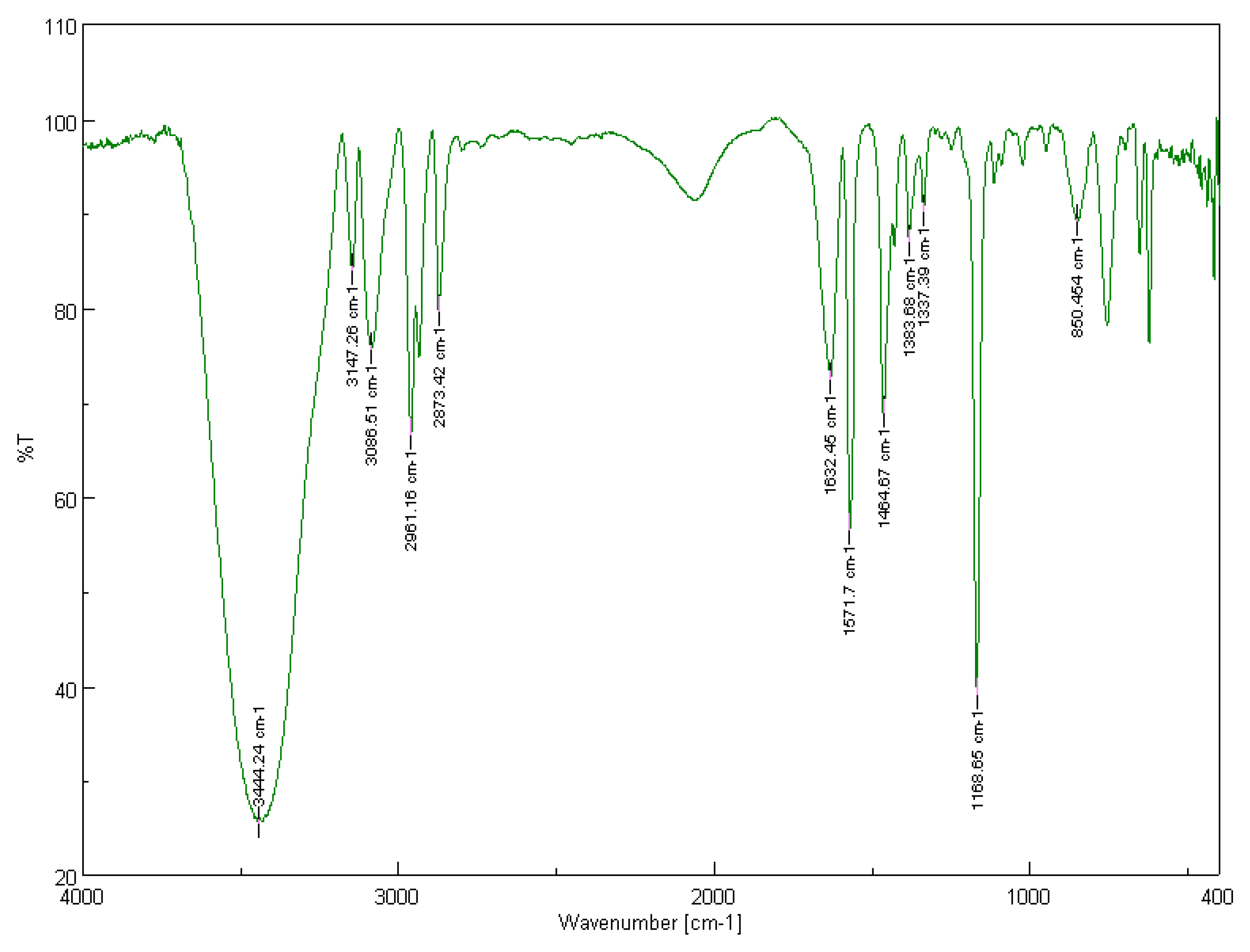
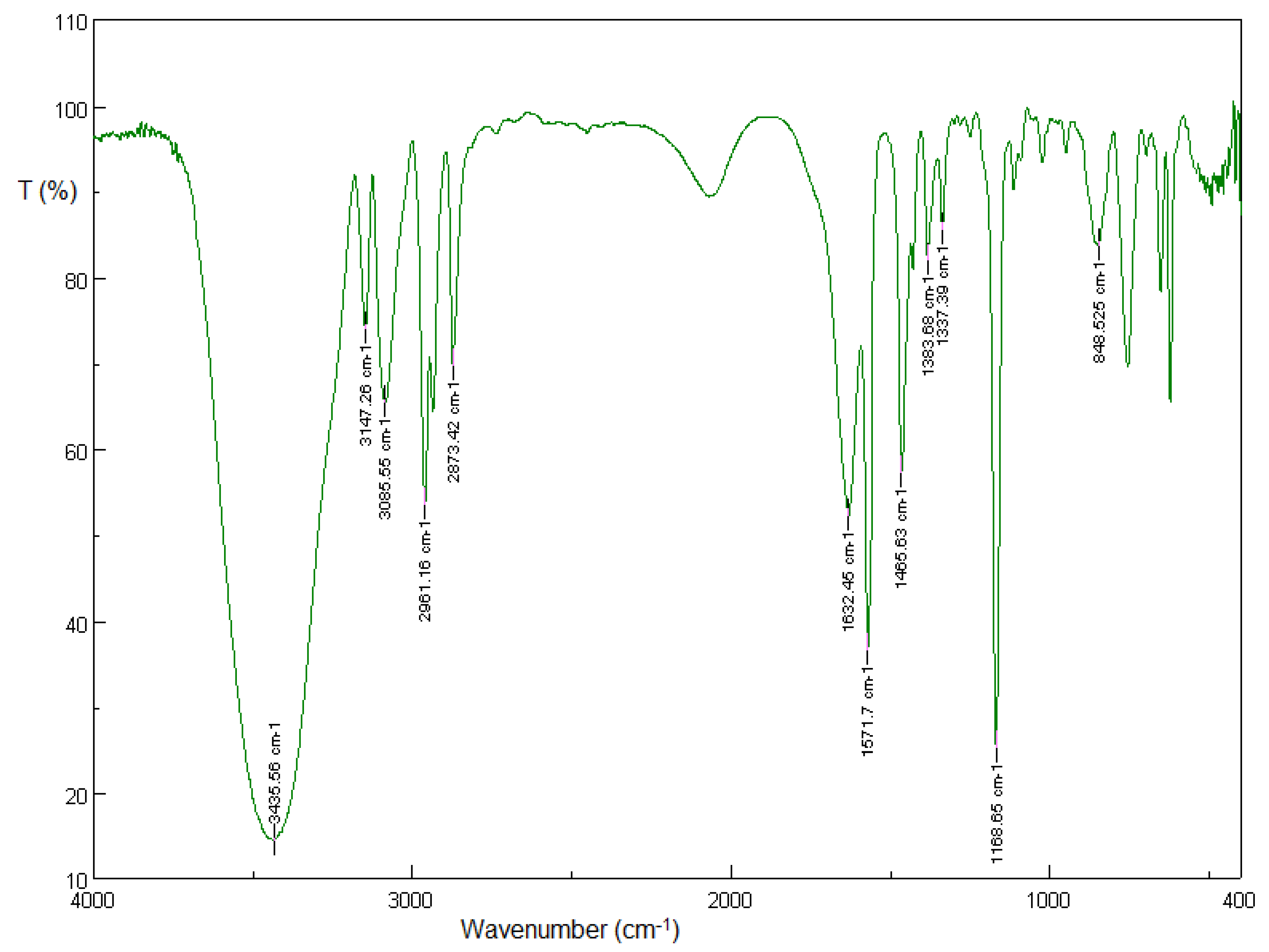
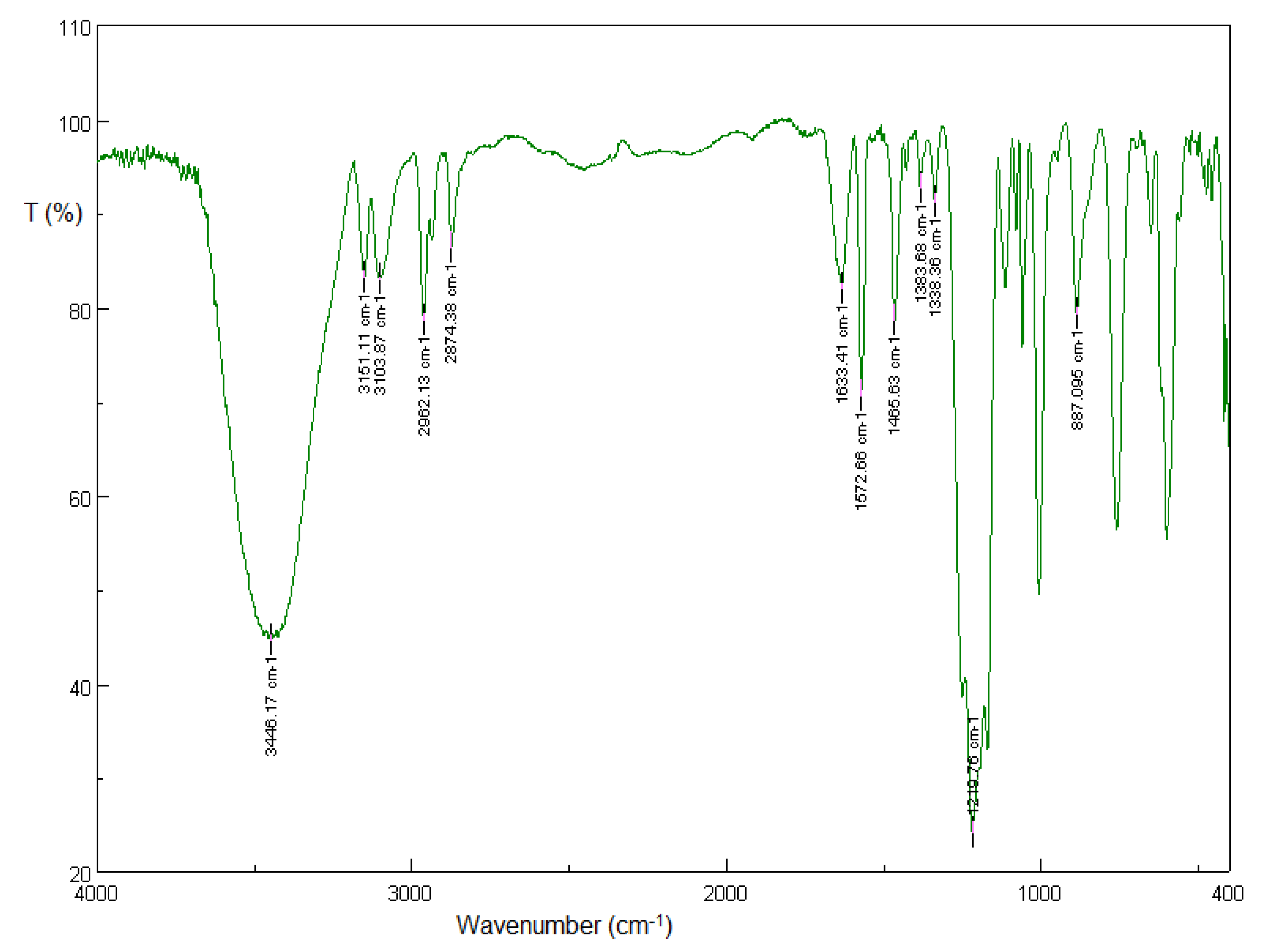
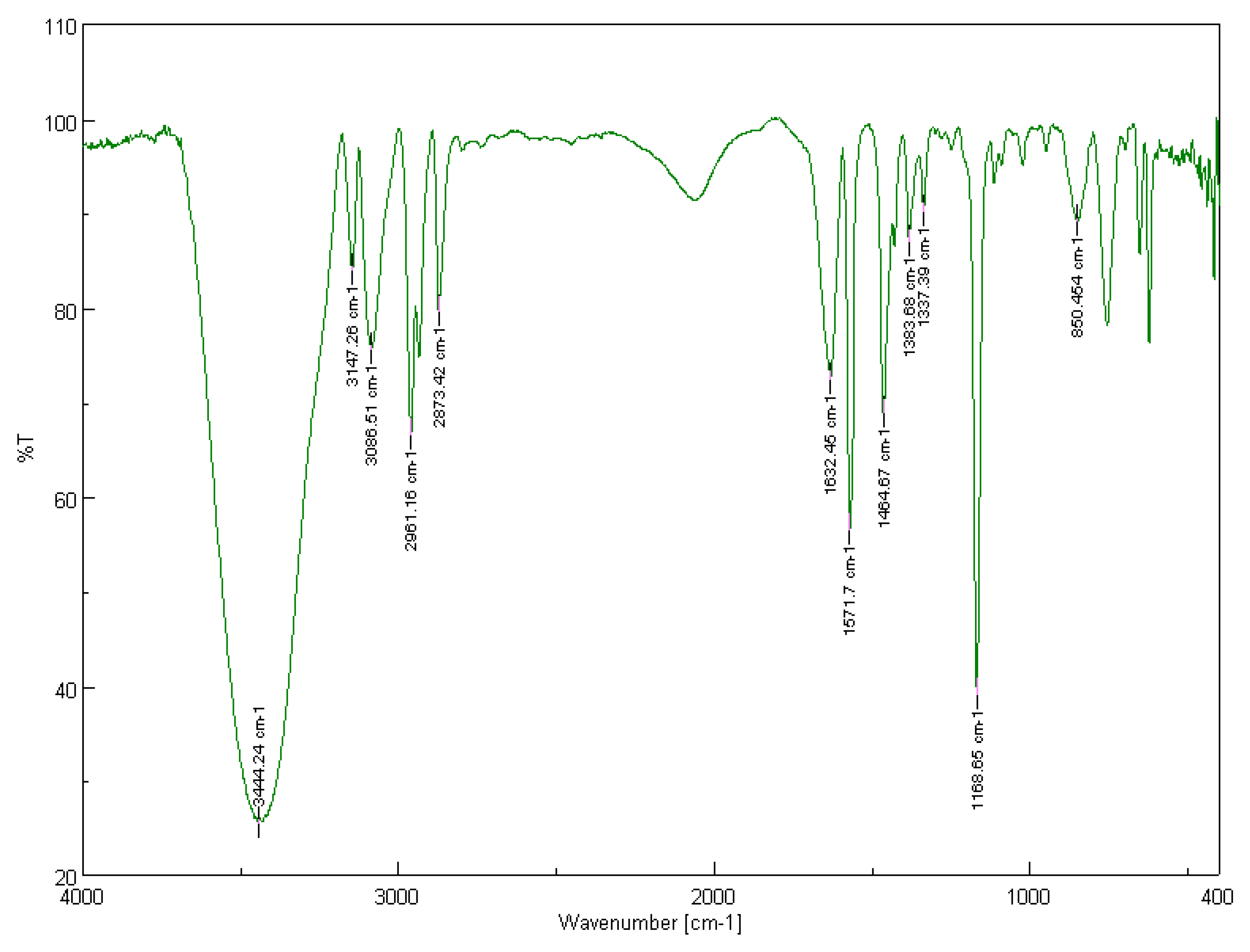
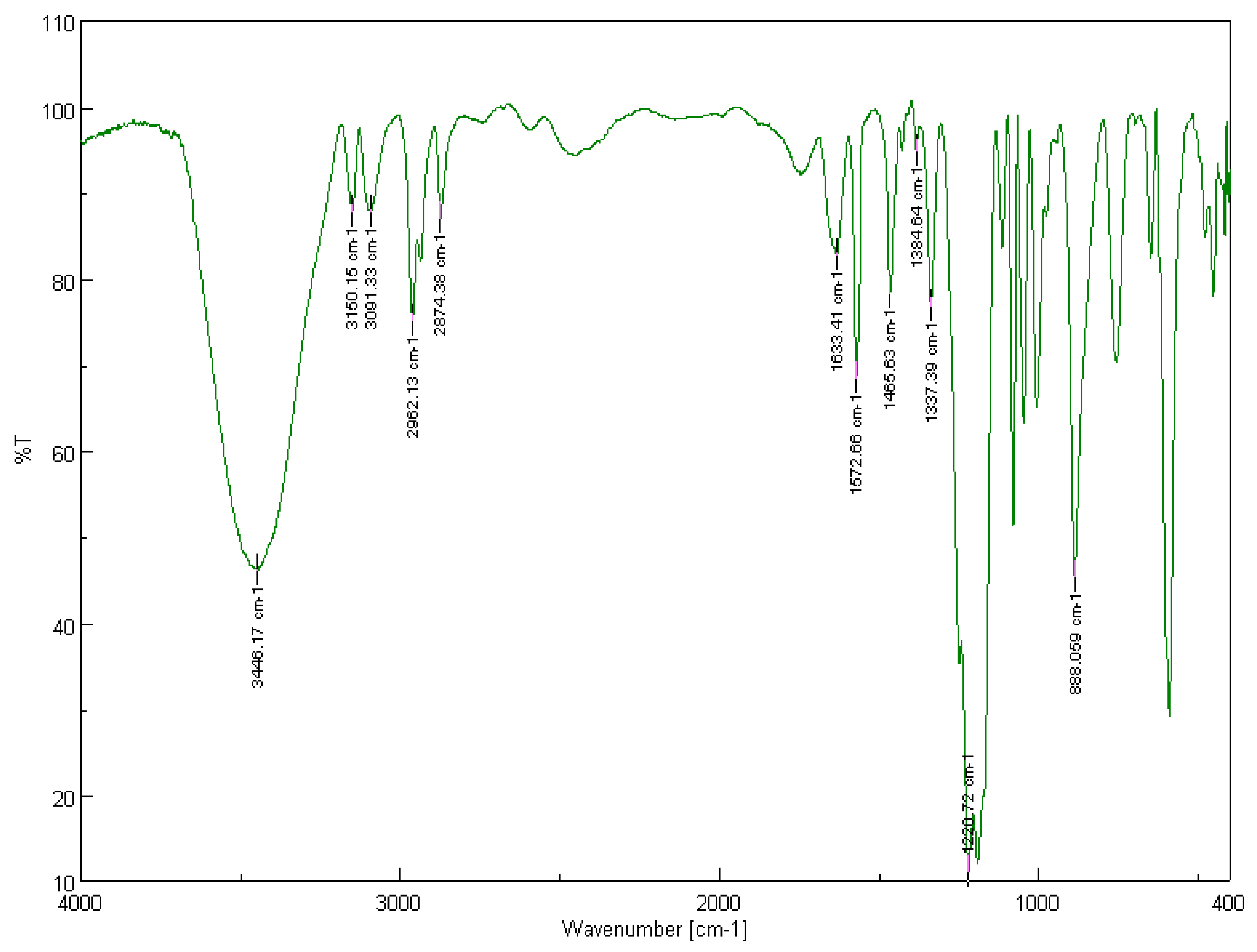
3.1.1. Solid-State FT-IR Spectroscopy
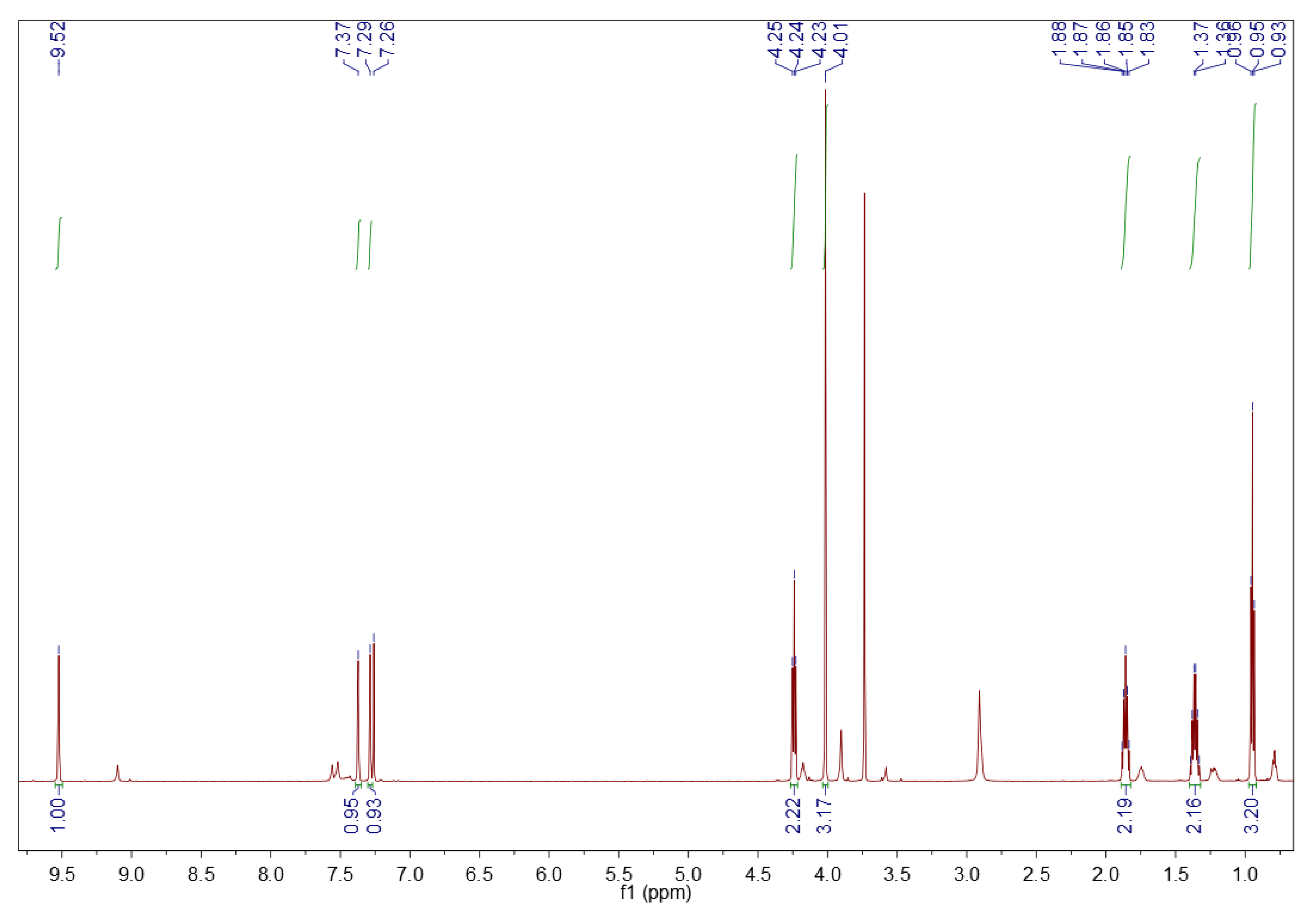
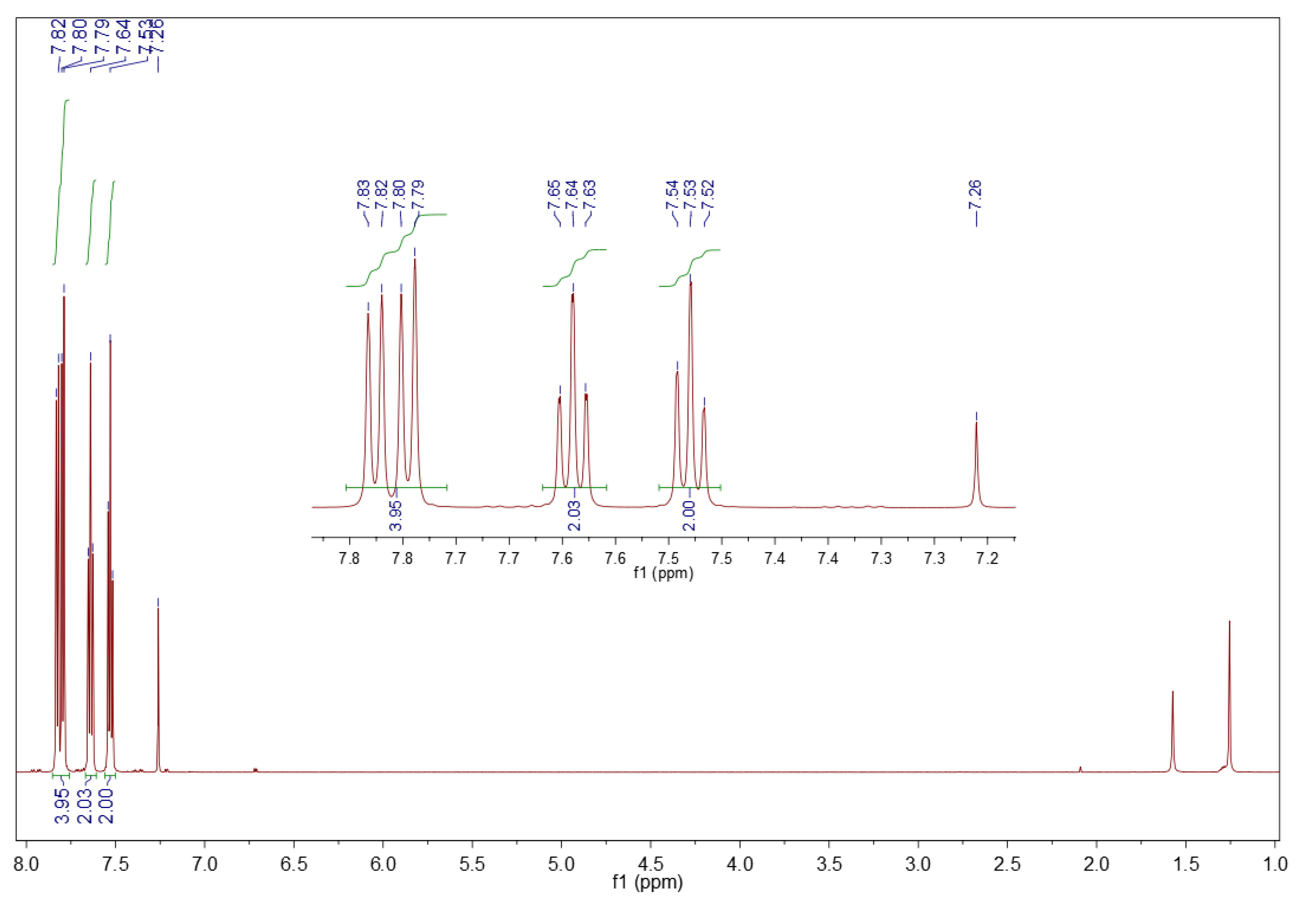
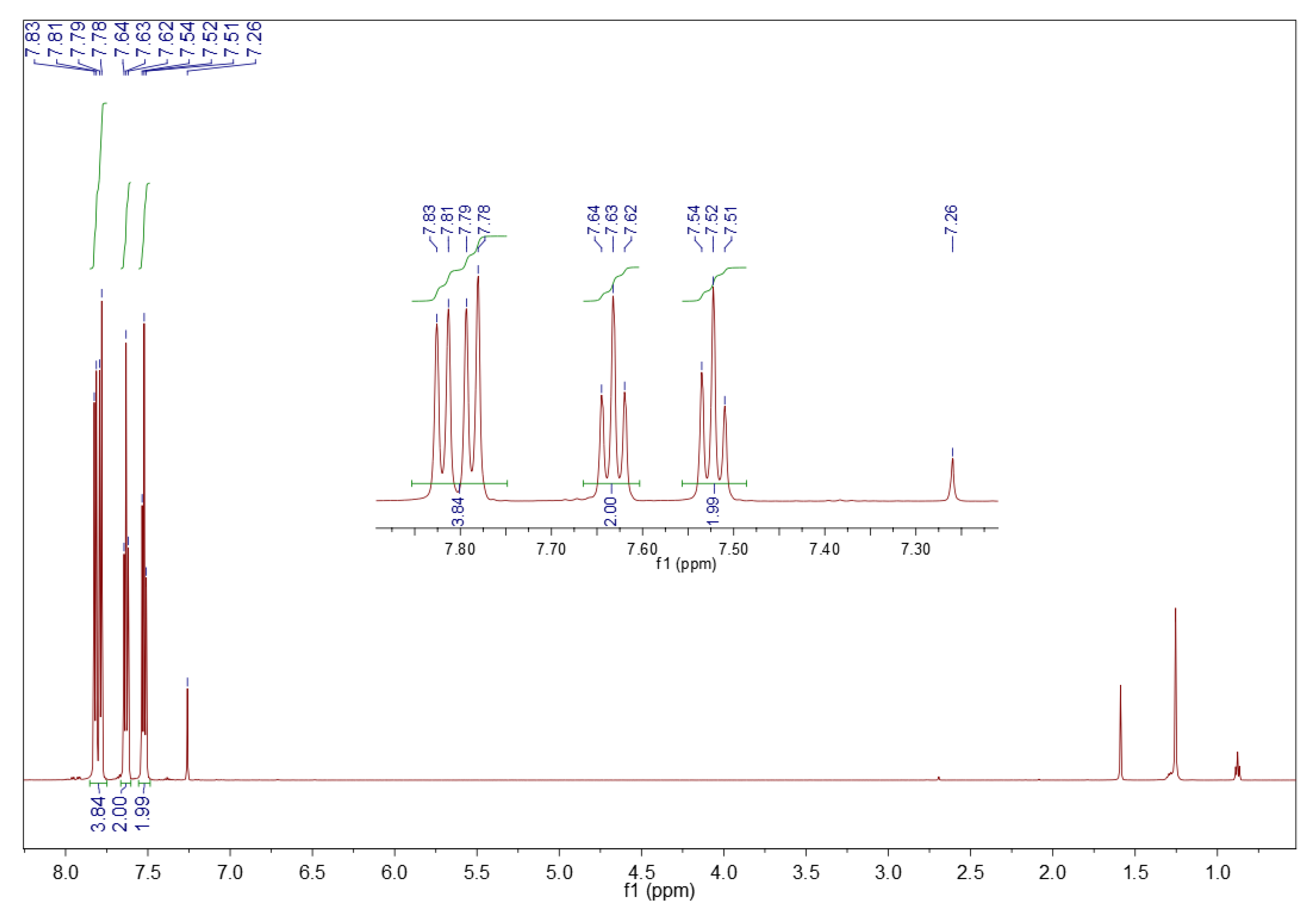
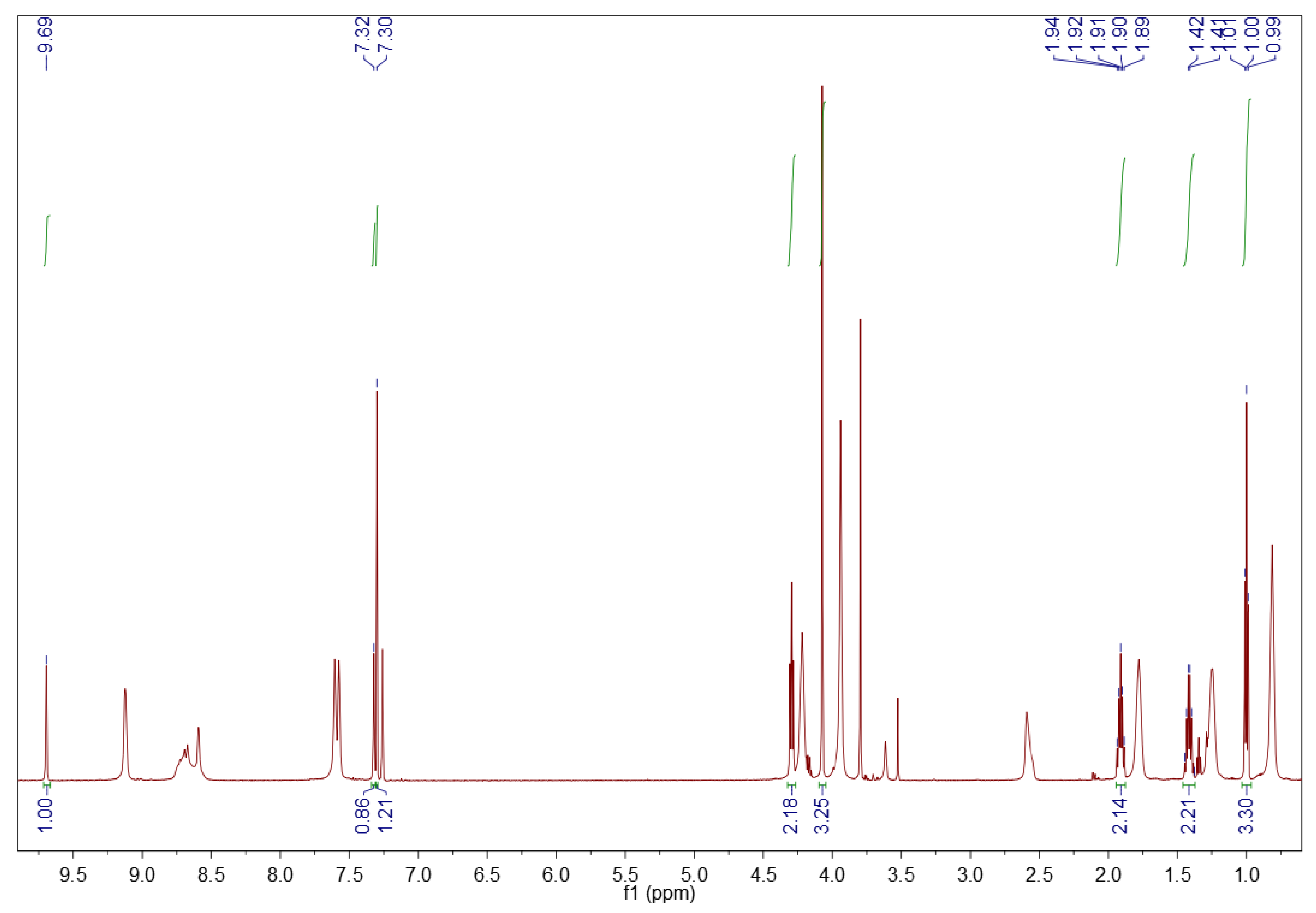
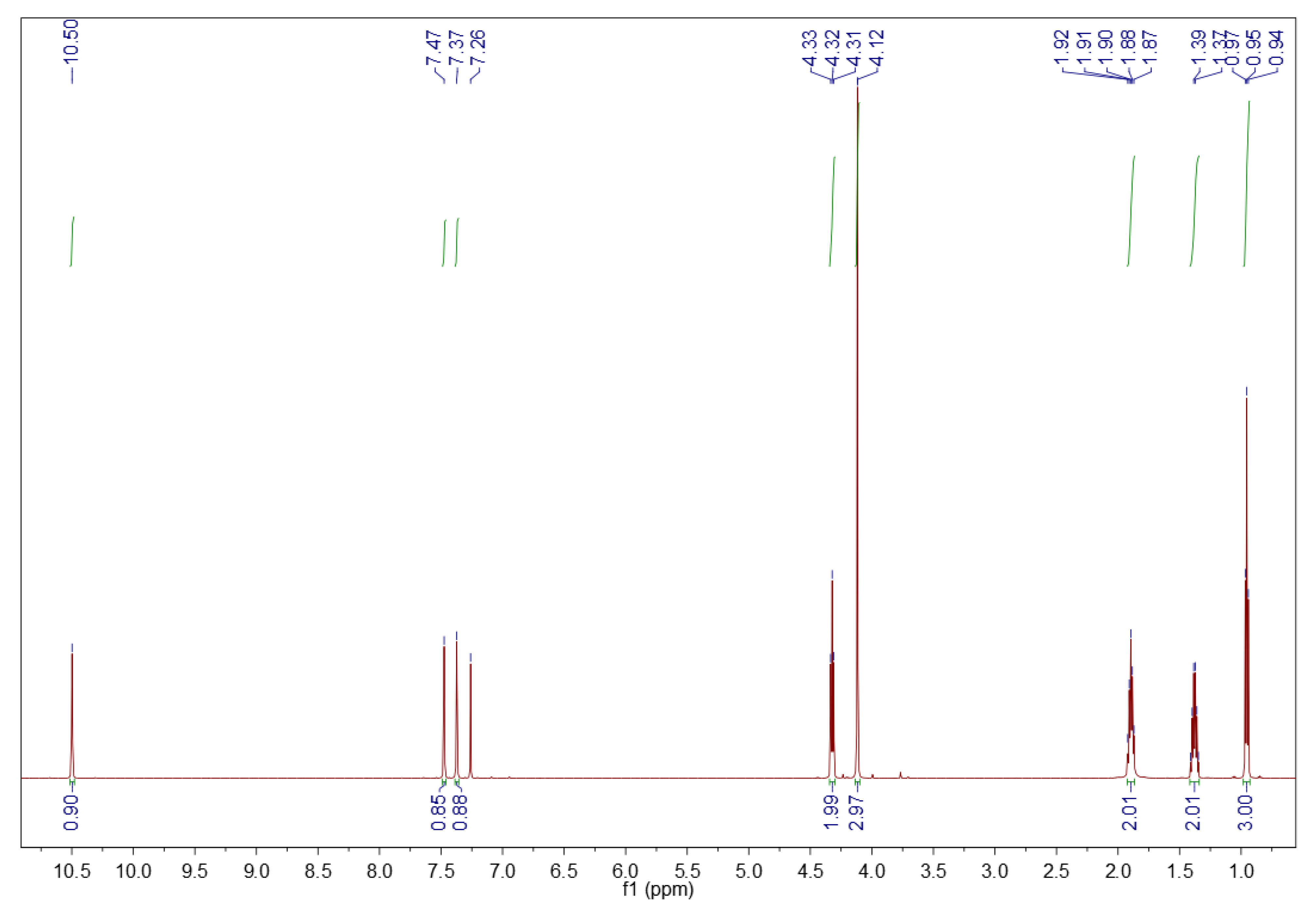
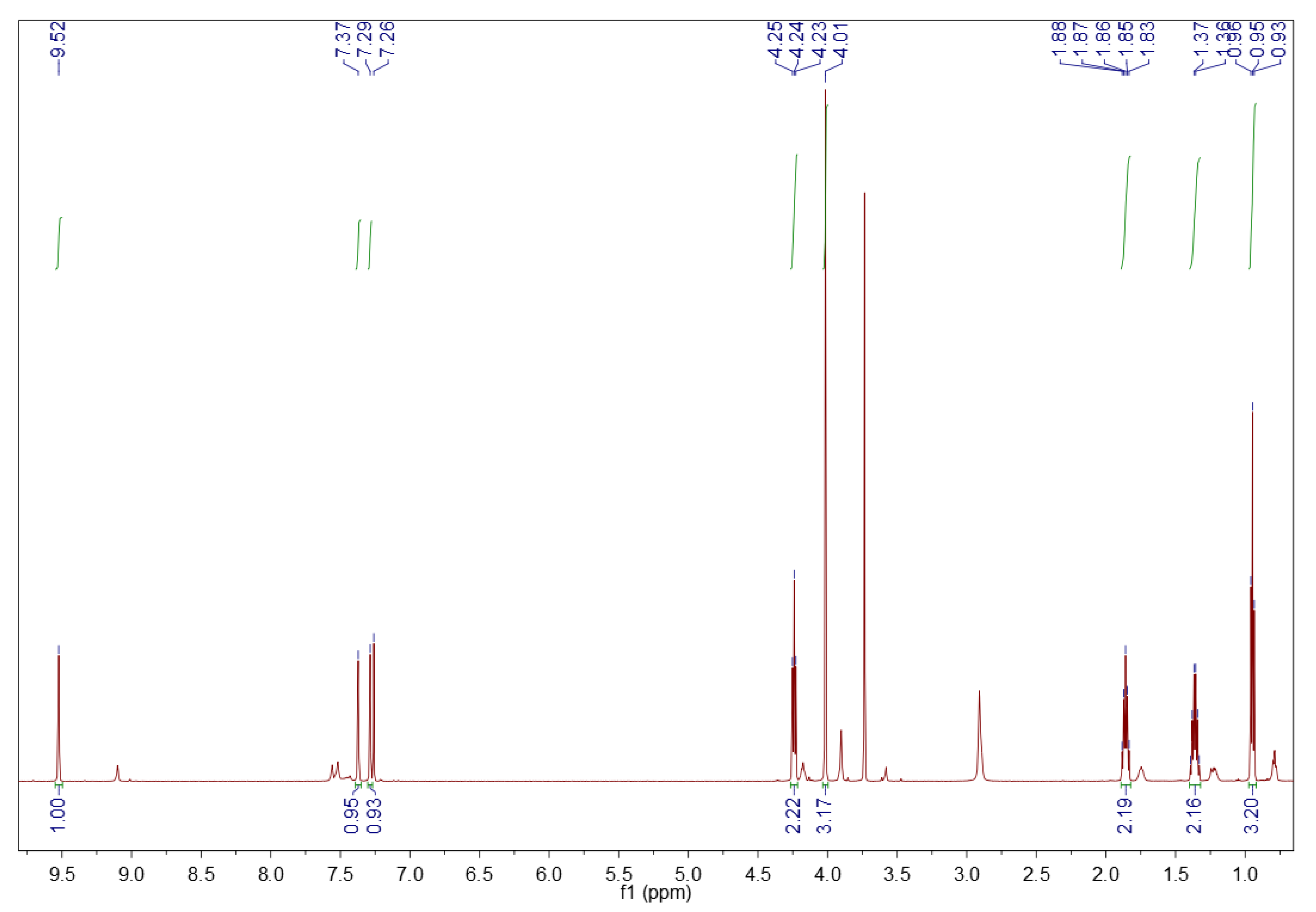
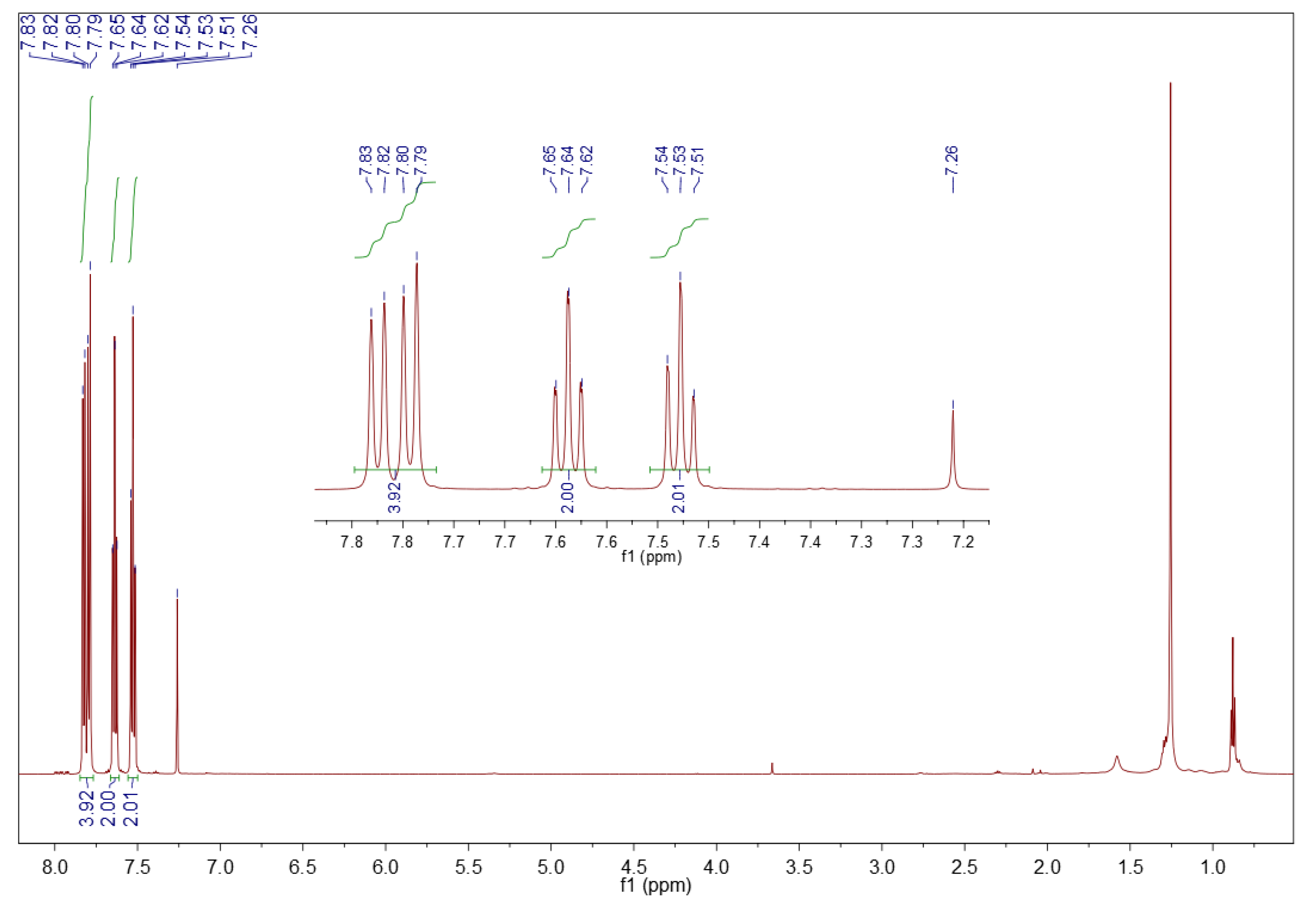
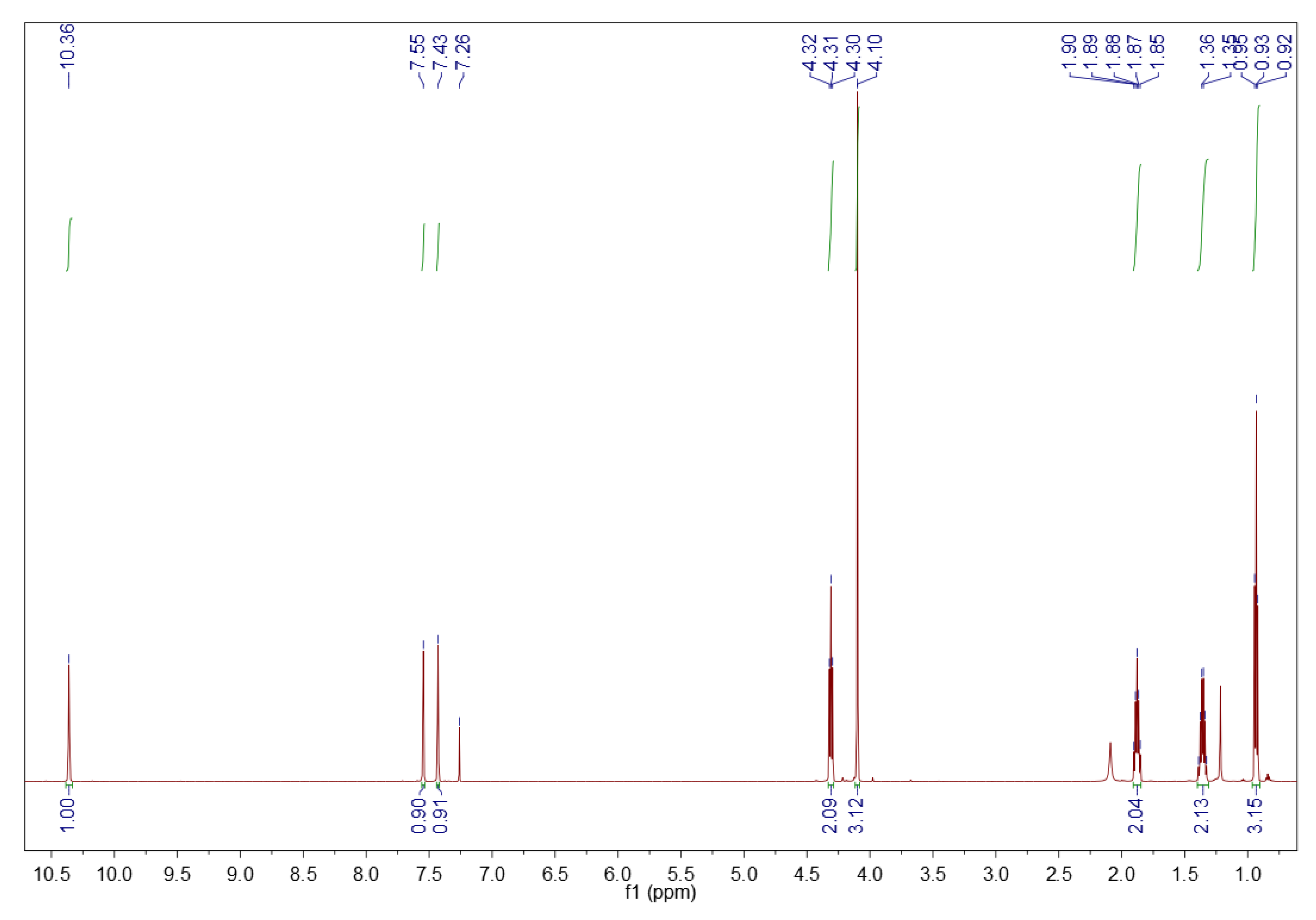
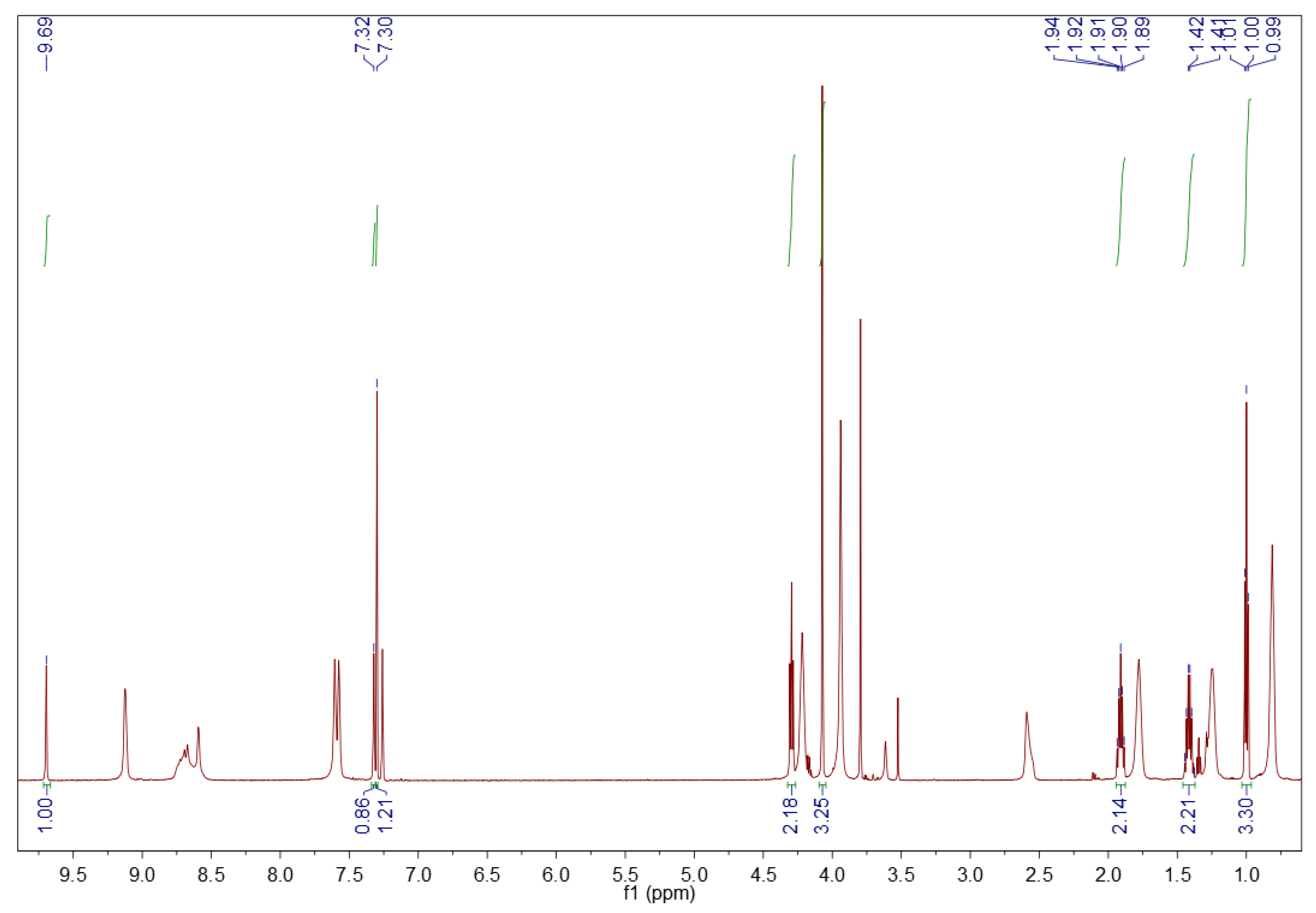
3.1.2. 1H NMR Spectroscopy
3.2. Characterization of the Precipitated Sediments
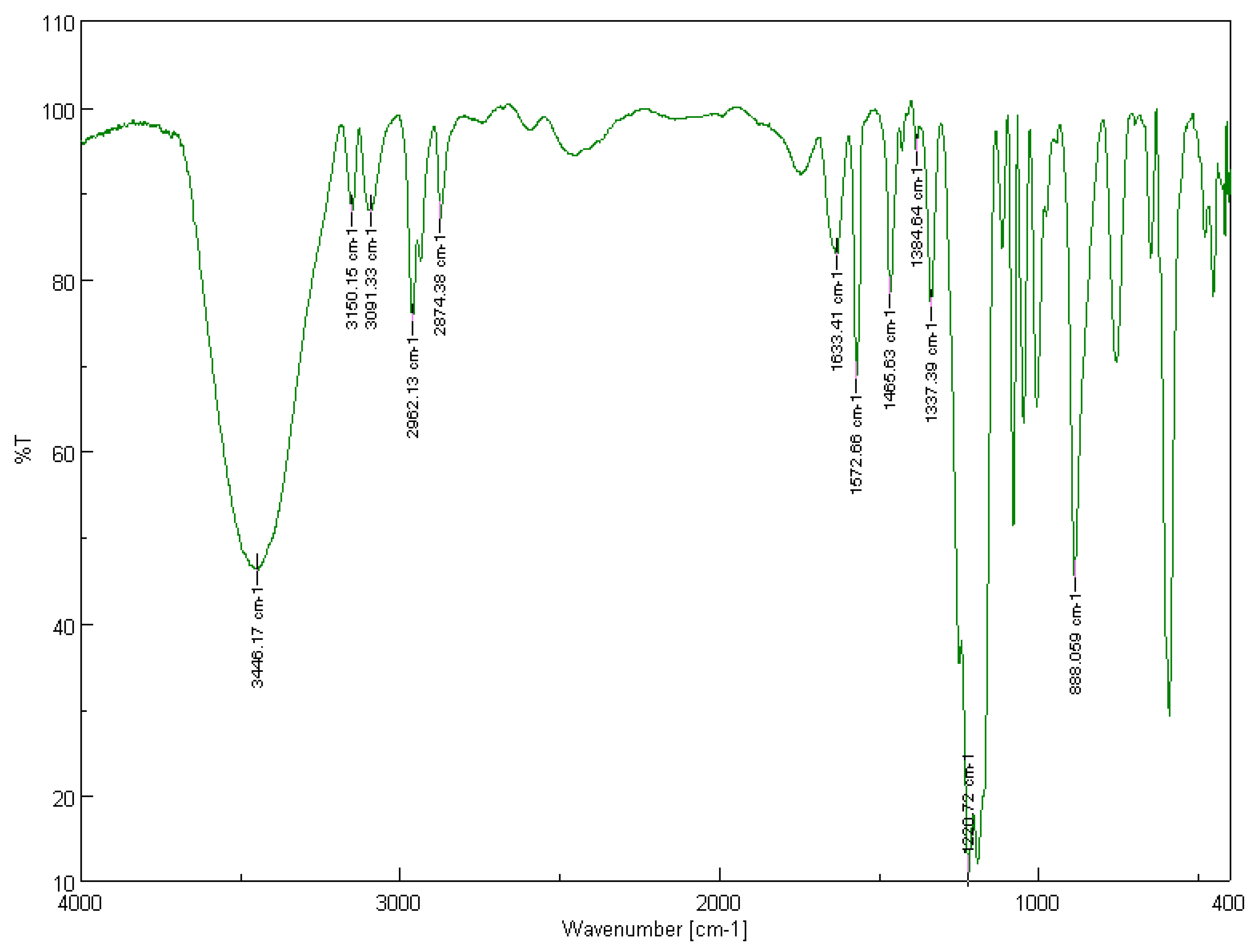
3.2.1. Solid-State FT-IR Spectroscopy
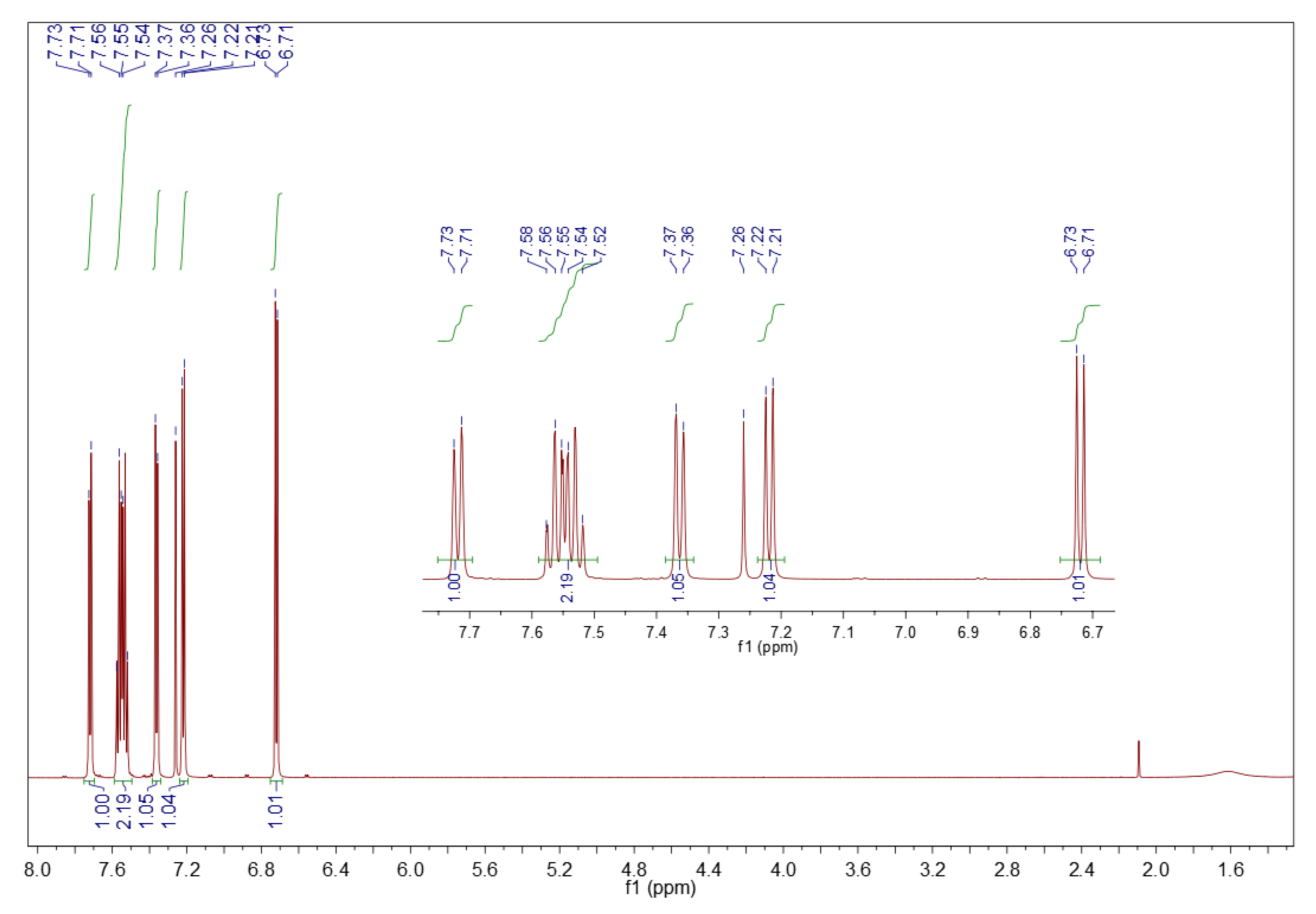
3.2.2. 1H NMR Spectroscopy
3.3. Desulfurization of the Surrogate Blends Using Conventional Solvents
3.3.1. Properties of the Raffinate Surrogate Products
3.3.2. Liquid FT-IR Spectroscopy
3.4. Desulfurization of the Oxidized Surrogate Blends Using Ionic Liquids
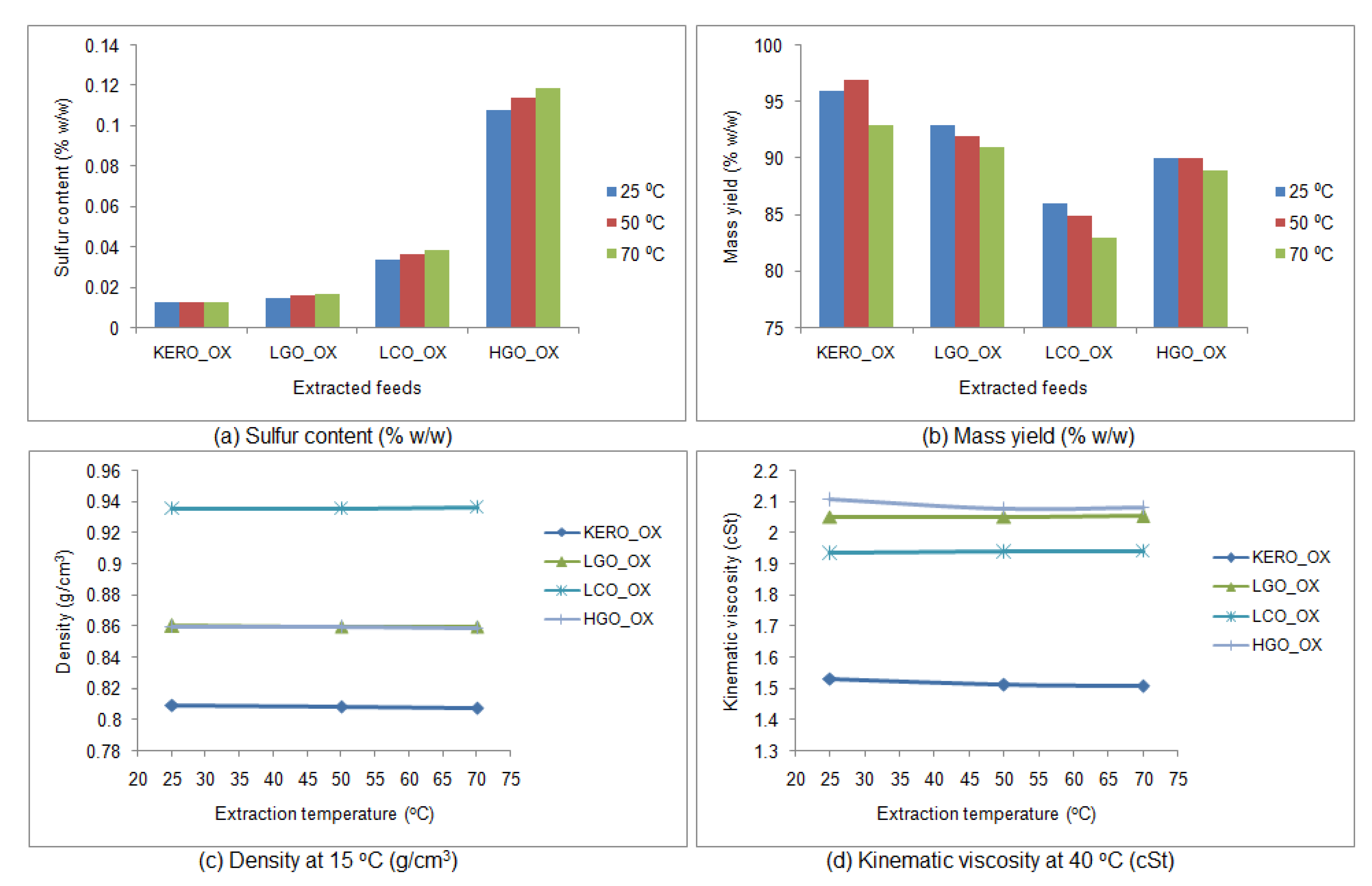
3.4.1. Influence of Temperature Extraction
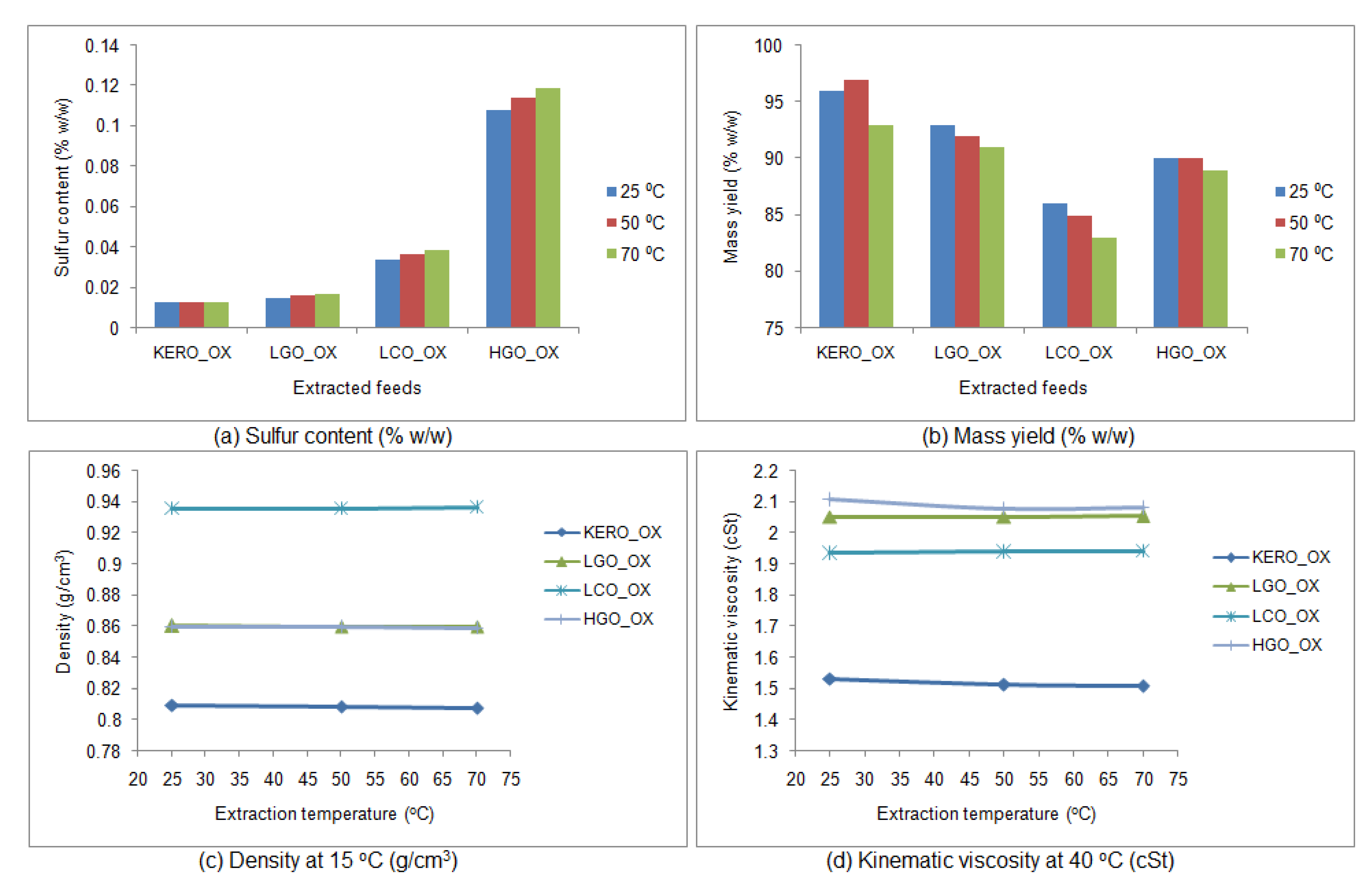
3.4.2. Properties of the Raffinate Surrogate Products
3.4.3. Liquid FT-IR Spectroscopy
3.5. Properties and Characterization of the Recycled Ionic Liquids
3.5.1. Solid-State FT-IR Spectroscopy
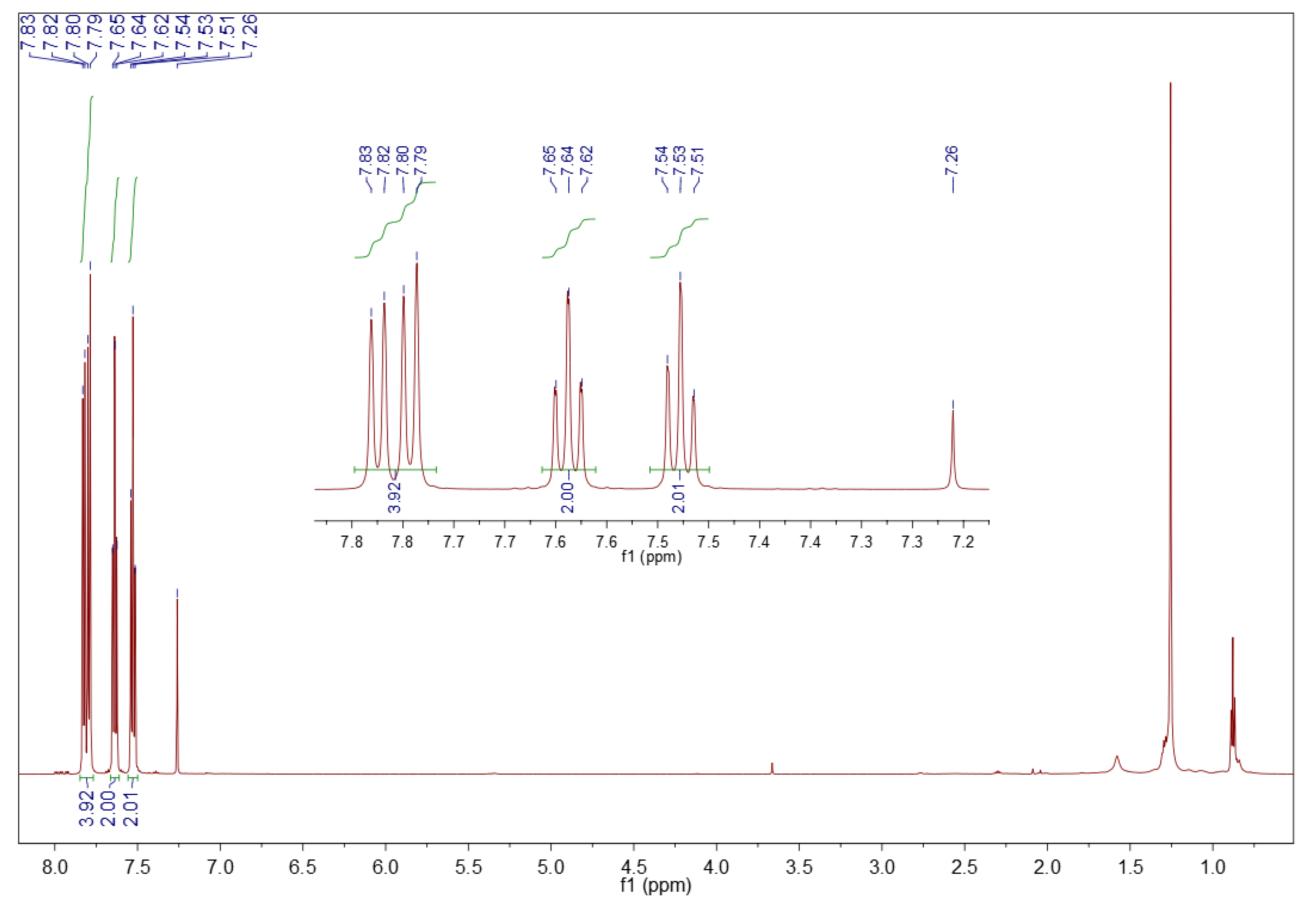
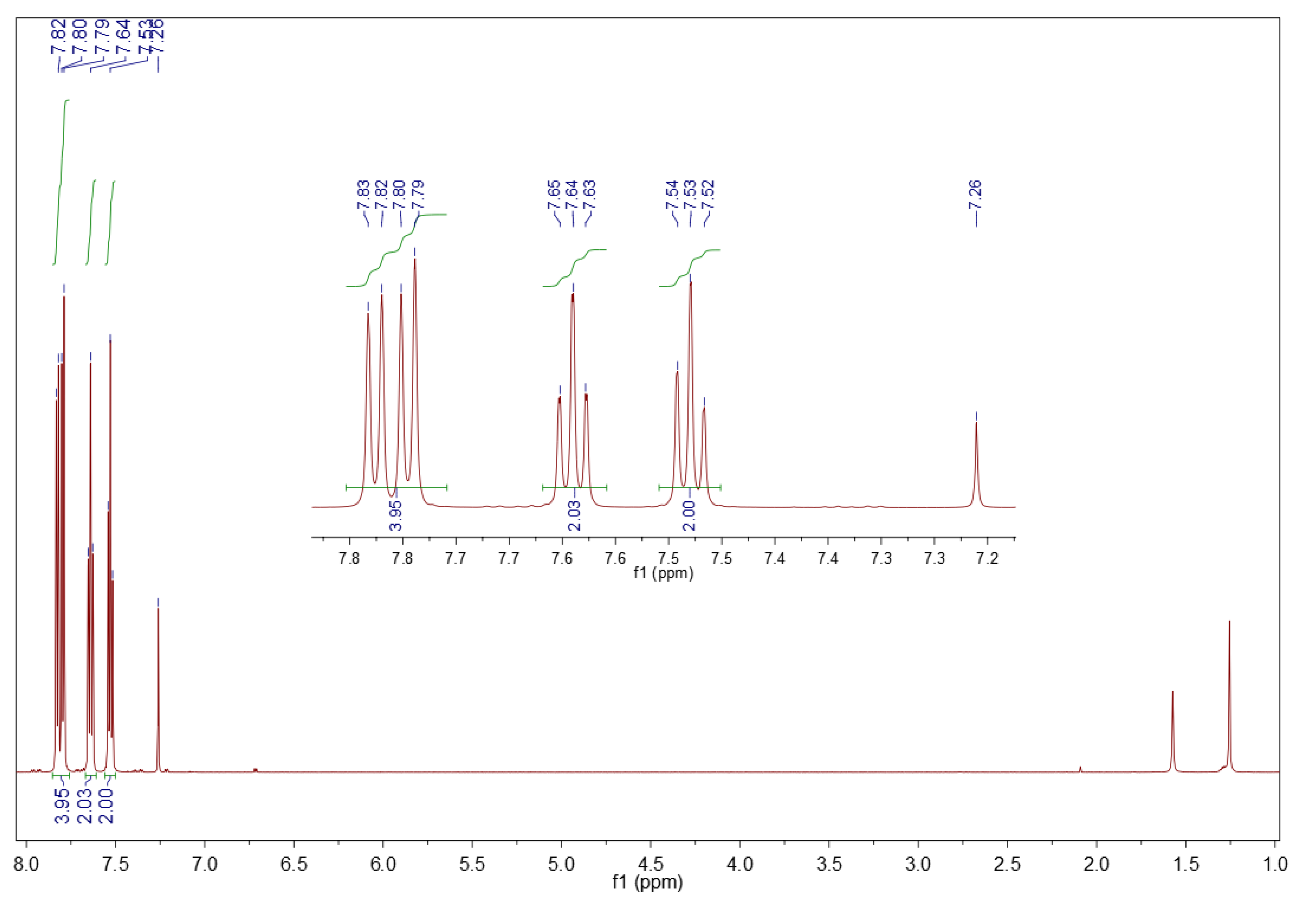
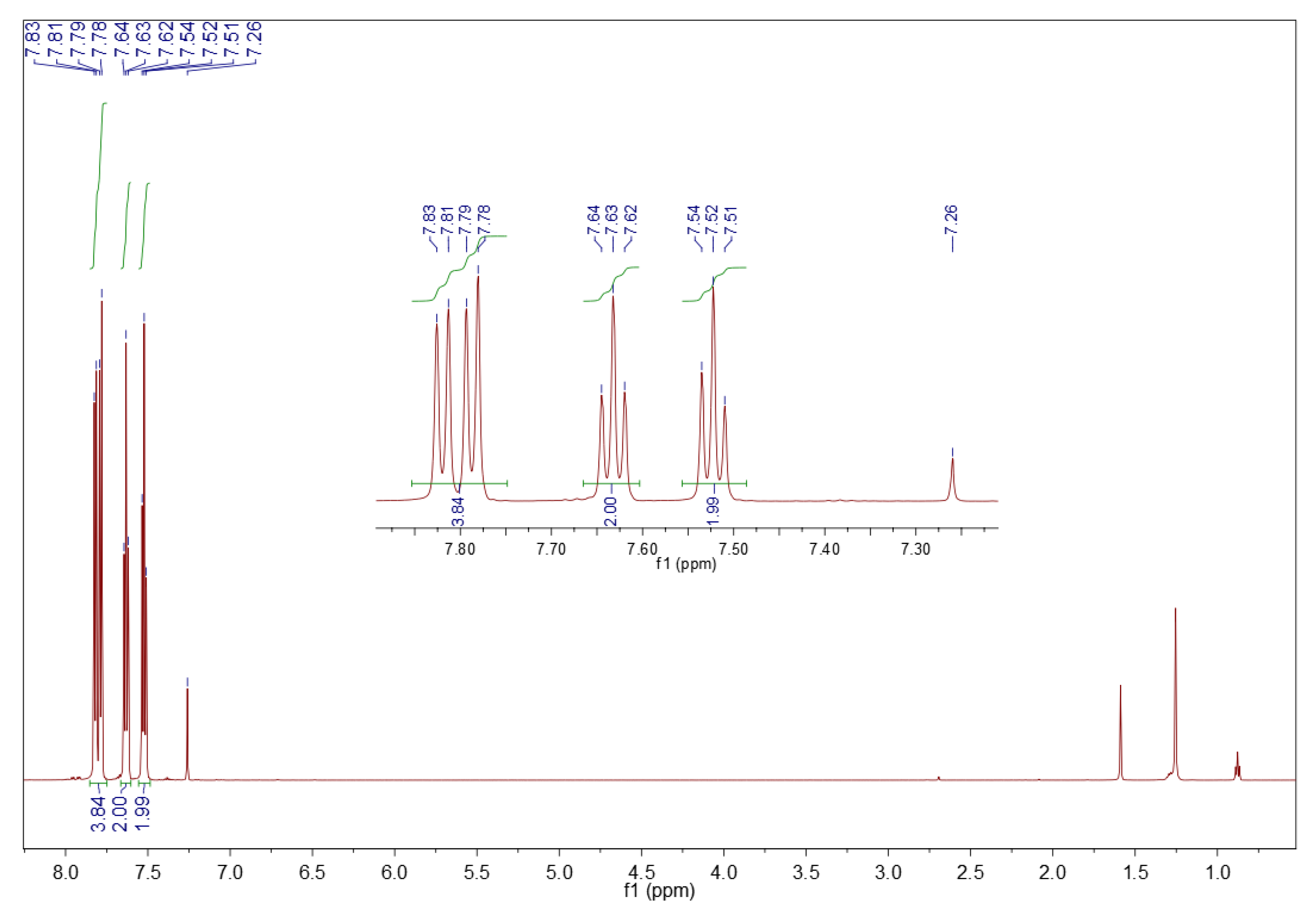
3.5.2. 1H NMR Spectroscopy
3.6. Desulfurization of the Oxidized Surrogate Blends Using Recycled Ionic Liquids
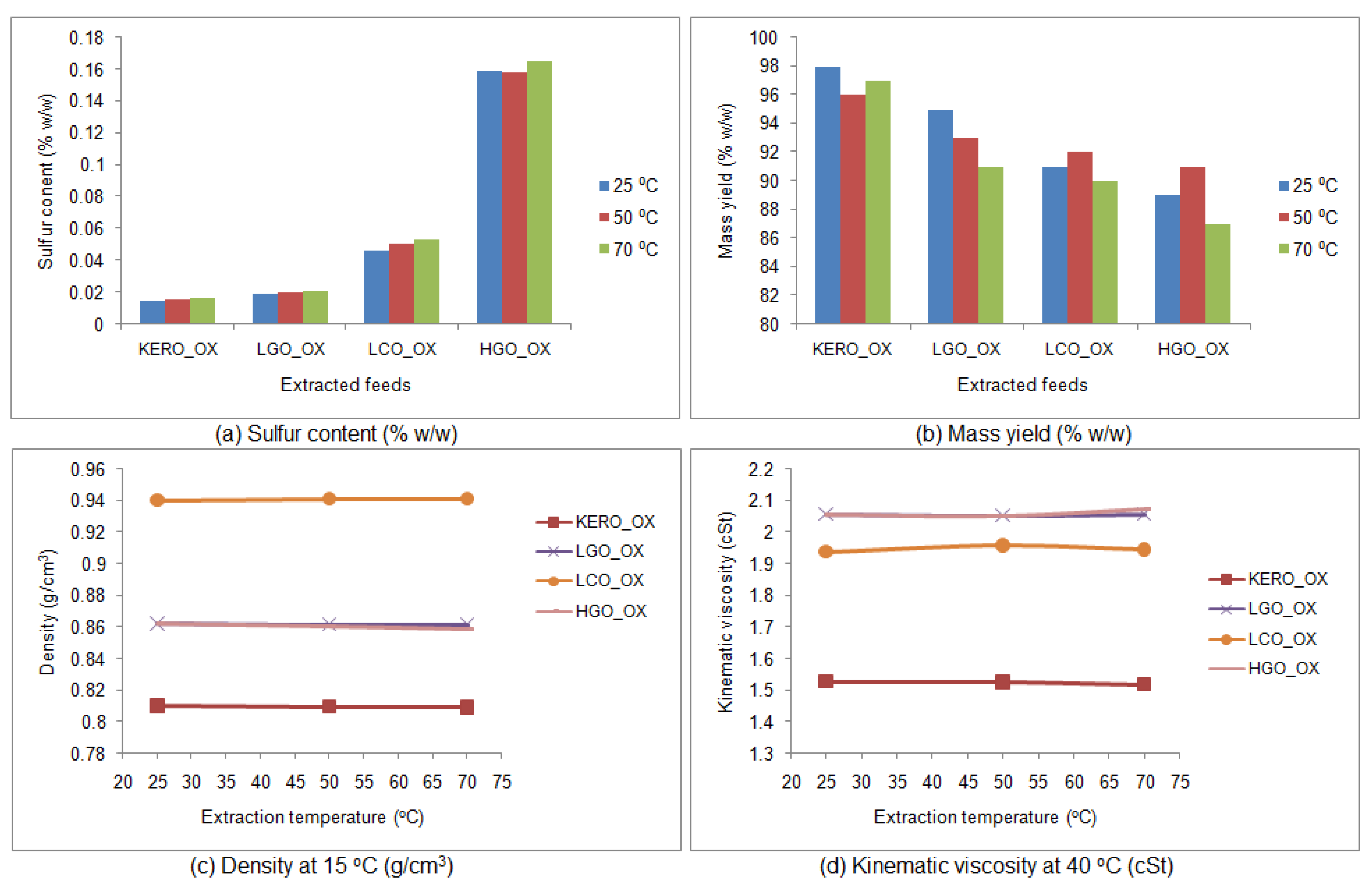
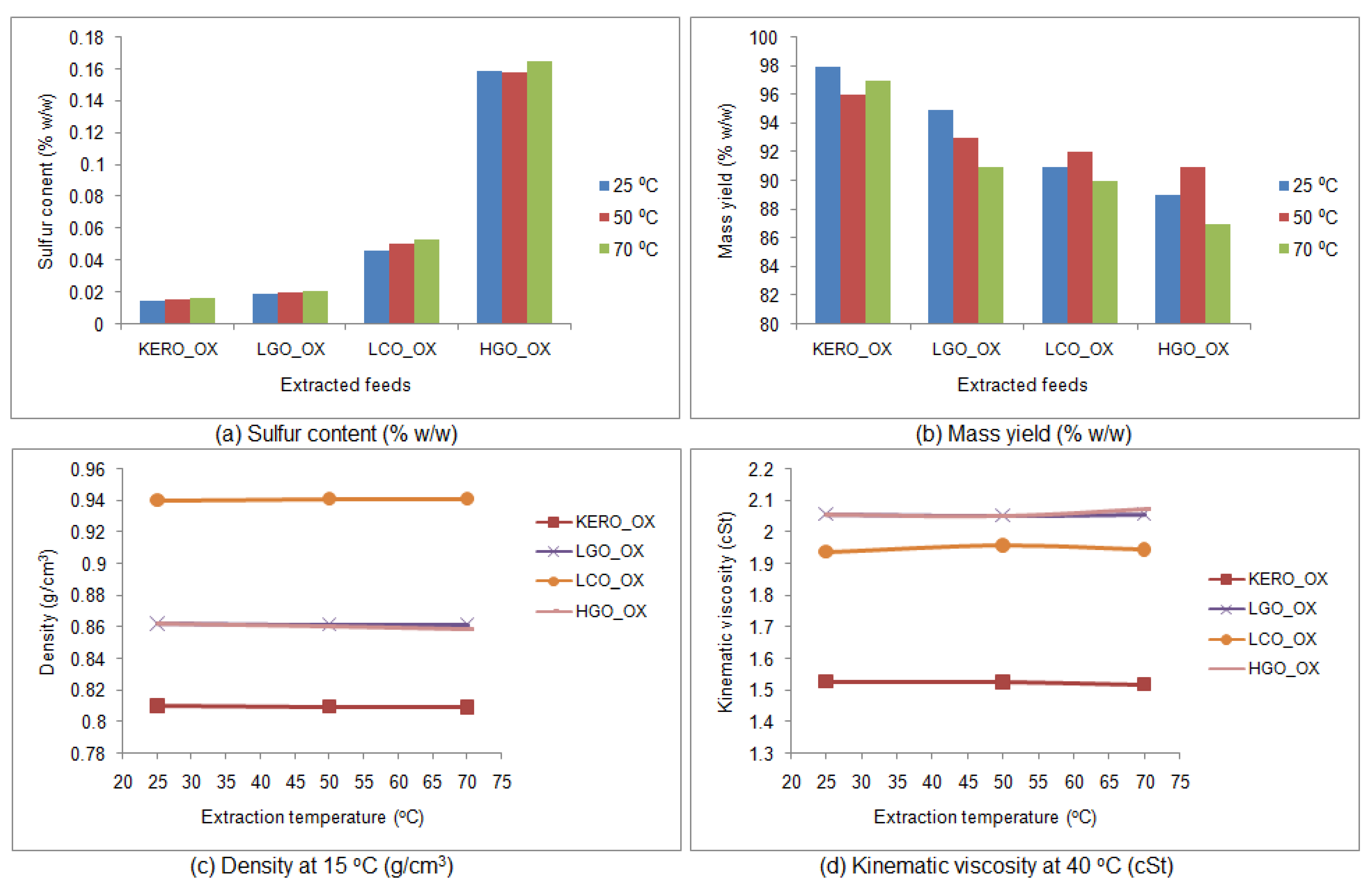
3.6.1. Properties of the Raffinate Surrogate Products
3.6.2. Liquid FT-IR Spectroscopy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Babich, I.V.; Moulijn, J.A. Science and Technology of Novel Processes for Deep Desulfurization of Oil Refinery Streams: A Review. Fuel 2003, 82, 607–631. [Google Scholar] [CrossRef]
- Ito, E.; van Veen, J.A.R. On Novel Processes for Removing Sulfur from Refinery Stream. Catal. Today 2006, 116, 446–460. [Google Scholar] [CrossRef]
- Saleh, T.A. Characterization, Determination and Elimination Technologies for Sulfur from Petroleum: Toward Cleaner Fuel and a Safe Environment. Trends Environ. Anal. Chem. 2020, 25, e00080. [Google Scholar] [CrossRef]
- Kumar, S.; Srivastava, V.C.; Nanoti, S.M. Extractive Desulfurization of Gas Oils: A Perspective Review for Use in Petroleum Refineries. Sep. Purif. Rev. 2017, 46, 319–347. [Google Scholar] [CrossRef]
- Dehghan, R.; Anbia, M. Zeolites for Adsorptive Desulfurization from Fuels: A Review. Fuel Process. Technol. 2017, 167, 99–116. [Google Scholar] [CrossRef]
- Wei, S.; He, H.; Cheng, Y.; Yang, C.; Zeng, G.; Qiu, L. Performances, Kinetics and Mechanisms of Catalytic Oxidative Desulfurization from Oils. RSC Adv. 2016, 6, 103253–103269. [Google Scholar] [CrossRef]
- Boniek, D.; Figueiredo, D.; Dos Santos, A.F.B.; Stoianoff, M.A.D.R. Biodesulfurization: A Mini Review about the Immediate Search for the Future Technology. Clean Technol. Environ. Policy 2015, 17, 29–37. [Google Scholar] [CrossRef]
- Mohebali, G.; Ball, A.S. Biodesulfurization of Diesel Fuels: Past, Present and Future Perspectives. Int. Biodeterior. Biodegrad. 2016, 110, 163–180. [Google Scholar] [CrossRef]
- Heimlich, B.N.; Wallace, T.J. Kinetics and Mechanism of the Oxidation of Dibenzothiophene in Hydrocarbon Solution. Tetrahedron 1966, 22, 3571–3579. [Google Scholar] [CrossRef]
- Aida, T.; Yamamoto, D. Oxidative desulfurization of liquid fuels. In Proceedings of the American Chemical Society (ACS) National Meeting, Washington, DC, USA, 21–26 August 1994; Volume 39, pp. 623–626. [Google Scholar]
- Zannikos, F.; Lois, E.; Stournas, S. Desulfurization of Petroleum Fractions by Oxidation and Solvent Extraction. Fuel Process. Technol. 1995, 42, 35–45. [Google Scholar] [CrossRef]
- Otsuki, S.; Nonaka, T.; Takashima, N.; Qian, W.; Ishihara, A.; Imai, A.T.; Kabe, T. Oxidative Desulfurization of Light Gas Oil and Vacuum Gas Oil by Oxidation and Solvent Extraction. Energy Fuels 2000, 14, 1232–1239. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Tachibana, K.; Hirai, T.; Komasawa, I. Desulfurization and Denitrogenation Process for Light Oils Based on Chemical Oxidation Followed by Liquid-Liquid Extraction. Ind. Eng. Chem. Res. 2002, 41, 4362–4375. [Google Scholar] [CrossRef]
- De Filippis, P.; Scarsella, M. Oxidative Desulfurization: Oxidation Reactivity of Sulfur Compounds in Different Organic Matrixes. Energy Fuels 2003, 17, 1452–1455. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Hirai, T. Desulfurization of Vacum Gas Based on Chemical Oxidation Followed by Liquid-Liquid Extraction. Energy Fuels 2004, 18, 37–40. [Google Scholar] [CrossRef]
- Ali, M.F.; Al-Malki, A.; El-Ali, B.; Martinie, G.; Siddiqui, M.N. Deep Desulfurization of Gasoline and Diesel Fuels Using Non-Hydrogen Consuming Techniques. Fuel 2006, 85, 1354–1363. [Google Scholar] [CrossRef]
- Dehkordi, A.M.; Sobati, M.A.; Nazem, M.A. Oxidative Desulfurization of Non-hydrotreated Kerosene Using Hydrogen Peroxide and Acetic Acid. Chin. J. Chem. Eng. 2009, 17, 869–874. [Google Scholar] [CrossRef]
- Ahmedzeki, N.S.; Ibrahem, B.J. Reduction of Sulfur Compounds from Petroleum Fraction Using Oxidation-Adsorption Technique. Iraqi J. Chem. Pet. Eng. 2015, 16, 35–48. [Google Scholar]
- Rakhmanov, E.V.; Domashkin, A.A.; Myltykbaeva, Z.K.; Kairbekov, Z.; Shigapova, A.A.; Akopyan, A.V.; Anisimov, A.V. Peroxide Oxidative Desulfurization of a Mixture of Nonhydrotreated Vacuum Gas Oil and Diesel Fraction. Pet. Chem. 2016, 56, 742–744. [Google Scholar] [CrossRef]
- Karonis, D.; Syntyhaki, E. Oxidative desulfurization of petroleum middle distillates using hydrogen peroxide/acetic acid and solvent extraction. In Proceedings of the 12th International Colloquium, Fuels—Conventional and Future Energy for Automobiles, Stuttgart, Germany, 25–26 June 2019; pp. 157–166. [Google Scholar]
- Wang, D.; Qian, E.W.; Amano, H.; Okata, K.; Ishihara, A.; Kabe, T. Oxidative desulfurization of fuel oil Part I. Oxidation of dibenzothiophenes using tert-butyl hydroperoxide. Appl. Catal. A Gen. 2003, 253, 91–99. [Google Scholar] [CrossRef]
- Ishihara, A.; Wang, D.; Dumeignil, F.; Amano, H.; Qian, E.W.; Kabe, T. Oxidative desulfurization and denitrogenation of a light gas oil using an oxidation/adsorption continuous flow process. Appl. Catal. A Gen. 2005, 279, 279–287. [Google Scholar] [CrossRef]
- Chica, A.; Corma, A.; Dómine, M.E. Catalytic oxidative desulfurization (ODS) of diesel fuel on a continuous fixed-bed reactor. J. Catal. 2006, 242, 299–308. [Google Scholar] [CrossRef]
- Bhadra, B.N.; Jhung, S.H. Oxidative Desulfurization and Denitrogenation of Fuels Using Metal-Organic Framework-Based/-Derived Catalysts. Appl. Catal. B Environ. 2019, 259, 118021–118045. [Google Scholar] [CrossRef]
- Juliao, D.; Mirante, F.; Ribeiro, S.O.; Gomes, A.C.; Valenca, R.; Ribeiro, J.C.; Pillinger, M.; de Castro, B.; Goncalves, I.S.; Balula, S.S. Deep Oxidative Desulfurization of Diesel Fuels Using Homogeneous and SBA-15-Supported Peroxophosphotungstate Catalysts. Fuel 2019, 241, 616–624. [Google Scholar] [CrossRef]
- Duarte, F.A.; Mello, P.D.A.; Bizzi, C.A.; Nunes, M.A.; Moreira, E.M.; Alencar, M.S.; Motta, H.N.; Dressler, V.L.; Flores, É.M. Sulfur Removal from Hydrotreated Petroleum Fractions Using Ultrasound-Assisted Oxidative Desulfurization Process. Fuel 2011, 90, 2158–2164. [Google Scholar] [CrossRef] [Green Version]
- Ja’fari, M.; Ebrahimi, S.L.; Khosravi-Nikou, M.R. Ultrasound-Assisted Oxidative Desulfurization and Denitrogenation of Liquid Hydrocarbon Fuels: A Critical Review. Ultrason. Sonochem. 2018, 40, 955–968. [Google Scholar] [CrossRef] [PubMed]
- More, N.S.; Gogate, P.R. Intensified Approach for Desulfurization of Simulated Fuel Containing Thiophene Based on Ultrasonic Flow Cell and Oxidizing Agents. Ultrason. Sonochem. 2019, 51, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Ramalingam, A. Desulfurization and Denitrification of Diesel Oil Using Ionic Liquids. Experiments and Quantum Chemical Predictions; Elsevier: Amsterdam, The Netherlands, 2015; p. 342. [Google Scholar]
- Zhao, H.; Xia, S.; Ma, P. Use of ionic liquids as ‘green’ solvents for extractions. Review. J. Chem. Technol. Biotechnol. 2005, 80, 1089–1096. [Google Scholar] [CrossRef]
- Zhao, H.; Baker, G.A. Oxidative desulfurization of fuels using ionic liquids: A review. Front. Chem. Sci. Eng. 2015, 9, 262–279. [Google Scholar] [CrossRef]
- Ibrahim, M.H.; Hayyan, M.; Hashim, M.A.; Hayyan, A. The role of ionic liquids in desulfurization of fuels: A review. Renew. Sustain. Energy Rev. 2017, 76, 1534–1549. [Google Scholar] [CrossRef]
- Gao, S.; Li, J.; Chen, X.; Abdeltawab, A.A.; Yakout, S.M.; Yu, G. A combination desulfurization method for diesel fuel: Oxidation by ionic liquid with extraction by solvent. Fuel 2018, 224, 545–551. [Google Scholar] [CrossRef]
- Zhao, D.; Sun, Z.; Li, F. Optimization of oxidative desulfurization of dibenzothiophene using acidic ionic liquid as catalytic solvent. J. Fuel Chem. Technol. 2009, 37, 194–198. [Google Scholar] [CrossRef]
- Gao, H.; Guo, C.; Xing, J.; Zhao, J.; Liu, H. Extraction and oxidative desulfurization of diesel fuel catalyzed by a Brønsted acidic ionic liquid at room temperature. Green Chem. 2010, 12, 1220–1224. [Google Scholar] [CrossRef]
- Chen, X.; Song, D.; Asumana, C.; Yu, G. Deep oxidative desulfurization of diesel fuels by Lewis acidic ionic liquids based on 1-n-butyl-3-methylimidazolium metal chloride. J. Mol. Catal. A Chem. 2012, 359, 8–13. [Google Scholar] [CrossRef]
- Nie, Y.; Dong, Y.; Bai, L.; Dong, H.; Zhang, X. Fast oxidative desulfurization of fuel oil using dialkylpyridinium. Fuel 2013, 103, 997–1002. [Google Scholar] [CrossRef]
- Chen, X.; Guan, Y.; Abdeltawab, A.A.; Al-Deyab, S.S.; Yuan, X.; Wang, C.; Yu, G. Using functional acidic ionic liquids as both extractant and catalyst in oxidative desulfurization of diesel fuel: An investigation of real feedstock. Fuel 2015, 146, 6–12. [Google Scholar] [CrossRef]
- Chen, X.; Guo, H.; Abdeltawab, A.A.; Guan, Y.; Al-Deyab, S.S.; Yu, G.; Yu, L. Brønsted–Lewis Acidic Ionic Liquids and Application in Oxidative Desulfurization of Diesel Fuel. Energy Fuels 2015, 29, 2998–3003. [Google Scholar] [CrossRef]
- Bösmann, A.; Datsevich, L.; Jess, A.; Lauter, A.; Schmitz, C.; Wasserscheid, P. Deep desulfurization of diesel fuel by extraction with ionic liquids. Chem. Comm. 2001, 23, 2494–2495. [Google Scholar] [CrossRef]
- Eßer, J.; Wasserscheid, P.; Jess, A. Deep desulfurization of oil refinery streams by extraction with ionic liquids. Green Chem. 2004, 6, 316–322. [Google Scholar] [CrossRef]
- Mai, N.L.; Ahn, K.; Koo, Y.-M. Methods for recovery of ionic liquids—A review. Process Biochem. 2014, 49, 872–881. [Google Scholar] [CrossRef]
- Kuzmina, O.; Hallett, J.P. (Eds.) Application, Purification, and Recovery of Ionic Liquids; Elsevier: Amsterdam, The Netherlands, 2016; p. 286. [Google Scholar]
- Sarathy, S.M.; Farooq, A.; Kalghatgi, G.T. Recent progress in gasoline surrogate fuels. Prog. Energy Combust. Sci. 2018, 65, 67–108. [Google Scholar] [CrossRef]
- Syntyhaki, E.; Karonis, D. Oxidative and extractive desulfurization of petroleum middle distillates, using imidazole ionic liquids. Fuel Comm. 2021, 7, 100011. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Tsanas, C.; Tzani, A.; Papadopoulos, A.; Detsi, A.; Voutsas, E. Ionic liquids as entrainers for the separation of the ethanol/water system. Fluid Phase Equilibria 2014, 379, 148–156. [Google Scholar] [CrossRef]
- Dharaskar, S.A.; Wasewar, K.L.; Varma, M.N.; Shende, D.Z. Imidazolium ionic liquid as energy efficient solvent for desulfurization of liquid fuel. Sep. Purif. Technol. 2015, 155, 101–109. [Google Scholar] [CrossRef]
- Tajika, H.; Niknam, K.; Parsa, F. Using Acidic Ionic Liquid 1-Butyl-3-methylimidazolium Hydrogen Sulfate in Selective Nitration of Phenols under Mild Conditions. J. Iran. Chem. Soc. 2009, 6, 159–164. [Google Scholar] [CrossRef]
- Singh, V.; Kaur, S.; Sapehiyia, V.; Singh, J.; Kad, G. Microwave accelerated preparation of [bmim][HSO4] ionic liquid: An acid catalyst for improved synthesis of coumarins. Catal. Commun. 2005, 6, 57–60. [Google Scholar] [CrossRef]
- Le Bui, T.T.; Nguyen, D.D.; Van Ho, S.; Nguyen, B.T.; Uong, H.T.N. Synthesis, characterization and application of some non-halogen ionic liquids as green solvents for deep desulfurization of diesel oil. Fuel 2017, 191, 54–61. [Google Scholar] [CrossRef]
- Seddon, K.R.; Stark, A.; Torre, M.-J. Viscosity and Density of 1-Alkyl-3-methylimidazolium Ionic Liquids. In Clean Solvents; (ACS Symposium Series); American Chemical Society: Washington, DC, USA, 2002; Volume 819, pp. 34–49. [Google Scholar]
- Tshibangu, P.N.; Ndwandwe, S.N.; Dikio, E.D. Density, Viscosity and Conductivity Study of 1-Butyl-3-Methylimidazolium Bromide. Int. J. Electrochem. Sci. 2011, 6, 2201–2213. [Google Scholar]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; John Wiley & Sons: West Essex, UK, 2000. [Google Scholar]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Spectrometric Identification of Organic Compounds, 7th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- De Filippis, P.; Scarsella, M.; Verdone, N. Oxidative Desulfurization I: Peroxyformic Acid Oxidation of Benzothiophene and Dibenzothiophene. Ind. Eng. Chem. Res. 2010, 49, 4594–4600. [Google Scholar] [CrossRef]
- Syntyhaki, E.; Karonis, D. Evaluation of Oxidative Desulfurization and Solvent Extraction of Model Sulfur Compounds, Present in Petroleum Middle Distillates, with Infrared and Nuclear Magnetic Resonance Spectroscopy. Anal. Lett. 2020, 54, 1470–1495. [Google Scholar] [CrossRef]
- Brown, K.N.; Espenson, J.H. Stepwise Oxidation of Thiophene and Its Derivatives by Hydrogen Peroxide Catalyzed by Methyltrioxorhenium(VII). Inorg. Chem. 1996, 35, 7211–7216. [Google Scholar] [CrossRef] [PubMed]
- Geneste, P.; Olive, J.L.; Ung, S.N.; Faghi, M.E.A.E.; Easton, J.W.; Beierbeck, H.; Saunders, J.K. Carbon-13 Nuclear Magnetic Resonance Study of Benzo[b]thiophenes and Benzo[b]thiophene S-Oxides and S,S-Dioxides. J. Org. Chem. 1979, 44, 2887–2892. [Google Scholar] [CrossRef]
- Thiemann, T.; Arima, K.; Kumazoe, K.; Mataka, S. Benzothiophene-S-Oxides—An Overview. Rep. Inst. Adv. Mater. Study 2000, 14, 139–142. [Google Scholar]
- Madec, D.; Mingoia, F.; Macovei, C.; Maitro, G.; Giambastiani, G.; Poli, G. New Enantiopure Bis(thioether) and Bis(sulfoxide) Ligands from Benzothiophene. Eur. J. Org. Chem. 2005, 2005, 552–557. [Google Scholar] [CrossRef]
- Yu, B.; Liu, A.-H.; He, L.-N.; Li, B.; Diao, Z.-F.; Li, Y.-N. Catalyst-free approach for solvent-dependent selective oxidation of organic sulfides with oxone. Green Chem. 2012, 14, 957–962. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | KERO | LGO | LCO | HGO |
|---|---|---|---|---|
| Sulfur content, % w/w | 0.206 | 0.781 | 0.470 | 1.272 |
| Density, g/cm3 (15 °C) | 0.7929 | 0.8350 | 0.9631 | 0.8637 |
| Kinematic viscosity, cSt (40 °C) | 1.117 | 2.592 | 4.088 | 6.001 |
| Freezing point, °C | −56.4 | − | − | − |
| Cloud point, °C | − | −16 | − | +6 |
| Pour point, °C | − | −22 | −10 | +8 |
| CFPP, °C | − | −16 | −4 | +7 |
| Mono-aromatics, % w/w | 15.4 | 14.4 | 20.1 | 14.4 |
| Di-aromatics, % w/w | 1.4 | 8.6 | 36.4 | 11.9 |
| Tri-aromatics, % w/w | − | 0.3 | 12.4 | 1.6 |
| Poly-aromatics, % w/w | − | 8.9 | 48.8 | 13.5 |
| Total aromatics, % w/w | 16.8 | 23.3 | 68.9 | 27.9 |
| Property | KERO | LGO | LCO | HGO |
|---|---|---|---|---|
| S content, % w/w | 0.207 | 0.787 | 0.471 | 1.301 |
| Density, g/cm3 (15 °C) | 0.8124 | 0.8743 | 0.9503 | 0.8831 |
| Kinematic viscosity, cSt (40 °C) | 1.512 | 1.999 | 1.955 | 2.026 |
| Property | KERO_OX | LGO_OX | LCO_OX | HGO_OX |
|---|---|---|---|---|
| S content, % w/w | 0.024 | 0.054 | 0.151 | 0.224 |
| Density, g/cm3 (15 °C) | 0.8108 | 0.8675 | 0.9479 | 0.8682 |
| Kinematic viscosity, cSt (40 °C) | 1.512 | 2.009 | 1.961 | 2.027 |
| Property | [BMIM][Br] | [BMIM][HSO4] | [BMIM][Br]recycled | [BMIM][HSO4]recycled |
|---|---|---|---|---|
| Sulfur content, % w/w | 0.005 | 7.420 | 0.009 | 7.409 |
| Density, g/cm3 (40 °C) | 1.2943 | 1.2341 | 1.2937 | 1.2449 |
| Density, g/cm3 (100 °C) | 1.2545 | 1.1978 | 1.2539 | 1.2090 |
| Dynamic viscosity, cP (40 °C) | 894.71 | 407.49 | 883.63 | 660.50 |
| Dynamic viscosity, cP (100 °C) | 34,394 | 34,455 | 34,346 | 47,011 |
| Kinematic viscosity, cSt (40 °C) | 691.29 | 330.19 | 683.05 | 530.58 |
| Kinematic viscosity, cSt (100 °C) | 27.416 | 28.766 | 27.392 | 38.885 |
| Surrogate Feeds | Oxidation Temperature | Solvent to Feed Ratio | Acetonitrile | Methanol | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mass Yield, % w/w | Sulfur Content, % w/w | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Mass Yield, % w/w | Sulfur Content, % w/w | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | |||
| KERO | 70 °C | 1.0 | 84 | 0.010 | 0.8033 | 1.516 | 84 | 0.011 | 0.8044 | 1.519 |
| 1.5 | 80 | 0.008 | 0.8008 | 1.520 | 74 | 0.011 | 0.8022 | 1.536 | ||
| 2.0 | 77 | 0.007 | 0.7986 | 1.524 | 64 | 0.010 | 0.7997 | 1.535 | ||
| LGO | 70 °C | 1.0 | 76 | 0.009 | 0.8467 | 2.144 | 76 | 0.013 | 0.8490 | 2.019 |
| 1.5 | 69 | 0.008 | 0.8411 | 2.195 | 69 | 0.010 | 0.8446 | 2.090 | ||
| 2.0 | 67 | 0.007 | 0.8375 | 2.229 | 64 | 0.010 | 0.8402 | 2.109 | ||
| LCO | 90 °C | 1.0 | 31 | 0.034 | 0.8821 | 1.592 | 56 | 0.066 | 0.9198 | 1.720 |
| 2.0 | 23 | 0.019 | 0.8579 | 1.873 | 28 | 0.039 | 0.8978 | 1.917 | ||
| HGO | 90 °C | 1.0 | 73 | 0.080 | 0.8451 | 2.014 | 79 | 0.119 | 0.8527 | 2.134 |
| 1.5 | 67 | 0.063 | 0.8393 | 2.083 | 74 | 0.099 | 0.8481 | 2.100 | ||
| 2.0 | 63 | 0.053 | 0.8358 | 2.099 | 70 | 0.086 | 0.8436 | 2.153 | ||
| Extraction Cycles | KERO | LGO | LCO | HGO | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | |
| 1st | 0.013 | 94 | 96 | 0.015 | 98 | 93 | 0.034 | 93 | 86 | 0.108 | 92 | 90 |
| 2nd | 0.011 | 95 | 97 | 0.013 | 98 | 93 | 0.021 | 96 | 89 | 0.062 | 95 | 90 |
| 3rd | 0.012 | 94 | 97 | 0.014 | 98 | 95 | 0.016 | 97 | 90 | 0.044 | 97 | 93 |
| 4th | − | − | − | − | − | − | 0.016 | 97 | 91 | 0.035 | 97 | 94 |
| 5th | − | − | − | − | − | − | − | − | − | 0.031 | 98 | 95 |
| 6th | − | − | − | − | − | − | − | − | − | 0.031 | 98 | 94 |
| Extraction Cycles | KERO | LGO | LCO | HGO | ||||
|---|---|---|---|---|---|---|---|---|
| Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | |
| 1st | 0.8094 | 1.532 | 0.8602 | 2.051 | 0.9354 | 1.935 | 0.8597 | 2.107 |
| 2nd | 0.8084 | 1.537 | 0.8551 | 2.114 | 0.9260 | 1.945 | 0.8521 | 2.141 |
| 3rd | 0.8073 | 1.538 | 0.8515 | 2.153 | 0.9180 | 1.964 | 0.8483 | 2.186 |
| 4th | − | − | − | − | 0.9109 | 2.004 | 0.8451 | 2.234 |
| 5th | − | − | − | − | − | − | 0.8423 | 2.269 |
| 6th | − | − | − | − | − | − | 0.8398 | 2.311 |
| Extraction Cycles | KERO | LGO | LCO | HGO | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | |
| 1st | 0.015 | 93 | 98 | 0.019 | 98 | 95 | 0.046 | 90 | 91 | 0.159 | 88 | 89 |
| 2nd | 0.013 | 94 | 98 | 0.016 | 98 | 96 | 0.046 | 90 | 90 | 0.109 | 92 | 94 |
| 3rd | 0.013 | 94 | 98 | 0.017 | 98 | 96 | 0.028 | 94 | 92 | 0.077 | 94 | 95 |
| 4th | − | − | − | − | − | − | 0.026 | 94 | 92 | 0.062 | 95 | 95 |
| 5th | − | − | − | − | − | − | 0.026 | 94 | 90 | 0.052 | 96 | 96 |
| 6th | − | − | − | − | − | − | − | − | − | 0.044 | 97 | 97 |
| Extraction Cycles | KERO | LGO | LCO | HGO | ||||
|---|---|---|---|---|---|---|---|---|
| Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | |
| 1st | 0.8098 | 1.525 | 0.8619 | 2.056 | 0.9400 | 1.938 | 0.8622 | 2.053 |
| 2nd | 0.8088 | 1.537 | 0.8578 | 2.089 | 0.9340 | 1.942 | 0.8568 | 2.107 |
| 3rd | 0.8078 | 1.536 | 0.8545 | 2.118 | 0.9285 | 1.946 | 0.8529 | 2.141 |
| 4th | − | − | − | − | 0.9223 | 1.971 | 0.8497 | 2.201 |
| 5th | − | − | − | − | 0.9162 | 2.001 | 0.8471 | 2.226 |
| 6th | − | − | − | − | − | − | 0.8448 | 2.264 |
| Extraction Cycles | KERO | LGO | LCO | HGO | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | |
| 1st | 0.014 | 93 | 98 | 0.018 | 98 | 94 | 0.038 | 92 | 86 | 0.106 | 92 | 93 |
| 2nd | 0.013 | 94 | 97 | 0.015 | 98 | 95 | 0.025 | 95 | 88 | 0.062 | 95 | 94 |
| 3rd | 0.012 | 94 | 97 | 0.017 | 98 | 86 | 0.020 | 96 | 89 | 0.043 | 97 | 97 |
| 4th | 0.013 | 94 | 98 | − | − | − | 0.019 | 96 | 91 | 0.035 | 97 | 96 |
| 5th | − | − | − | − | − | − | 0.020 | 96 | 90 | 0.031 | 98 | 96 |
| 6th | − | − | − | − | − | − | − | − | − | 0.030 | 98 | 96 |
| Extraction Cycles | KERO | LGO | LCO | HGO | ||||
|---|---|---|---|---|---|---|---|---|
| Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | |
| 1st | 0.8095 | 1.517 | 0.8605 | 2.057 | 0.9359 | 1.929 | 0.8599 | 2.067 |
| 2nd | 0.8084 | 1.525 | 0.8552 | 2.106 | 0.9263 | 1.944 | 0.8545 | 2.129 |
| 3rd | 0.8068 | 1.538 | 0.8490 | 2.200 | 0.9173 | 1.967 | 0.8505 | 2.176 |
| 4th | 0.8057 | 1.546 | − | − | 0.9086 | 2.004 | 0.8476 | 2.195 |
| 5th | − | − | − | − | 0.9013 | 2.062 | 0.8452 | 2.235 |
| 6th | − | − | − | − | − | − | 0.8429 | 2.281 |
| Extraction Cycles | KERO | LGO | LCO | HGO | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | Sulfur Content, % w/w | Desulf. Yield, % w/w | Mass Yield, % w/w | |
| 1st | 0.016 | 92 | 98 | 0.022 | 97 | 96 | 0.053 | 89 | 93 | 0.141 | 89 | 95 |
| 2nd | 0.016 | 92 | 98 | 0.018 | 98 | 96 | 0.036 | 92 | 93 | 0.099 | 92 | 96 |
| 3rd | 0.017 | 92 | 99 | 0.018 | 98 | 96 | 0.032 | 93 | 93 | 0.080 | 94 | 97 |
| 4th | − | − | − | − | − | − | 0.030 | 94 | 93 | 0.065 | 95 | 96 |
| 5th | − | − | − | − | − | − | 0.031 | 93 | 94 | 0.054 | 96 | 96 |
| 6th | − | − | − | − | − | − | − | − | − | 0.050 | 96 | 95 |
| Extraction Cycles | KERO | LGO | LCO | HGO | ||||
|---|---|---|---|---|---|---|---|---|
| Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | Density, g/cm3 (15 °C) | Kinematic Viscosity, cSt (40 °C) | |
| 1st | 0.8101 | 1.574 | 0.8633 | 2.100 | 0.9417 | 1.980 | 0.8634 | 2.089 |
| 2nd | 0.8094 | 1.524 | 0.8597 | 2.065 | 0.9369 | 1.934 | 0.8596 | 2.081 |
| 3rd | 0.8087 | 1.534 | 0.8565 | 2.102 | 0.9324 | 1.952 | 0.8564 | 2.114 |
| 4th | − | − | − | − | 0.9280 | 1.963 | 0.8535 | 2.158 |
| 5th | − | − | − | − | 0.9231 | 1.995 | 0.8509 | 2.201 |
| 6th | − | − | − | − | − | − | 0.8485 | 2.235 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syntyhaki, E.; Karonis, D. Synthesis of Surrogate Blends Corresponding to Petroleum Middle Distillates, Oxidative and Extractive Desulfurization Using Imidazole Ionic Liquids. Fuels 2022, 3, 44-74. https://doi.org/10.3390/fuels3010004
Syntyhaki E, Karonis D. Synthesis of Surrogate Blends Corresponding to Petroleum Middle Distillates, Oxidative and Extractive Desulfurization Using Imidazole Ionic Liquids. Fuels. 2022; 3(1):44-74. https://doi.org/10.3390/fuels3010004
Chicago/Turabian StyleSyntyhaki, Eleni, and Dimitrios Karonis. 2022. "Synthesis of Surrogate Blends Corresponding to Petroleum Middle Distillates, Oxidative and Extractive Desulfurization Using Imidazole Ionic Liquids" Fuels 3, no. 1: 44-74. https://doi.org/10.3390/fuels3010004
APA StyleSyntyhaki, E., & Karonis, D. (2022). Synthesis of Surrogate Blends Corresponding to Petroleum Middle Distillates, Oxidative and Extractive Desulfurization Using Imidazole Ionic Liquids. Fuels, 3(1), 44-74. https://doi.org/10.3390/fuels3010004