
Molecular Structure of M(N13) Compounds with 12-Membered Nitrogen-Containing Cycle and Axial Nitrogen Atom (M = Mn, Fe): Quantum-Chemical Design by DFT Method



Abstract
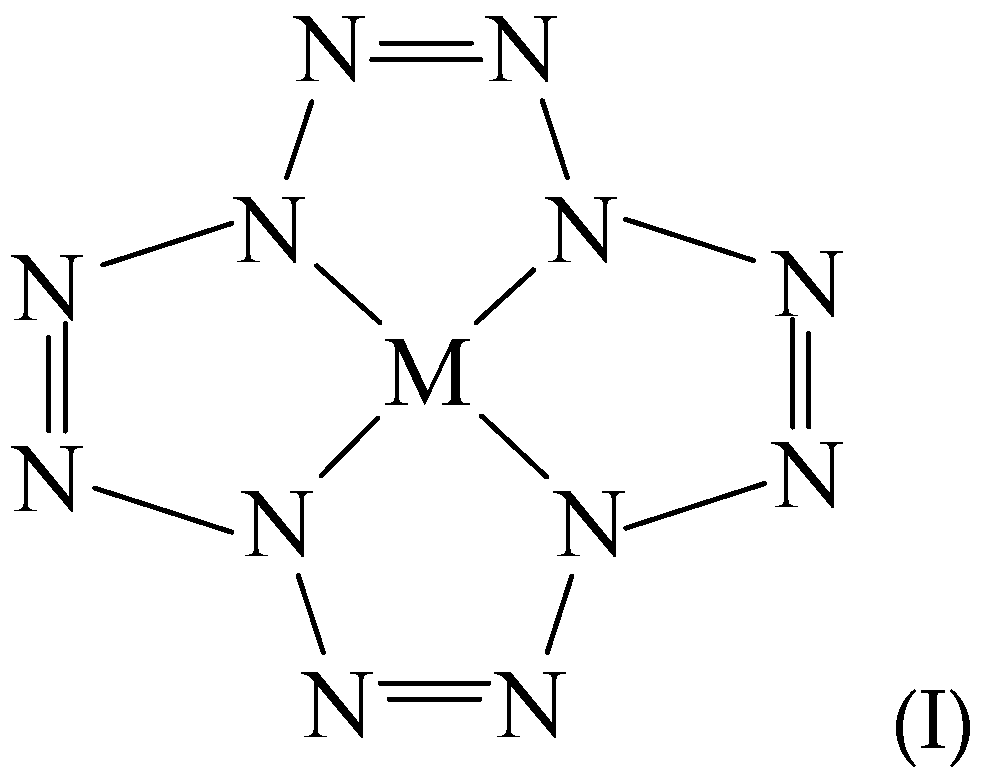
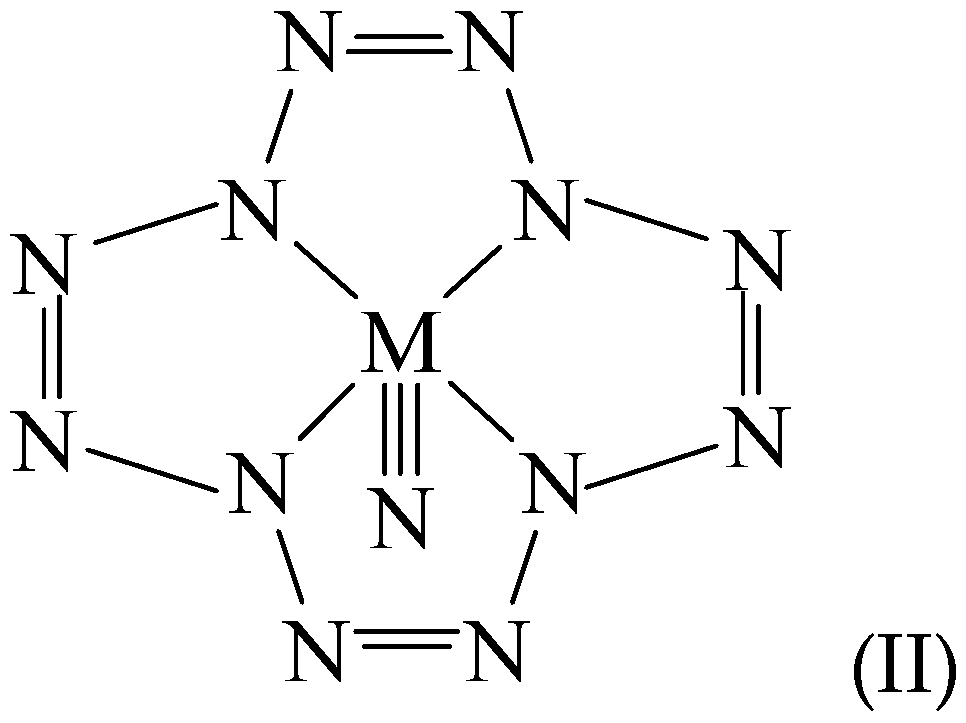
1. Introduction
2. Method
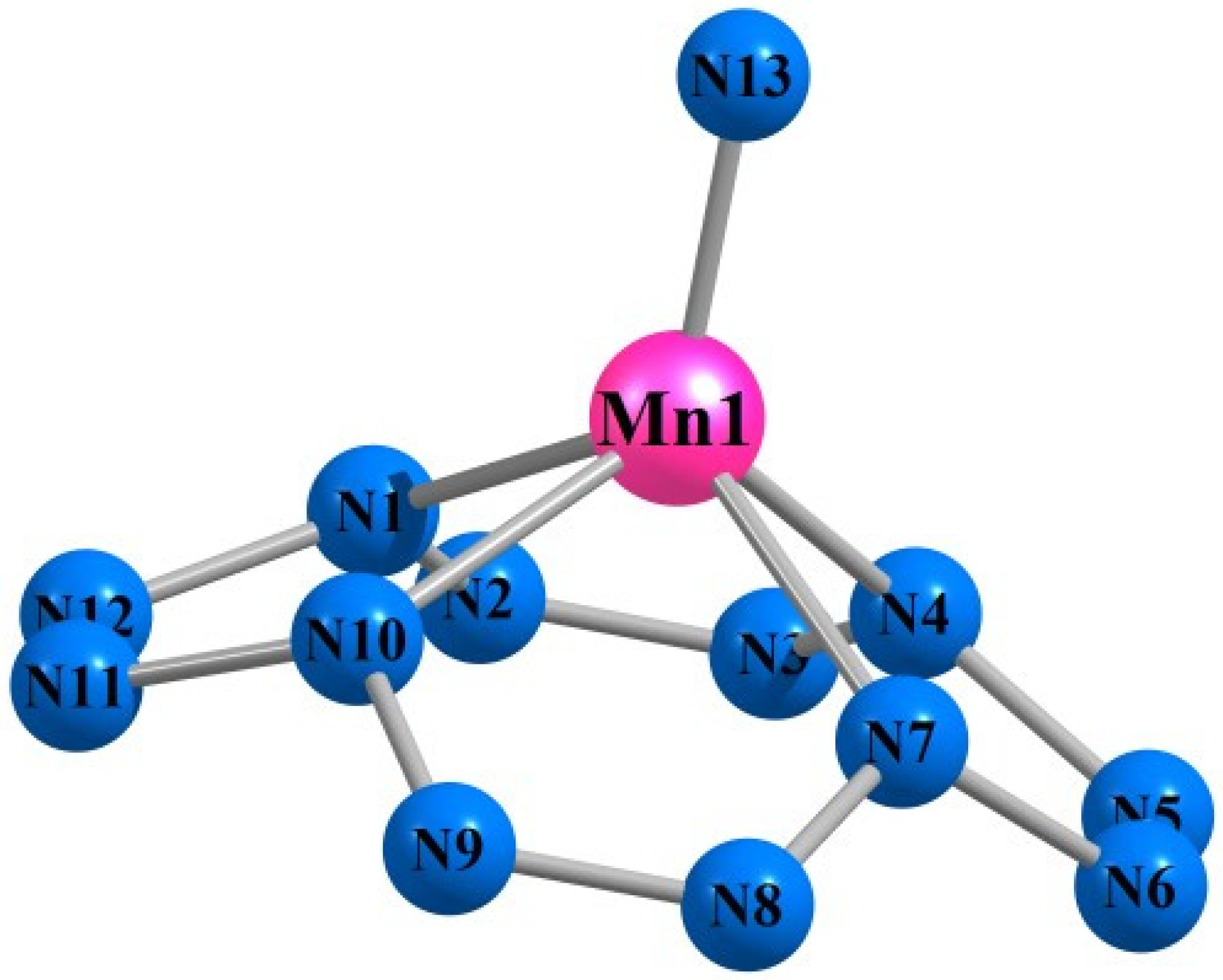
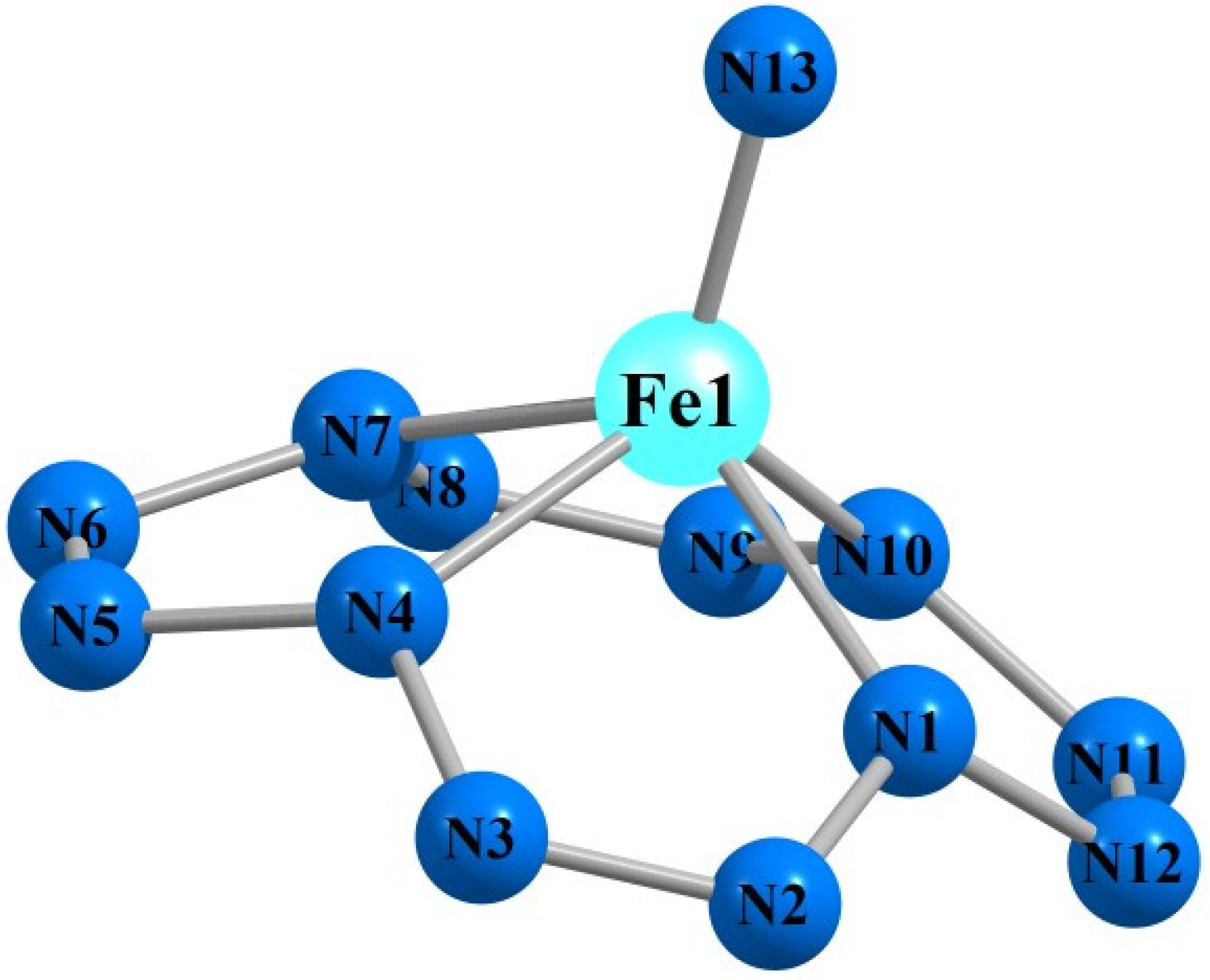
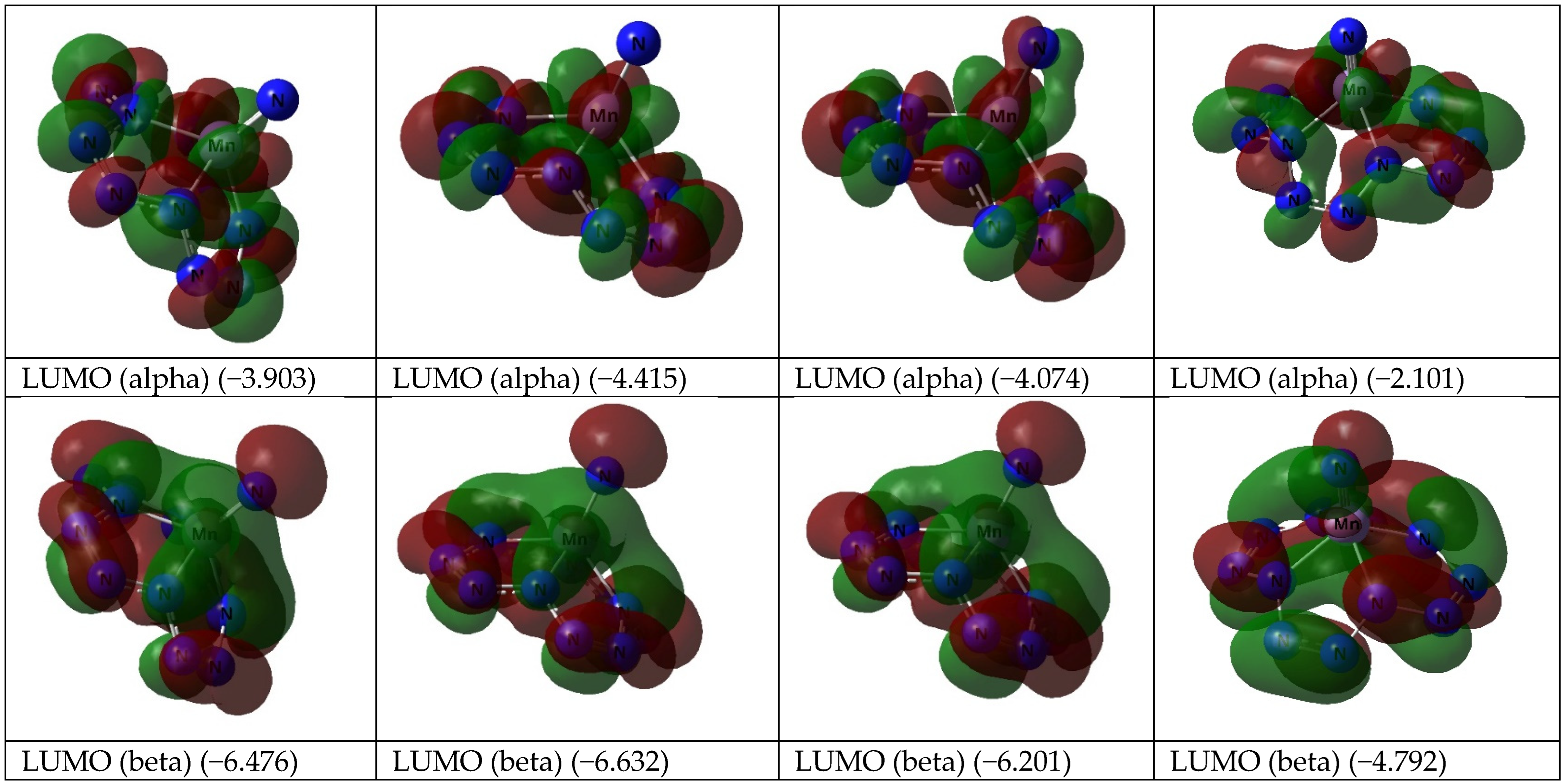
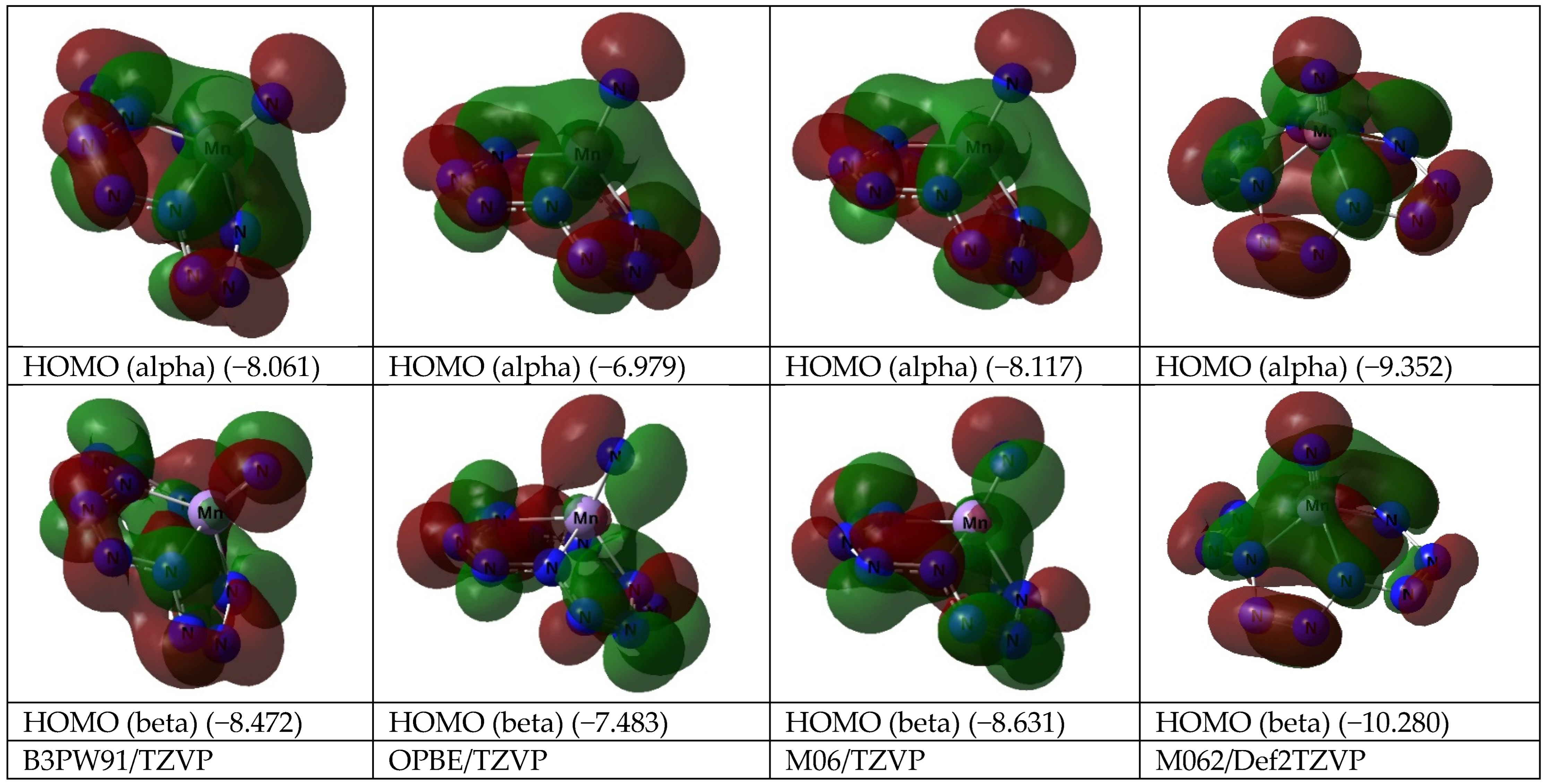
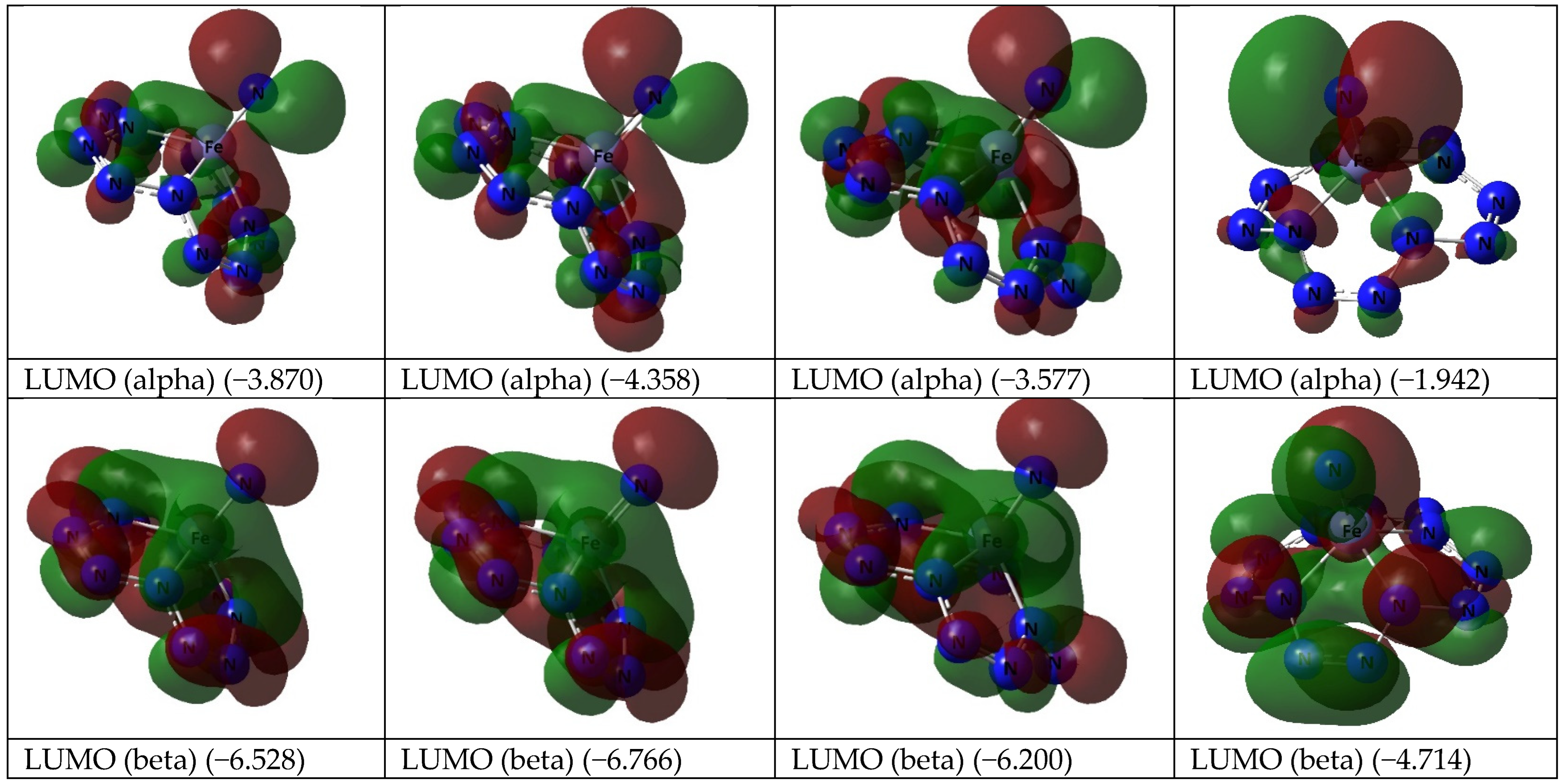
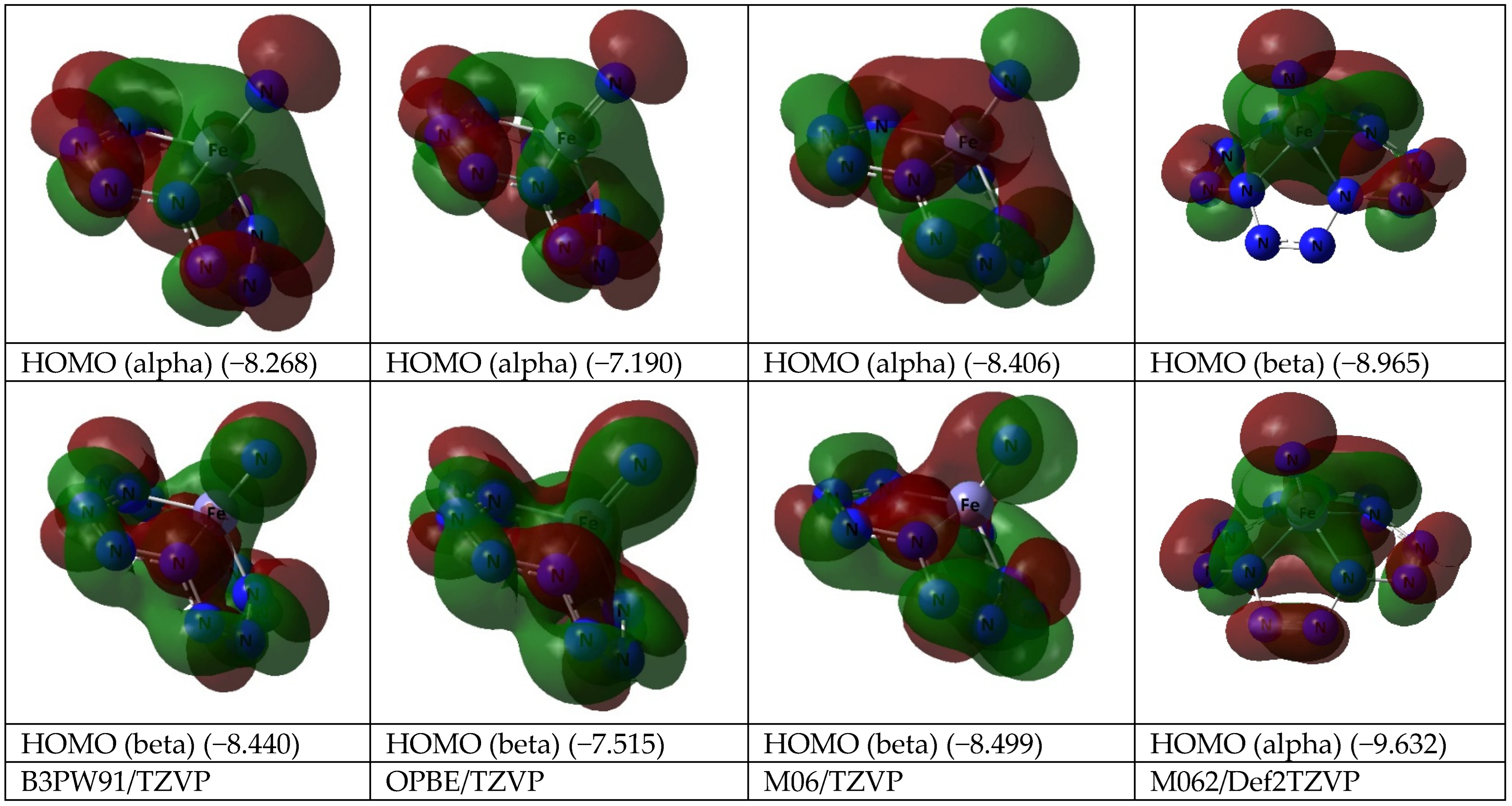
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mikhailov, O.V.; Chachkov, D.V. Twelve-Nitrogen-Atom Cyclic Structure Stabilized by 3d-Element Atoms: Quantum Chemical Modeling. Int. J. Mol. Sci. 2022, 23, 6560. [Google Scholar] [CrossRef] [PubMed]
- Klapötke, T.M.; Harcourt, R.D. The interconversion of N12 to N8 and two equivalents of N2. J. Mol. Struct. (Theochem) 2001, 541, 237–242. [Google Scholar] [CrossRef]
- Olah, G.A.; Prakash, G.K.S.; Rasul, G. N62+ and N42+ Dications and Their N12 and N10 Azido Derivatives: DFT/GIAO-MP2 Theoretical Studies. J. Am. Chem. Soc. 2001, 123, 3308–3310. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.S.; Zhao, J.F. Theoretical Study of Potential Energy Surfaces for N12 Clusters. J. Phys. Chem. A 2002, 106, 5367–5372. [Google Scholar] [CrossRef]
- Bruney, L.Y.; Bledson, T.M.; Strout, D.L. What Makes an N12 Cage Stable? Inorg. Chem. 2003, 42, 8117–8120. [Google Scholar] [CrossRef]
- Samartzis, P.C.; Woodtke, A.M. All-nitrogen chemistry: How far are we from N60? Intern. Revs. Phys. Chem. 2006, 25, 1952–2005. [Google Scholar] [CrossRef]
- Greschner, M.J.; Zhang, M.; Majumdar, A.; Liu, H.; Peng, F.; Tse, J.S.; Yao, Y. A New Allotrope of Nitrogen as High-Energy Density Material. J. Phys. Chem. A 2016, 120, 2920–2925. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Molecular structures and thermodynamics of stable N4, N6 and N8 neutral polynitrogens according to data of QCISD(T)/TZVP method. Chem. Phys. Lett. 2020, 753, 137594. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Tetra-, hexa-, and octanitrogen molecules: A quantum chemical design and thermodynamic properties. Russ. Chem. Bull. 2020, 69, 2067–2072. [Google Scholar] [CrossRef]
- Mikhailov, O.V. Molecular and Electronic Structures of Neutral Polynitrogens: Review on the Theory and Experiment in 21st Century. Int. J. Mol. Sci. 2022, 23, 2841. [Google Scholar] [CrossRef]
- Straka, M. N6 ring as a planar hexagonal ligand in novel M(η6-N6) species. Chem. Phys. Lett. 2002, 358, 531–536. [Google Scholar] [CrossRef]
- Choi, C.; Yoo, H.-W.; Goh, E.M.; Cho, S.G.; Jung, Y.S. Ti(N5)4 as a Potential Nitrogen-Rich Stable High-Energy Density Material. J. Phys. Chem. A 2016, 120, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Brathwaite, A.D.; Abbott-Lyon, H.L.; Duncan, M.A. Distinctive Coordination of CO vs N2 to Rhodium Cations: An Infrared and Computational Study. J. Phys. Chem. A 2016, 120, 7659–7670. [Google Scholar] [CrossRef]
- Ding, K.; Xu, H.; Yang, Y.; Li, T.; Chen, Z.; Ge, Z.; Zhu, W.; Zheng, W. Mass Spectrometry and Theoretical Investigation of VNn+ (n = 8, 9, and 10) Clusters. J. Phys. Chem. A 2018, 122, 4687–4695. [Google Scholar] [CrossRef] [PubMed]
- Bykov, M.; Bykova, E.; Koemets, E.; Fedotenko, T.; Aprilis, G.; Glazyrin, K.; Liermann, H.-P.; Ponomareva, A.V.; Tidholm, J.; Tasnadi, F.; et al. High-pressure synthesis of a nitrogen-rich inclusion compound ReN8 xN2 with conjugated polymeric nitrogen chains. Angew. Chem. Int. Ed. 2018, 57, 9048–9053. [Google Scholar] [CrossRef]
- Ding, K.; Chen, H.; Xu, H.; Yang, B.; Ge, Z.; Lu, C.; Zheng, W. Identification of octahedral coordinated ZrN12+ cationic clusters by mass spectrometry and structure searches. Dalton Trans. 2021, 50, 10187–10192. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Revs. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Revs. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. DFT Quantum-Chemical Modeling Molecular Structures of Cobalt Macrocyclic Complexes with Porphyrazine or Its Benzo-Derivatives and Two Oxygen Acido Ligands. Int. J. Mol. Sci. 2020, 21, 9085. [Google Scholar] [CrossRef]
- Hoe, W.M.; Cohen, A.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, H.; Duelund, L.; Winkler, H.; Toftlund, H.; Trautwein, A.X. Free Energy of Spin-Crossover Complexes Calculated with Density Functional Methods. Inorg. Chem. 2001, 40, 2201–2203. [Google Scholar] [CrossRef]
- Swart, M.; Groenhof, A.R.; Ehlers, A.W.; Lammertsma, K. Validation of Exchange—Correlation Functionals for Spin States of Iron Complexes. J. Phys. Chem. A 2004, 108, 5479–5483. [Google Scholar] [CrossRef]
- Swart, M.; Ehlers, A.W.; Lammertsma, K. Performance of the OPBE exchange-correlation functional. Mol. Phys. 2004, 102, 2467–2474. [Google Scholar] [CrossRef]
- Swart, M. Metal–ligand bonding in metallocenes: Differentiation between spin state, electrostatic and covalent bonding. Inorg. Chim. Acta 2007, 360, 179–189. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, UK, 2009; Available online: https://www.scienceopen.com/document?vid=7625a2b3-85a4-4746-8a93-fb3335021944 (accessed on 1 January 2023).
- Cioslowski, J. A New Population Analysis Based on Atomic Polar Tensors. J. Am. Chem. Soc. 1989, 111, 8333–8336. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2000. [Google Scholar]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mn(N13) | Fe(N13) | |||||||
|---|---|---|---|---|---|---|---|---|
| Structural Parameter | B3PW91/ TZVP | OPBE/ TZVP | M06/ TZVP | M062X/ Def2TZVP | B3PW91/ TZVP | OPBE/ TZVP | M06/ TZVP | M062X/ Def2TZVP |
| M–N bond lengths in the MN4 chelate node, pm | ||||||||
| M1N1 | 188.4 | 188.5 | 190.7 | 191.1 | 183.0 | 182.6 | 184.5 | 184.9 |
| M1N4 | 188.4 | 188.5 | 189.3 | 191.1 | 183.0 | 182.6 | 183.1 | 188.8 |
| M1N7 | 188.4 | 188.5 | 189.3 | 191.1 | 183.0 | 182.6 | 183.1 | 188.8 |
| M1N10 | 188.4 | 188.5 | 190.7 | 191.1 | 183.0 | 182.6 | 184.5 | 184.9 |
| M–N bond lengths between M and nitride N atom, pm | ||||||||
| M1N13 | 151.0 | 152.5 | 150.9 | 147.3 | 150.9 | 151.5 | 150.0 | 154.4 |
| Nitrogen-nitrogen bond lengths in macrocycle, pm | ||||||||
| N1N2 | 134.9 | 134.9 | 138.4 | 142.1 | 134.5 | 134.9 | 138.8 | 136.2 |
| N2N3 | 127.2 | 128.2 | 125.9 | 123.4 | 127.5 | 128.2 | 125.8 | 123.8 |
| N3N4 | 134.9 | 134.9 | 134.0 | 142.1 | 134.5 | 134.9 | 134.0 | 142.9 |
| N4N5 | 134.9 | 134.9 | 137.6 | 129.9 | 134.5 | 134.9 | 137.6 | 130.0 |
| N5N6 | 127.2 | 128.2 | 124.6 | 132.3 | 127.5 | 128.2 | 124.6 | 131.3 |
| N6N7 | 134.9 | 134.9 | 137.6 | 130.0 | 134.5 | 134.9 | 137.6 | 130.0 |
| N7N8 | 134.9 | 134.9 | 134.0 | 142.1 | 134.5 | 134.9 | 134.0 | 142.9 |
| N8N9 | 127.2 | 128.2 | 125.9 | 123.4 | 127.5 | 128.2 | 125.8 | 123.8 |
| N9N10 | 134.9 | 134.9 | 138.4 | 142.1 | 134.5 | 134.9 | 138.8 | 136.2 |
| N10N11 | 134.9 | 134.9 | 132.0 | 129.9 | 134.5 | 134.9 | 131.4 | 137.9 |
| N11N12 | 127.2 | 128.2 | 129.4 | 132.3 | 127.5 | 128.2 | 129.9 | 124.2 |
| N12N1 | 134.9 | 134.9 | 132.0 | 130.0 | 134.5 | 134.9 | 131.4 | 137.9 |
| Bond angles in the MN4 grouping, deg | ||||||||
| N1M1N4 | 76.3 | 76.0 | 76.0 | 74.6 | 77.7 | 77.7 | 77.4 | 76.2 |
| N4M1N7 | 76.3 | 76.0 | 75.7 | 75.6 | 77.7 | 77.7 | 77.1 | 78.0 |
| N7M1N10 | 76.3 | 76.0 | 76.0 | 74.6 | 77.7 | 77.7 | 77.4 | 76.2 |
| N10M1N1 | 76.3 | 76.0 | 76.3 | 75.6 | 77.7 | 77.7 | 78.0 | 76.0 |
| Bond angles sum (BAS), deg | 305.2 | 304.0 | 304.0 | 300.4 | 310.8 | 310.8 | 309.9 | 306.4 |
| Deviation from coplanarity, deg | 54.8 | 56.0 | 56.0 | 59.6 | 49.2 | 49.2 | 50.1 | 53.6 |
| Non-bond angles in the MN4 grouping, deg | ||||||||
| N1N4N7 | 90.0 | 90.0 | 90.4 | 90.0 | 90.0 | 90.0 | 90.5 | 88.8 |
| N4N7N10 | 90.0 | 90.0 | 90.4 | 90.0 | 90.0 | 90.0 | 90.5 | 88.8 |
| N7N10N1 | 90.0 | 90.0 | 89.6 | 90.0 | 90.0 | 90.0 | 89.5 | 91.2 |
| N10N1N4 | 90.0 | 90.0 | 89.6 | 90.0 | 90.0 | 90.0 | 89.5 | 91.2 |
| Non-bond angles sum (NBAS), deg | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 |
| Deviation from coplanarity, deg | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Bond angles in 5-membered cycles, deg | ||||||||
| M1N1N2 | 117.8 | 118.2 | 117.4 | 117.7 | 117.8 | 118.1 | 117.4 | 119.1 |
| N1N2N3 | 113.1 | 112.6 | 111.3 | 112.4 | 112.3 | 112.0 | 110.4 | 114.9 |
| N2N3N4 | 113.0 | 112.6 | 115.4 | 112.4 | 112.3 | 112.0 | 114.5 | 110.1 |
| N3N4M1 | 117.8 | 118.2 | 118.0 | 117.7 | 117.8 | 118.1 | 118.2 | 116.9 |
| M1N4N5 | 117.8 | 118.2 | 118.0 | 119.1 | 117.8 | 118.1 | 118.3 | 116.9 |
| N4N5N6 | 113.0 | 112.6 | 113.0 | 113.1 | 112.3 | 112.0 | 112.1 | 114.1 |
| N5N6N7 | 113.1 | 112.6 | 113.0 | 113.1 | 112.3 | 112.0 | 112.1 | 114.1 |
| N6N7M1 | 117.8 | 118.2 | 118.0 | 119.1 | 117.8 | 118.1 | 118.3 | 116.9 |
| M1N7N8 | 117.8 | 118.2 | 118.0 | 117.7 | 117.8 | 118.1 | 118.2 | 116.8 |
| N7N8N9 | 113.1 | 112.6 | 115.4 | 112.4 | 112.3 | 112.0 | 114.5 | 110.1 |
| N8N9N10 | 113.0 | 112.6 | 111.3 | 112.4 | 112.3 | 112.0 | 110.4 | 114.9 |
| N9N10M1 | 117.8 | 118.2 | 117.4 | 117.7 | 117.8 | 118.1 | 117.4 | 119.1 |
| M1N10N11 | 117.8 | 118.2 | 117.7 | 119.1 | 117.8 | 118.1 | 117.7 | 119.1 |
| N10N11N12 | 113.0 | 112.6 | 113.7 | 113.1 | 112.3 | 112.0 | 112.9 | 112.0 |
| N11N12N1 | 113.1 | 112.6 | 113.7 | 113.1 | 112.3 | 112.0 | 112.9 | 112.0 |
| N12N1M1 | 117.8 | 118.2 | 117.7 | 119.1 | 117.8 | 118.1 | 117.7 | 119.1 |
| N–M–N bond lengths between N donor atom, M, and nitride N atom, pm | ||||||||
| N1M1N13 | 119.1 | 119.5 | 120.4 | 120.5 | 117.4 | 117.4 | 117.3 | 124.0 |
| N4M1N13 | 119.1 | 119.5 | 118.5 | 120.5 | 117.4 | 117.4 | 118.1 | 113.5 |
| N7M1N13 | 119.1 | 119.5 | 118.5 | 120.5 | 117.4 | 117.4 | 118.1 | 113.5 |
| N10M1N13 | 119.1 | 119.5 | 120.4 | 120.5 | 117.4 | 117.4 | 117.3 | 124.0 |
| APT Analysis Data | ||||||||
| M | Effective charge on an atom, units electron charge ē | |||||||
| M1 | N1 (N10) | N2 (N9) | N5 (N6) | N4 (N7) | N3 (N8) | N11(N12) | N13 | |
| Mn | +0.377 | −0.151 (−0.151) | +0.079 (+0.079) | +0.025 (+0.024) | −0.151 (−0.151) | +0.079 (+0.079) | +0.025 (+0.024) | −0.185 |
| Fe | +0.115 | −0.209 (−0.209) | +0.131 (+0.131) | −0.014 (−0.014) | −0.050 (−0.050) | −0.050 (−0.050) | +0.029 (+0.029) | +0.211 |
| NBO Analysis Data | ||||||||
| M | Effective charge on an atom, units electron charge ē | |||||||
| M1 | N1 (N10) | N2 (N9) | N5 (N6) | N4 (N7) | N3 (N8) | N11(N12) | N13 | |
| Mn | −0.065 | −0.113 (−0.113) | +0.057 (+0.057) | +0.012 (+0.012) | −0.113 (−0.113) | +0.057 (+0.057) | +0.012 (+0.012) | +0.239 |
| Fe | +0.056 | −0.167 (−0.167) | +0.075 (+0.075) | +0.023 (+0.023) | −0.040 (−0.040) | +0.004 (+0.004) | +0.021 (+0.021) | +0.111 |
| Compound | Calculation Method | ΔfH0, kJ/mol | S0, J/mol∙K | ΔfG0, kJ/mol |
|---|---|---|---|---|
| Mn(N13) | DFT B3PW91/TZVP | 1704.9 | 424.4 | 1956.4 |
| DFT OPBE/TZVP | 1411.2 | 429.5 | 1661.2 | |
| DFT M06/TZVP | 1834.5 | 425.7 | 2085.6 | |
| Fe(N13) | DFT B3PW91/TZVP | 1821.2 | 422.7 | 2071.8 |
| DFT OPBE/TZVP | 1486.3 | 416.8 | 1738.6 | |
| DFT M06/TZVP | 1980.1 | 419.8 | 2231.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhailov, O.V.; Chachkov, D.V. Molecular Structure of M(N13) Compounds with 12-Membered Nitrogen-Containing Cycle and Axial Nitrogen Atom (M = Mn, Fe): Quantum-Chemical Design by DFT Method. Quantum Rep. 2023, 5, 282-293. https://doi.org/10.3390/quantum5010019
Mikhailov OV, Chachkov DV. Molecular Structure of M(N13) Compounds with 12-Membered Nitrogen-Containing Cycle and Axial Nitrogen Atom (M = Mn, Fe): Quantum-Chemical Design by DFT Method. Quantum Reports. 2023; 5(1):282-293. https://doi.org/10.3390/quantum5010019
Chicago/Turabian StyleMikhailov, Oleg V., and Denis V. Chachkov. 2023. "Molecular Structure of M(N13) Compounds with 12-Membered Nitrogen-Containing Cycle and Axial Nitrogen Atom (M = Mn, Fe): Quantum-Chemical Design by DFT Method" Quantum Reports 5, no. 1: 282-293. https://doi.org/10.3390/quantum5010019
APA StyleMikhailov, O. V., & Chachkov, D. V. (2023). Molecular Structure of M(N13) Compounds with 12-Membered Nitrogen-Containing Cycle and Axial Nitrogen Atom (M = Mn, Fe): Quantum-Chemical Design by DFT Method. Quantum Reports, 5(1), 282-293. https://doi.org/10.3390/quantum5010019