
Assessing NO2-Hydrocarbon Interactions during Combustion of NO2/Alkane/Ar Mixtures in a Shock Tube Using CO Time Histories



Abstract
:1. Introduction
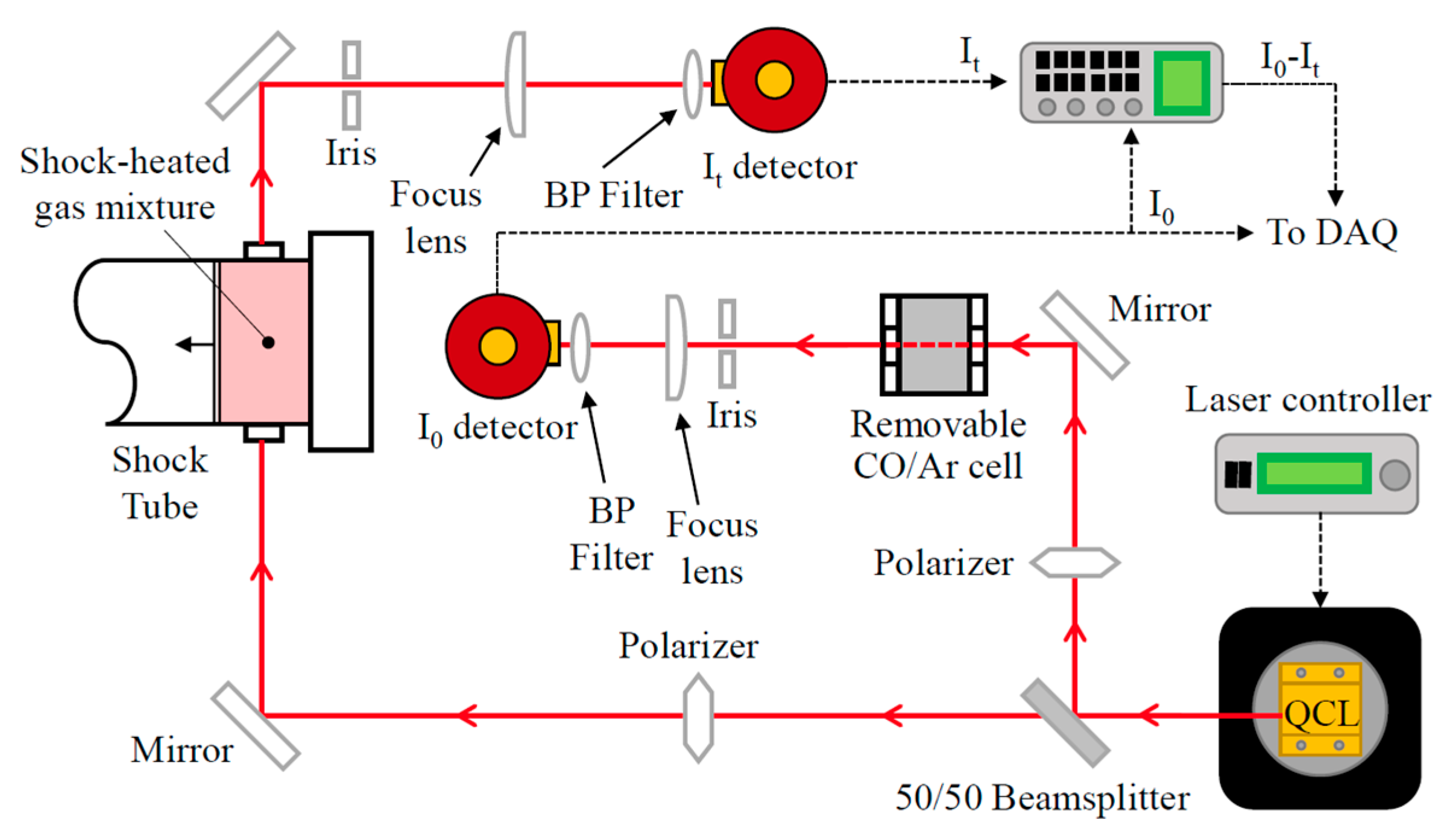
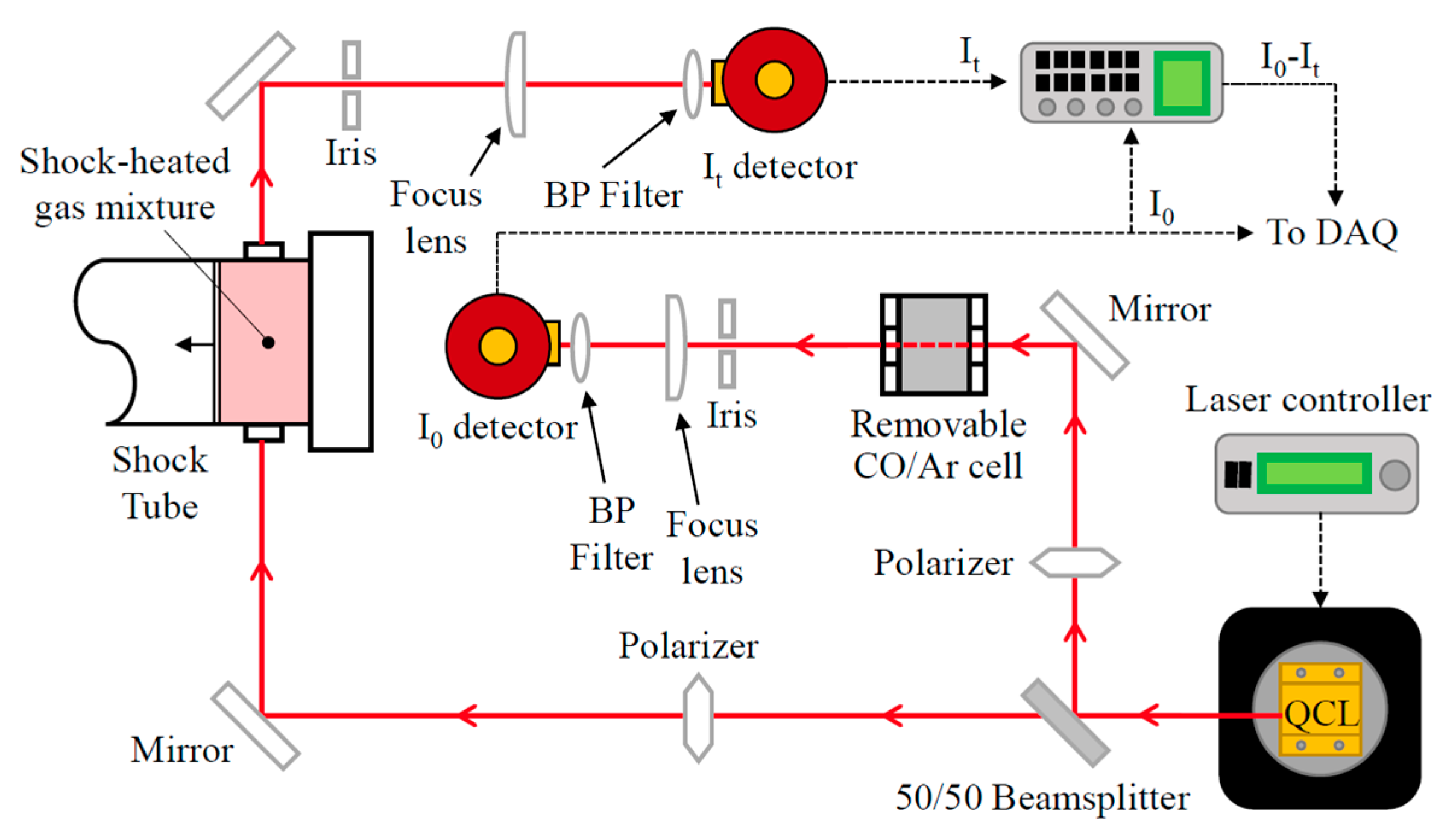
2. Experimental Setup
3. Modeling
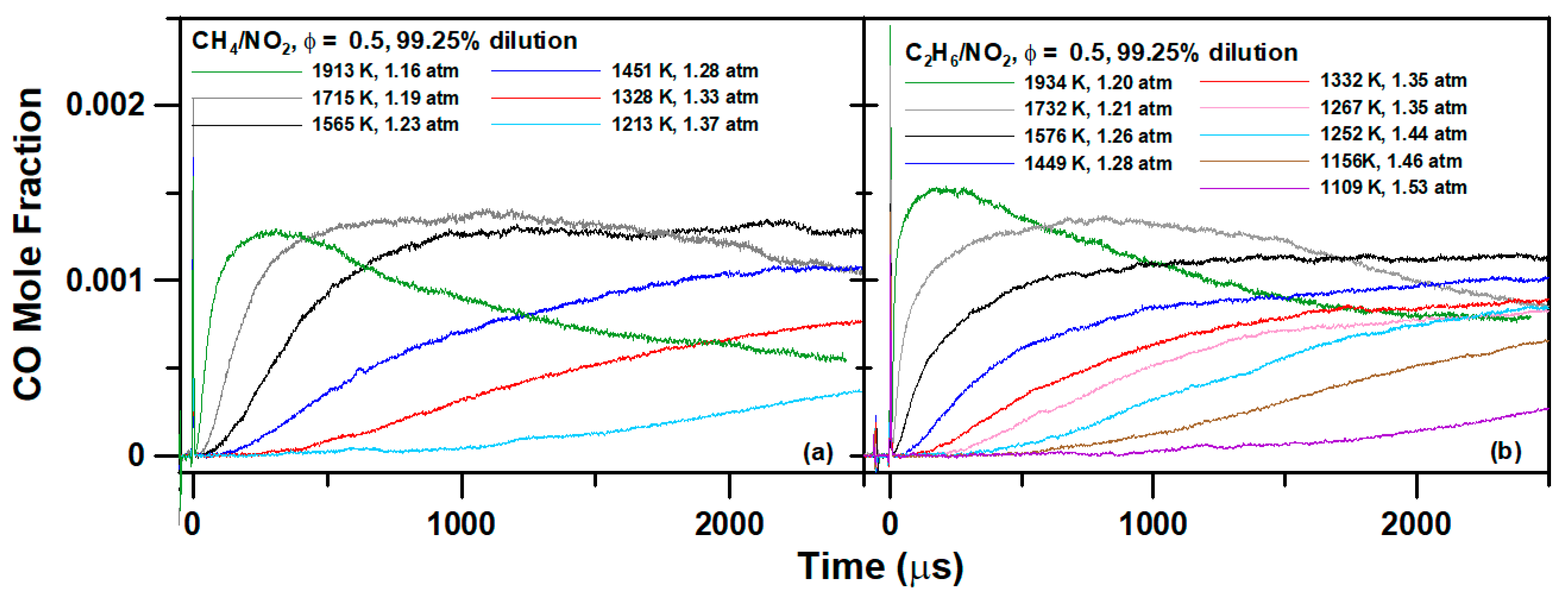
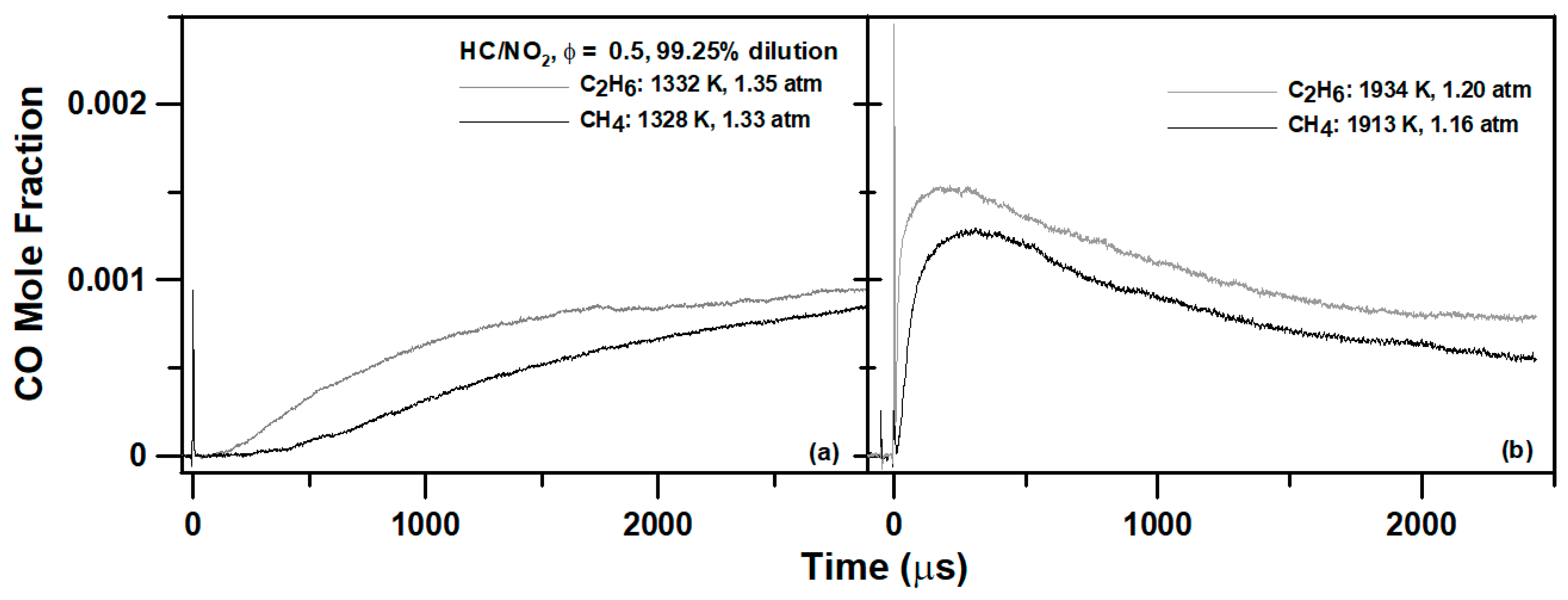
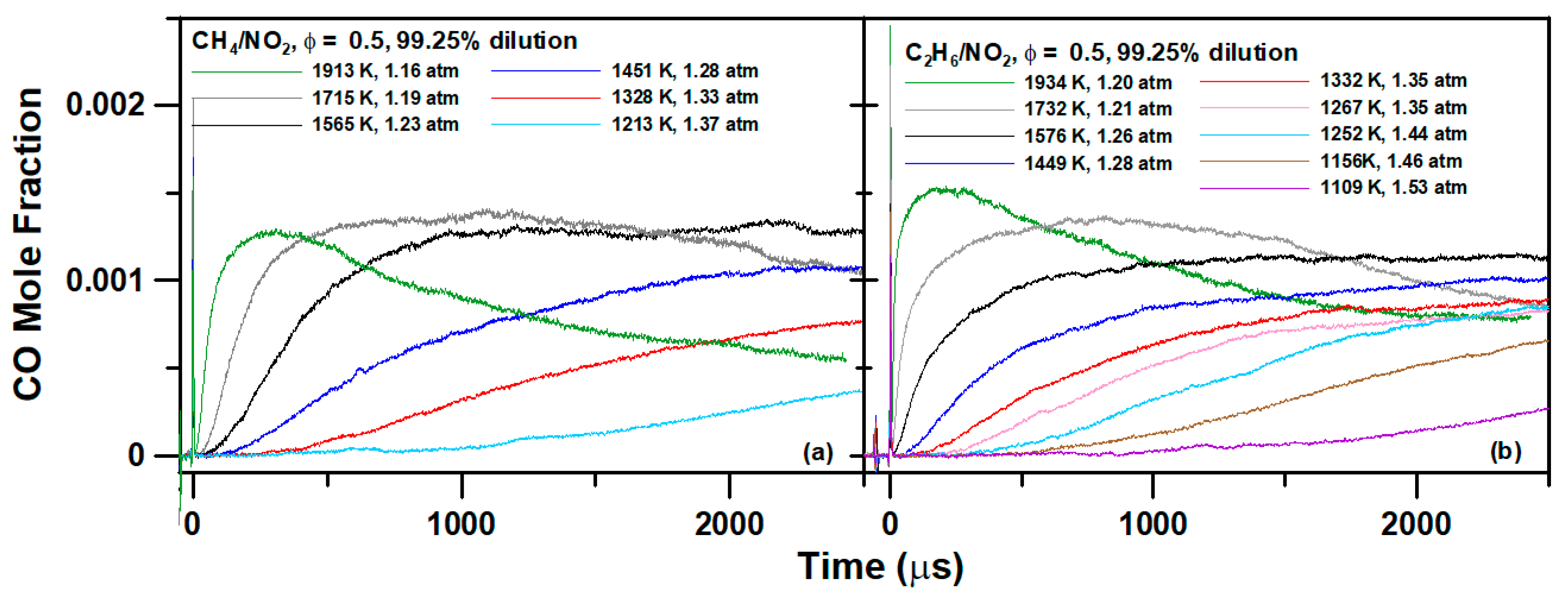
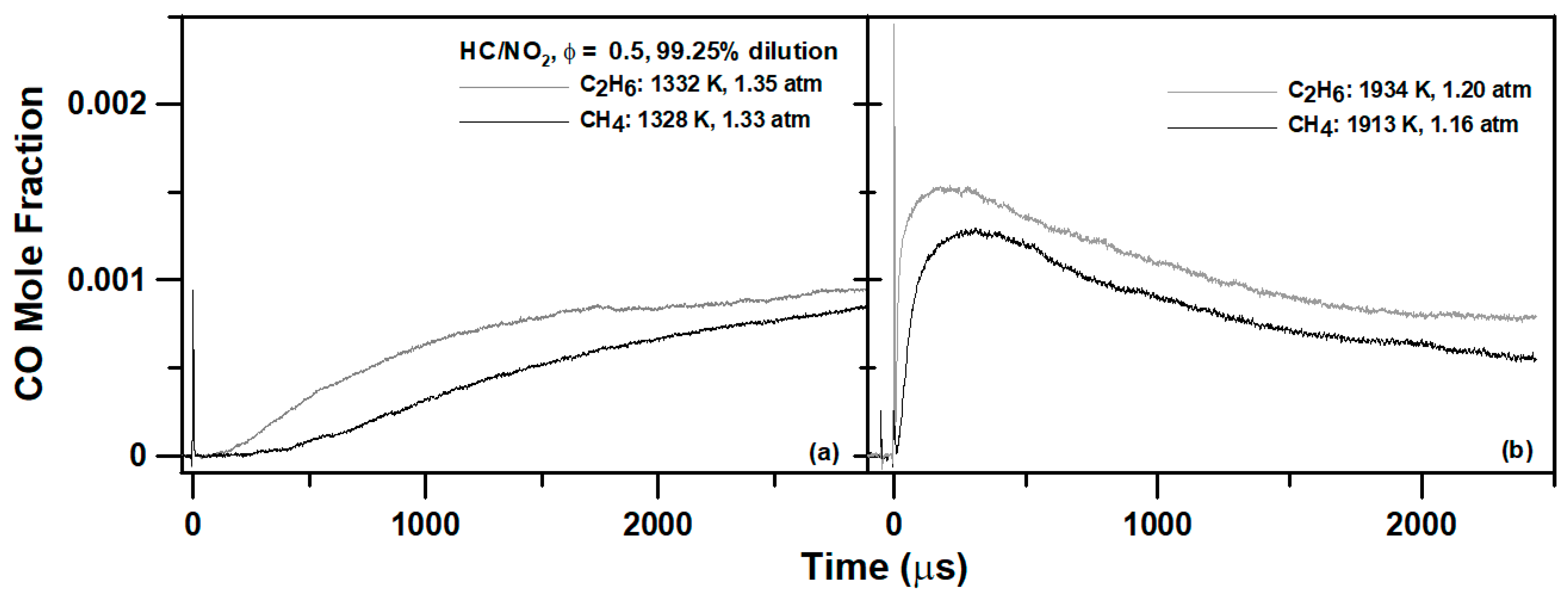
4. Experimental Results
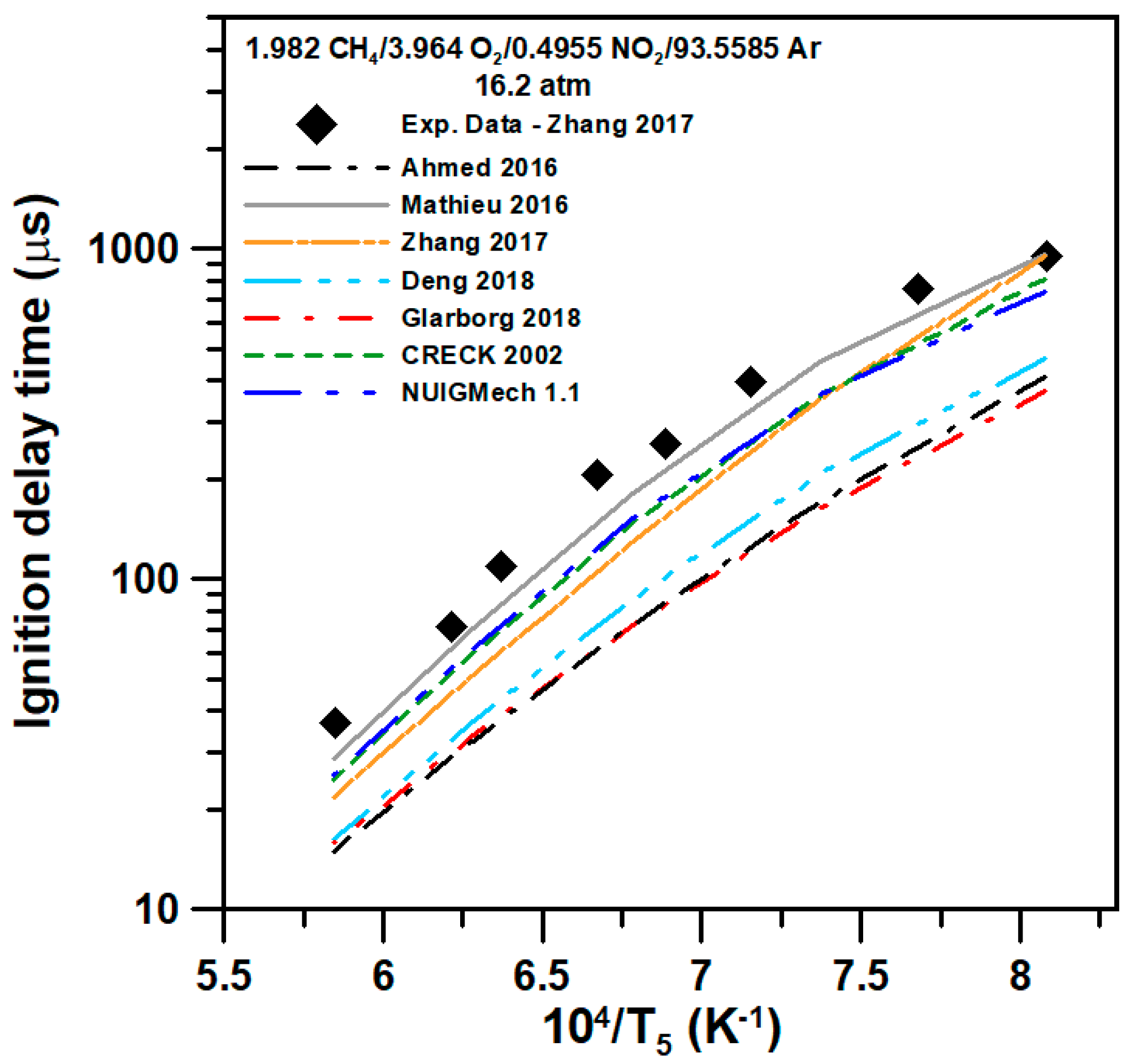
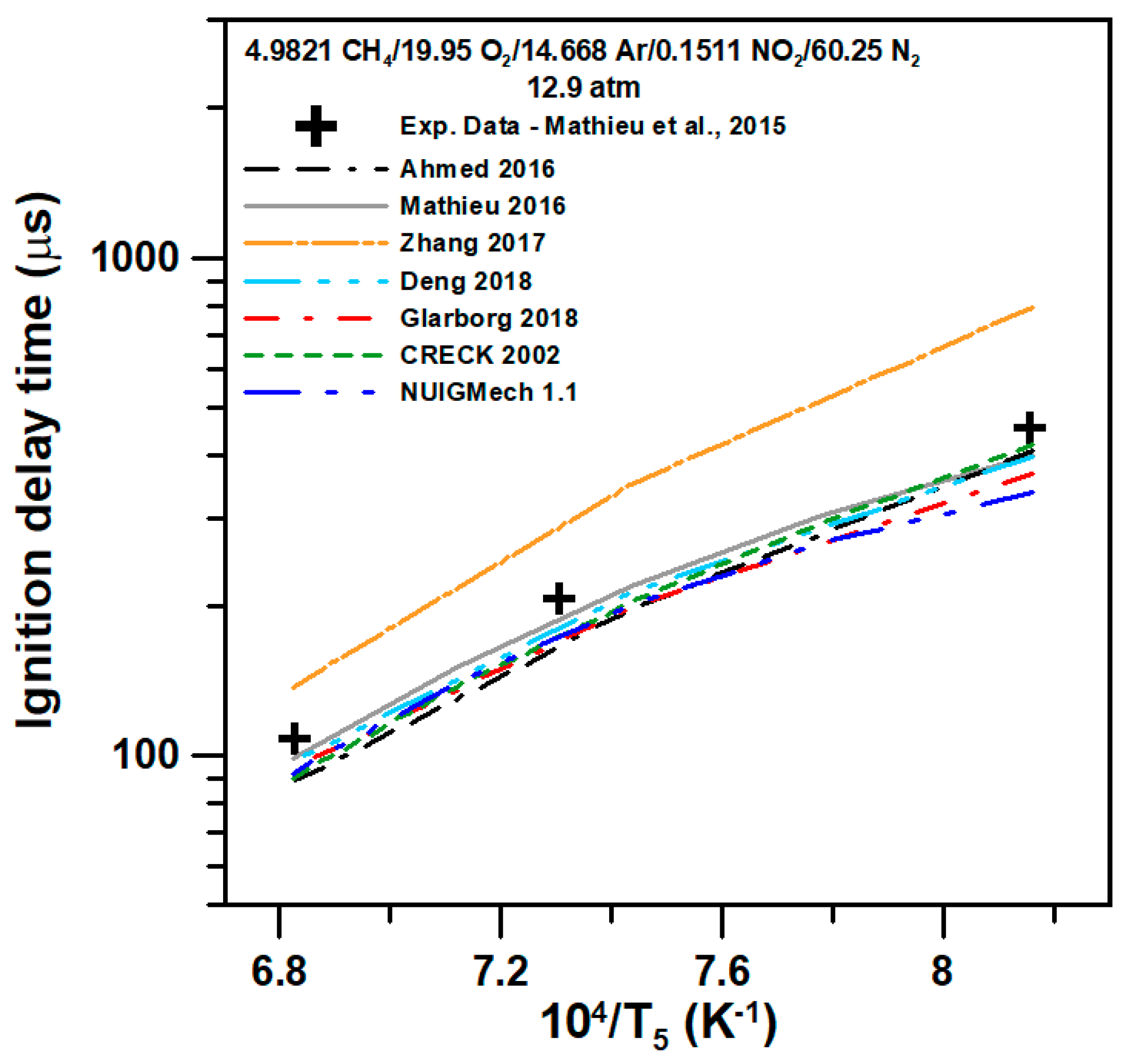
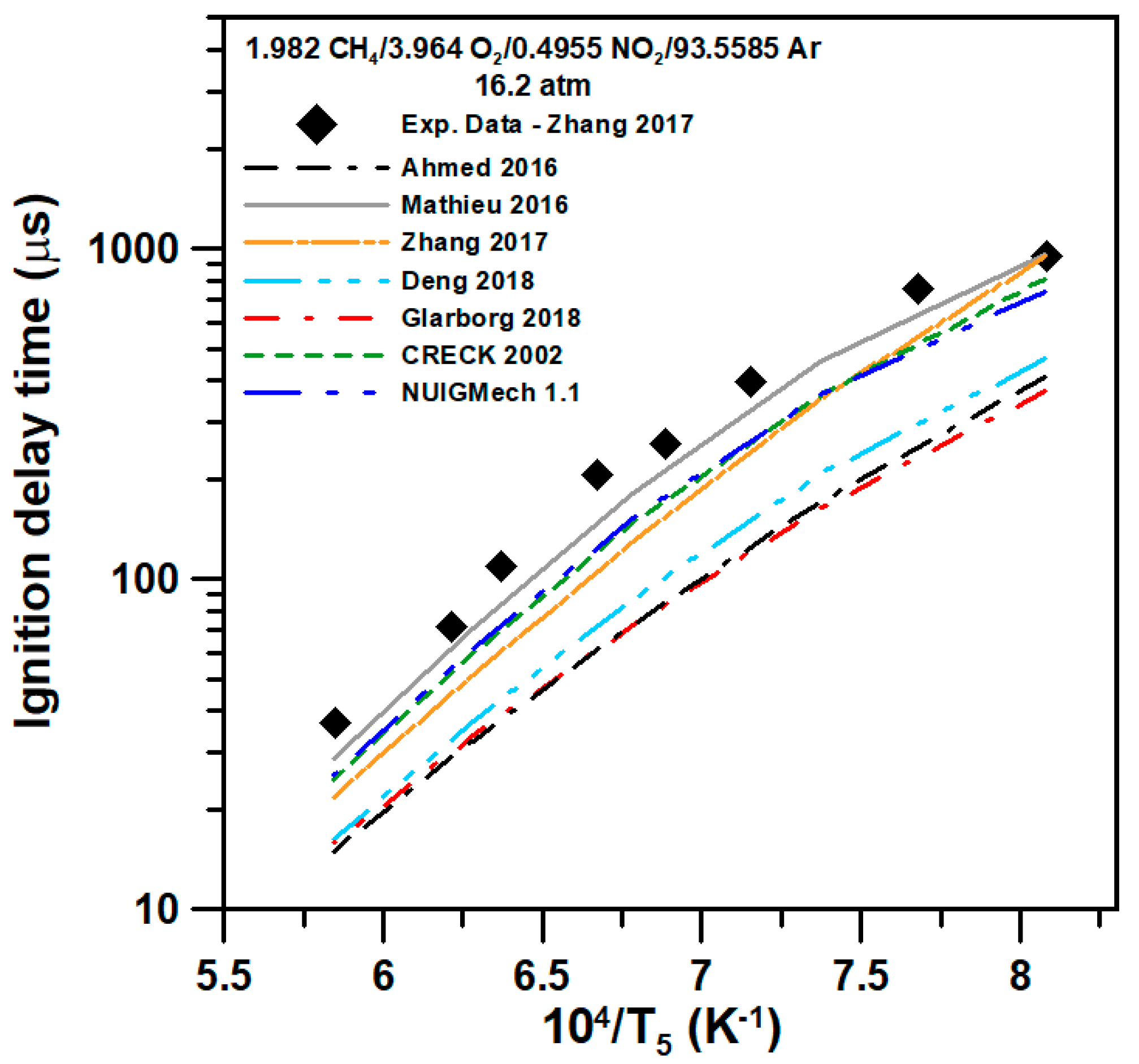
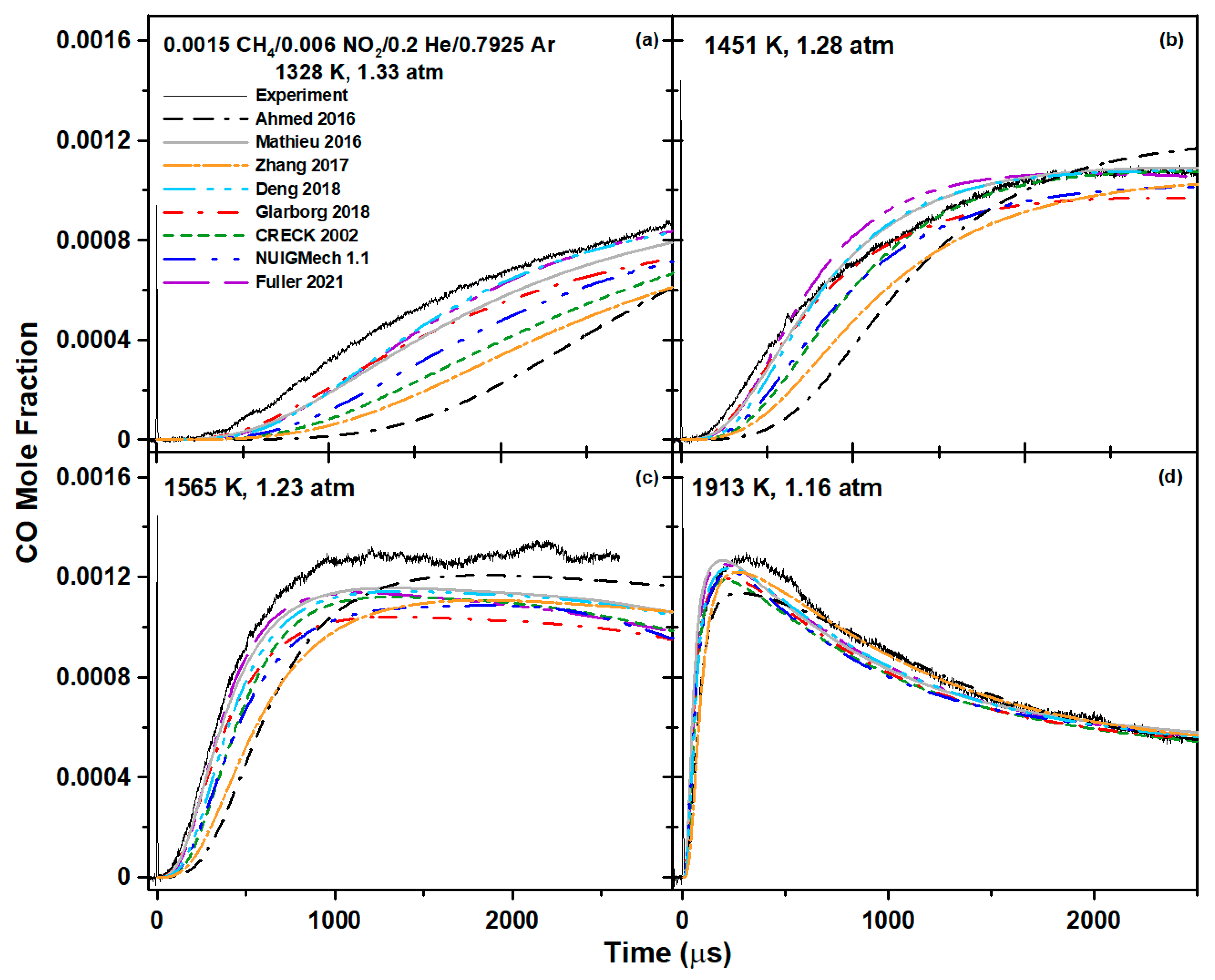
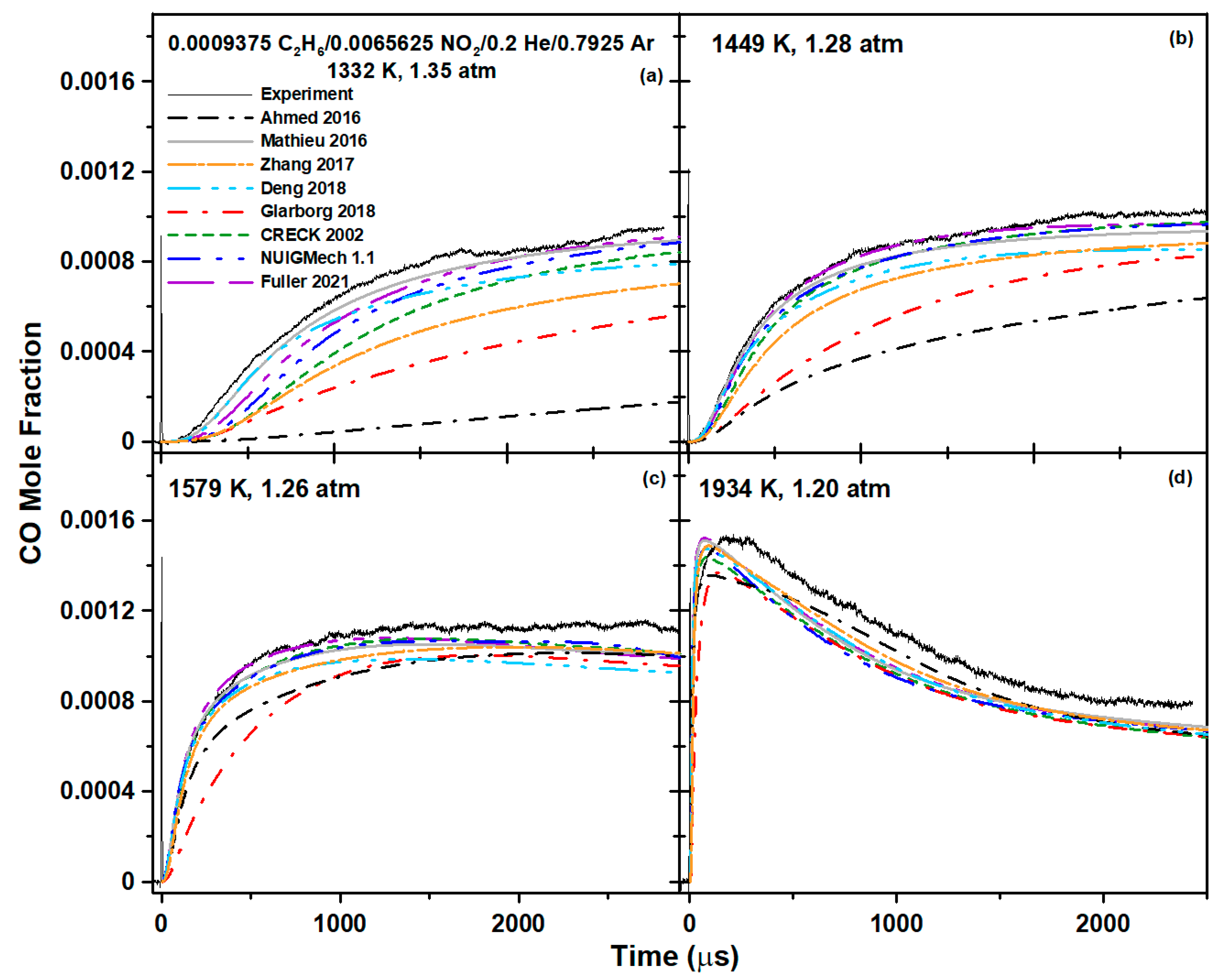
5. Model Comparison
6. Discussion
6.1. Methane Mixture
6.2. Ethane Mixture
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santner, J.; Ahmed, S.F.; Farouk, T.; Dryer, F.L. Computational Study of NOx Formation at Conditions Relevant to Gas Turbine Operation: Part 1. Energy Fuels 2016, 30, 6745–6755. [Google Scholar] [CrossRef]
- Glarborg, P.; Miller, J.A.; Ruscic, B.; Klippenstein, S.J. Modeling nitrogen chemistry in combustion. Prog. Energy Combust. Sci. 2018, 67, 31–68. [Google Scholar] [CrossRef] [Green Version]
- Dayma, G.; Dagaut, P. Effects of air contamination on the combustion of hydrogen—effect of NO and NO2 addition on hydrogen ignition and oxidation kinetics. Combust. Sci. Technol. 2006, 178, 1999–2024. [Google Scholar] [CrossRef]
- Mével, R.; Javoy, S.; Lafosse, F.; Chaumeix, N.; Dupré, G.; Paillard, C.-E. Hydrogen–nitrous oxide delay times: Shock tube experimental study and kinetic modelling. Proc. Combust. Inst. 2009, 32, 359–366. [Google Scholar] [CrossRef]
- Mével, R.; Lafosse, F.; Chaumeix, N.; Dupré, G.; Paillard, C.-E. Spherical expanding flames in H2–N2O–Ar mixtures: Flame speed measurements and kinetic modeling. Int. J. Hydrogen Energy 2009, 34, 9007–9018. [Google Scholar] [CrossRef]
- Mathieu, O.; Levacque, A.; Petersen, E.L. Effects of N2O additions on the ignition of H2-O2 mixtures: Experimental and detailed kinetic modeling study. Int. J. Hydrogen Energy 2012, 37, 15393–15405. [Google Scholar] [CrossRef]
- Mathieu, O.; Levacque, A.; Petersen, E.L. Effects of NO2 addition on hydrogen ignition behind reflected shock waves. Proc. Combust. Inst. 2013, 34, 633–640. [Google Scholar] [CrossRef]
- Mulvihill, C.R.; Mathieu, O.; Petersen, E.L. The Unimportance of the Reaction H2 + N2O = H2O + N2: A Shock-tube Study Using H2O Time Histories and Ignition Delay Times. Combust. Flame 2018, 196, 478–486. [Google Scholar] [CrossRef]
- Mulvihill, C.R.; Mathieu, O.; Petersen, E.L. H2O time histories in the H2-NO2 system for validation of NOx hydrocarbon kinetics mechanisms. Int. J. Chem. Kin. 2019, 51, 669–678. [Google Scholar] [CrossRef]
- Dagaut, P.; Nicole, A. Experimental study and detailed kinetic modeling of the effect of exhaust gas on fuel combustion: Mutual sensitization of the oxidation of nitric oxide and methane over extended temperature and pressure ranges. Combust. Flame 2005, 140, 161–171. [Google Scholar] [CrossRef]
- Rasmussen, C.L.; Rasmussen, A.E.; Glarborg, P. Sensitizing effects of NOx on CH4 oxidation at high pressure. Combust. Flame 2008, 154, 529–545. [Google Scholar] [CrossRef]
- Gersen, S.; Mokhov, A.V.; Darmeveil, J.H.; Levinsky, H.B.; Glarborg, P. Ignition-promoting effect of NO2 on methane, ethane and methane/ethane mixtures in a rapid compression machine. Proc. Combust. Inst. 2011, 33, 433–440. [Google Scholar] [CrossRef]
- Mathieu, O.; Pemelton, J.M.; Bourque, G.; Petersen, E.L. Shock-Induced Ignition of Methane Sensitized by NO2 and N2O. Combust. Flame 2015, 162, 3053–3070. [Google Scholar] [CrossRef] [Green Version]
- Marrodán, S.Y.L.; Vina, N.; Herbinet, O.; Assaf, E.; Fittschen, C.; Stagni, A.; Faravelli, T.; Alzueta, M.U.; Battin-Leclerc, F. The sensitizing effects of NO2 and NO on methane low temperature oxidation in a jet stirred reactor. Proc. Combust. Inst. 2019, 37, 667–675. [Google Scholar]
- Amrit BSahu, A.B.; El-Sabor Mohamed, A.A.; Panigrahy, S.; Saggese, C.; Patel, V.; Bourque, G.; Pitz, W.J.; Curran, H.J. An experimental and kinetic modeling study of NOx sensitization on methane autoignition and oxidation. Combust. Flame 2021, 111746, in press. [Google Scholar] [CrossRef]
- Dagaut, P.; Mathieu, O.; Nicolle, A.; Dayma, G. Experimental study and detailed kinetic modeling of the mutual sensitization of the oxidation of nitric oxide, ethylene and ethane. Combust. Sci. Technol. 2005, 177, 1767–1791. [Google Scholar] [CrossRef]
- Giménez-López, J.; Alzueta, M.U.; Rasmussen, C.T.; Marshall, P.; Glarborg, P. High pressure oxidation of C2H4/NO mixtures. Proc. Combust. Inst. 2011, 33, 449–457. [Google Scholar] [CrossRef]
- Herzler, J.; Naumann, C. Shock Tube Study of the Influence of NOx on the Ignition Delay Times of Natural Gas at High Pressure. Combust. Sci. Technol. 2012, 184, 1635–1650. [Google Scholar] [CrossRef] [Green Version]
- Mével, R.; Shepherd, J.E. gnition delay-time behind reflected shock waves of small hydrocarbons–nitrous oxide(–oxygen) mixtures. Shock Waves 2015, 25, 217–229. [Google Scholar] [CrossRef]
- Deng, F.; Zhang, Y.; Sun, W.; Huang, W.; Zhao, Q.; Qin, X.; Yang, F.; Huang, Z. Towards a Kinetic Understanding of the NOx Sensitization Effect on Unsaturation Hydrocarbons: A Case Study of Ethylene/Nitrogen Dioxide Mixtures. Proc. Combust. Inst. 2018, 37, 719–726. [Google Scholar] [CrossRef]
- Marshall, P.; Leung, C.; Gimenez-Lopez, J.; Rasmussen, C.T.; Hashemi, H.; Glarborg, P.; Abian, M.; Alzueta, M.U. The C2H2 + NO2 reaction: Implications for high pressure oxidation of C2H2/NOx mixtures. Proc. Combust. Inst. 2019, 37, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, O.; Mulvihill, C.R.; Petersen, E.L.; Curran, H.J. NOx-hydrocarbon kinetics model validation using measurements of H2O in shock-heated CH4/C2H6 mixtures with NO2 as oxidant. J. Eng. Gas Turb. Pow. 2019, 141, 41007. [Google Scholar] [CrossRef]
- Fuller, M.E.; Morsch, P.; Goldsmith, F.; Heufer, K.A. The Impact of NOx Addition on the Ignition Behavior of N-Pentane. Chem. Eng. Ind. Chem. 2021, in press. [Google Scholar] [CrossRef]
- Mathieu, O.; Giri, B.; Agard, A.R.; Adams, T.N.; Mertens, J.D.; Petersen, E.L. Nitromethane ignition behind reflected shock waves: Experimental and numerical study. Fuel 2016, 182, 597–612. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.F.; Santner, J.; Dryer, F.L.; Padak, B.; Farouk, T.I. Computational Study of NOx Formation at Conditions Relevant to Gas Turbine Operation, Part 2: NOx in High Hydrogen Content Fuel Combustion at Elevated Pressure. Energy Fuels 2016, 30, 7691–7703. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, W.; Shi, J.C.; Wu, X.J.; Zhang, R.T.; Luo, S.N. Shock-Induced Ignition of Methane, Ethane, and Methane/Ethane Mixtures Sensitized by NO2. Energy Fuels 2017, 31, 12780–12790. [Google Scholar] [CrossRef]
- Baigmohammadi, M.; Patel, V.; Nagaraja, S.; Ramalingam, A.; Martinez, S.; Panigrahy, S.; Mohamed, A.; Somers, K.P.; Burke, U.; Heufer, K.A.; et al. Comprehensive experimental and simulation study of the ignition delay time characteristics of binary blended methane, ethane, and ethylene over a wide range of temperature, pressure, equivalence ratio, and dilution. Energy Fuels 2020, 34, 8808–8823. [Google Scholar] [CrossRef]
- Ranzi, E.; Cavallotti, C.; Cuoci, A.; Frassoldati, A.; Pelucchi, M.; Faravelli, T. New reaction classes in the kinetic modeling of low temperature oxidation of n-alkanes. Combust. Flame 2015, 162, 1679–1691. [Google Scholar] [CrossRef]
- Mathieu, O.; Chaumeix, N.; Yamamoto, Y.; Abid, S.; Paillard, C.-E.; Tezuka, T.; Nakamura, H.; Mulvihill, C.R.; Petersen, E.L. Nitromethane Pyrolysis in Shock Tubes and a Micro Flow Reactor with a Controlled Temperature Profile. Proc. Combust. Inst. 2021, 38, 1007–1015. [Google Scholar] [CrossRef]
- Annesley, C.J.; Randazzo, J.B.; Klippenstein, S.J.; Harding, L.B.; Jasper, A.W.; Georgievskii, Y.; Ruscic, B.; Tranter, R.S. Thermal Dissociation and Roaming Isomerization of Nitromethane: Experiment and Theory. J. Phys. Chem. A 2015, 119, 7872–7893. [Google Scholar] [CrossRef]
- Mathieu, O.; Mulvihill, C.R.; Petersen, E.L. Shock tube water time-histories and ignition delay time measurements for H2S near atmospheric pressure. Proc. Combust. Inst. 2017, 36, 4019–4027. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, O.; Mulvihill, C.R.; Petersen, E.L. Assessment of modern detailed kinetics mechanisms to predict CO formation from methane combustion using shock-tube laser-absorption measurements. Fuel 2019, 236, 1164–1180. [Google Scholar] [CrossRef]
- Nauclér, J.D.; Nilsson, E.J.K.; Konnov, A.A. Laminar burning velocity of nitromethane + air flames: A comparison of flat and spherical flames. Combust. Flame 2015, 162, 3803–3809. [Google Scholar] [CrossRef]
- Ren, W.; Farooq, A.; Davidson, D.F.; Hanson, R.K. CO concentration and temperature sensor for combustion gases using quantum-cascade laser absorption near 4.7 μm. App. Phys. B 2012, 107, 849–860. [Google Scholar] [CrossRef]
- Mulvihill, C.R.; Alturaifi, S.A.; Petersen, E.L. High-temperature He- and O2-broadening of the R(12) line in the 1←0 band of carbon monoxide. J. Quant. Spectrosc. Radiat. Transf. 2018, 217, 432–439. [Google Scholar] [CrossRef]
- Rothman, L.S.; Jacquemart, D.; Barbe, A.; Benner, D.C.; Birk, M.; Brown, L.R.; Carleer, M.R.; Chackerian, C.; Chance, K.; Coudert, L.H.; et al. The HITRAN 2004 molecular spectroscopic database. J. Quant. Spectrosc. Radiat. Transf. 2005, 96, 139–204. [Google Scholar] [CrossRef] [Green Version]
- Alturaifi, S.A.; Mulvihill, C.R.; Mathieu, O.; Petersen, E.L. Speciation measurements in shock tubes for validation of complex kinetics mechanisms: Application to 2-Methyl-2-Butene oxidation. Combust. Flame 2021, 225, 196–213. [Google Scholar] [CrossRef]
- Mulvihill, C.R.; Keesee, C.L.; Sikes, T.; Teixeira, R.S.; Mathieu, O.; Petersen, E.L. Ignition delay times, laminar flame speeds, and species time-histories in the H2S/CH4 system at atmospheric pressure. Proc. Combust. Inst. 2019, 37, 735–742. [Google Scholar] [CrossRef]
- Mathieu, O.; Cooper, S.P.; Alturaifi, S.A.; Mulvihill, C.R.; Atherley, T.M.; Petersen, E.L. Shock-Tube Laser Absorption Measurements of CO and H2O during Iso-Octane Combustion. Energy Fuels 2020, 34, 7533–7544. [Google Scholar] [CrossRef]

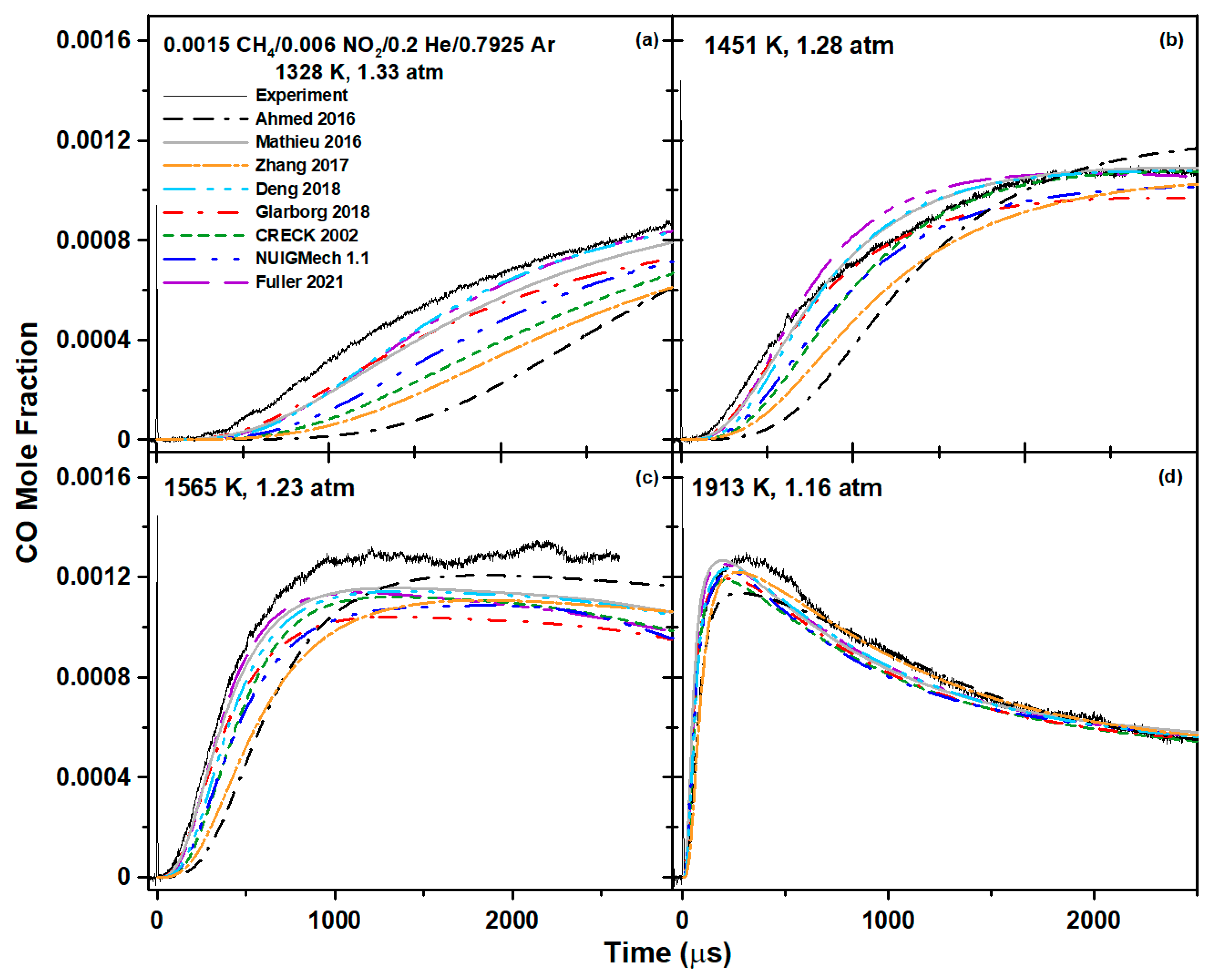


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mixture Composition (Mol. Fraction) | T5 (K) | P5 (atm) |
|---|---|---|
| 0.0015 CH4/0.006 NO2/0.2 He/0.7925 Ar | 1213–1913 | 1.16–1.37 |
| 0.0009375 C2H6/0.0065625 NO2/0.2 He/0.7925 Ar | 1109–1934 | 1.20–1.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathieu, O.; Cooper, S.P.; Alturaifi, S.A.; Petersen, E.L. Assessing NO2-Hydrocarbon Interactions during Combustion of NO2/Alkane/Ar Mixtures in a Shock Tube Using CO Time Histories. Fuels 2022, 3, 1-14. https://doi.org/10.3390/fuels3010001
Mathieu O, Cooper SP, Alturaifi SA, Petersen EL. Assessing NO2-Hydrocarbon Interactions during Combustion of NO2/Alkane/Ar Mixtures in a Shock Tube Using CO Time Histories. Fuels. 2022; 3(1):1-14. https://doi.org/10.3390/fuels3010001
Chicago/Turabian StyleMathieu, Olivier, Sean P. Cooper, Sulaiman A. Alturaifi, and Eric L. Petersen. 2022. "Assessing NO2-Hydrocarbon Interactions during Combustion of NO2/Alkane/Ar Mixtures in a Shock Tube Using CO Time Histories" Fuels 3, no. 1: 1-14. https://doi.org/10.3390/fuels3010001
APA StyleMathieu, O., Cooper, S. P., Alturaifi, S. A., & Petersen, E. L. (2022). Assessing NO2-Hydrocarbon Interactions during Combustion of NO2/Alkane/Ar Mixtures in a Shock Tube Using CO Time Histories. Fuels, 3(1), 1-14. https://doi.org/10.3390/fuels3010001