

Comparison of the Molecular Properties of Euglobals Differing by the Mutual Positions of the Two R–C=O Groups (R = H and CH2CH(CH3)2): A Computational Study

Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
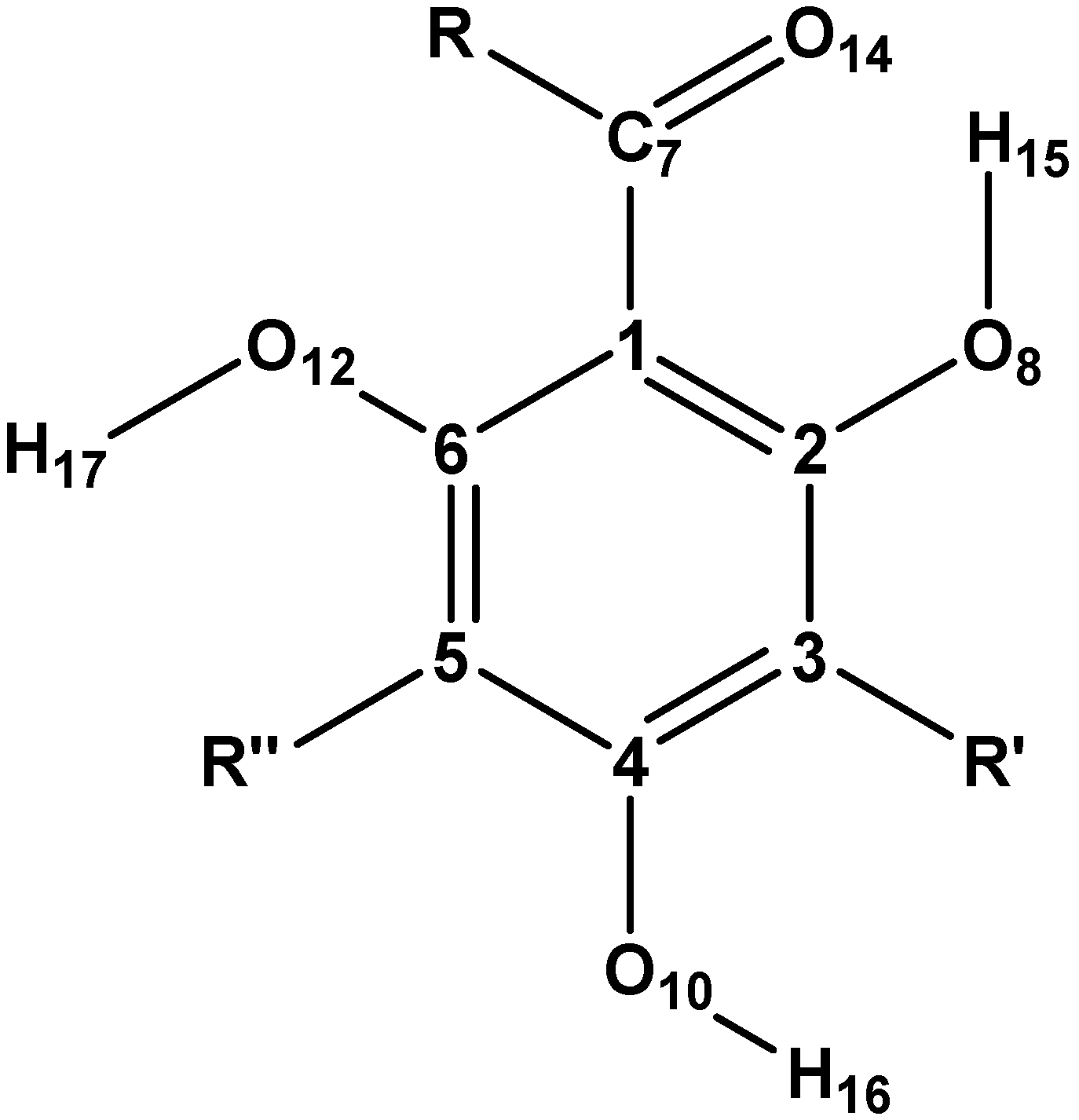
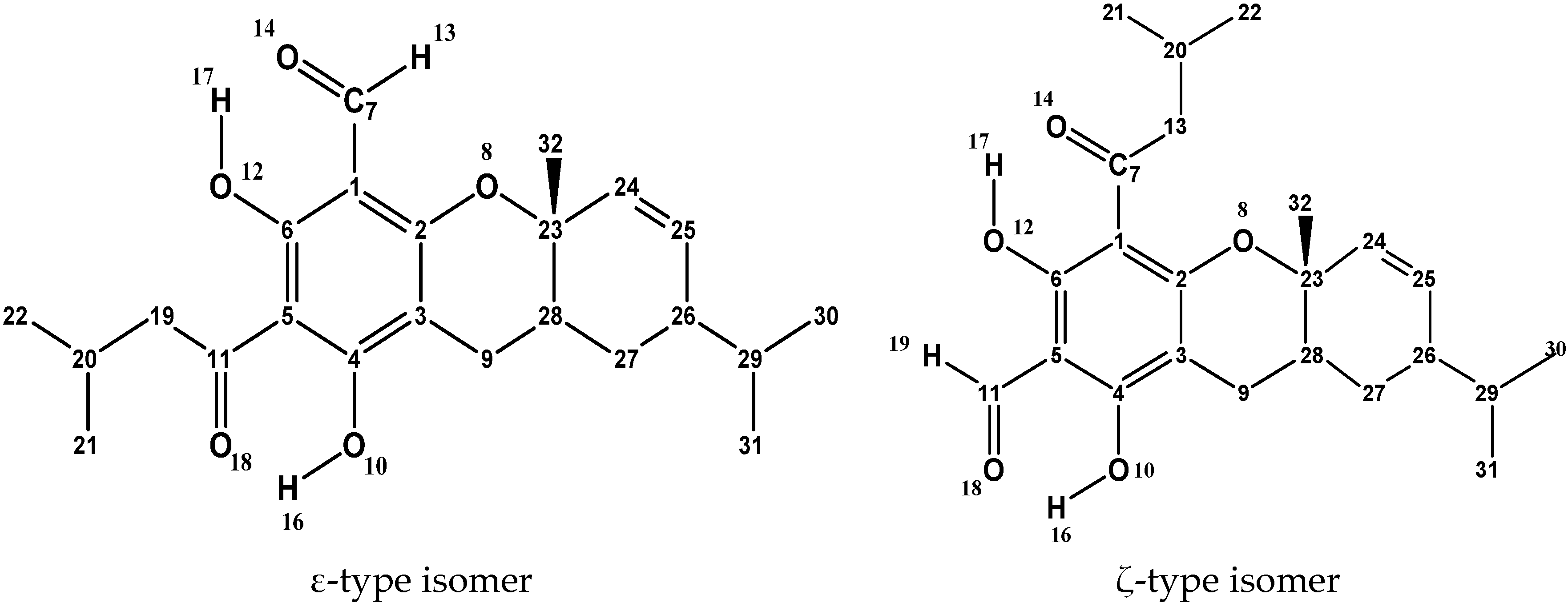
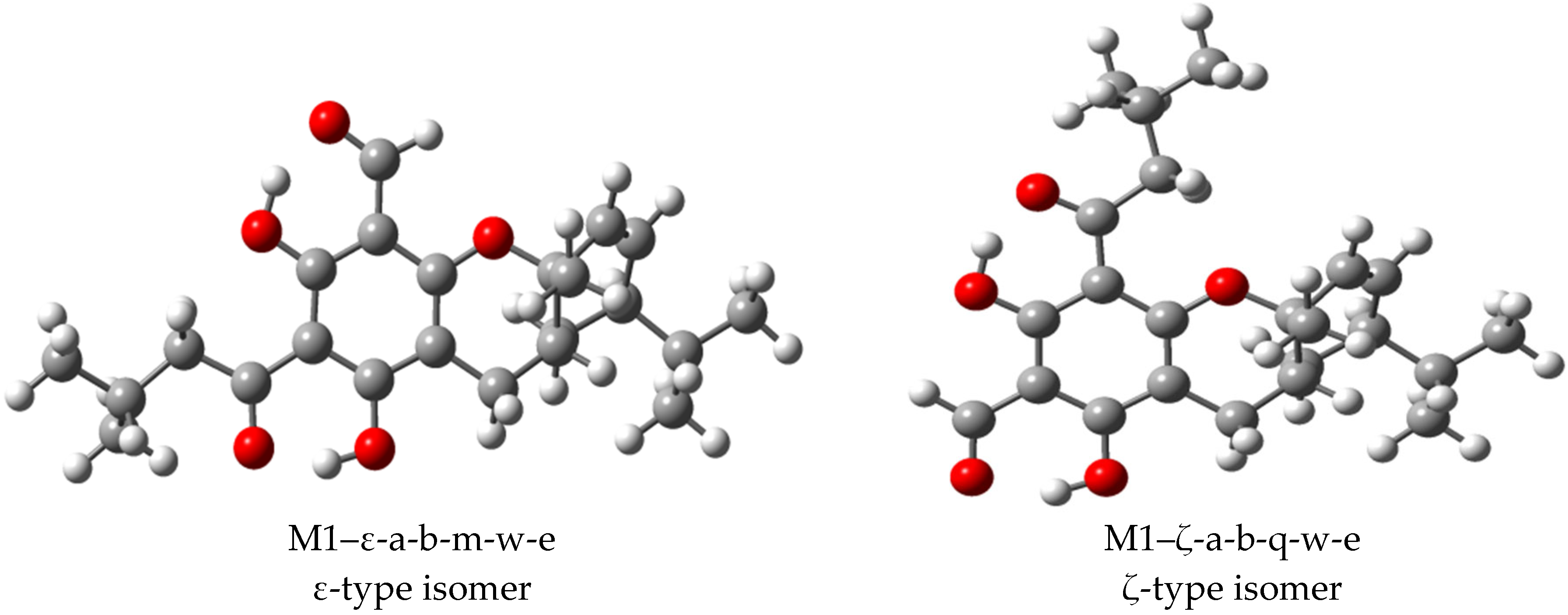
3.1. Selection of Structures and Naming of Structures and Conformers
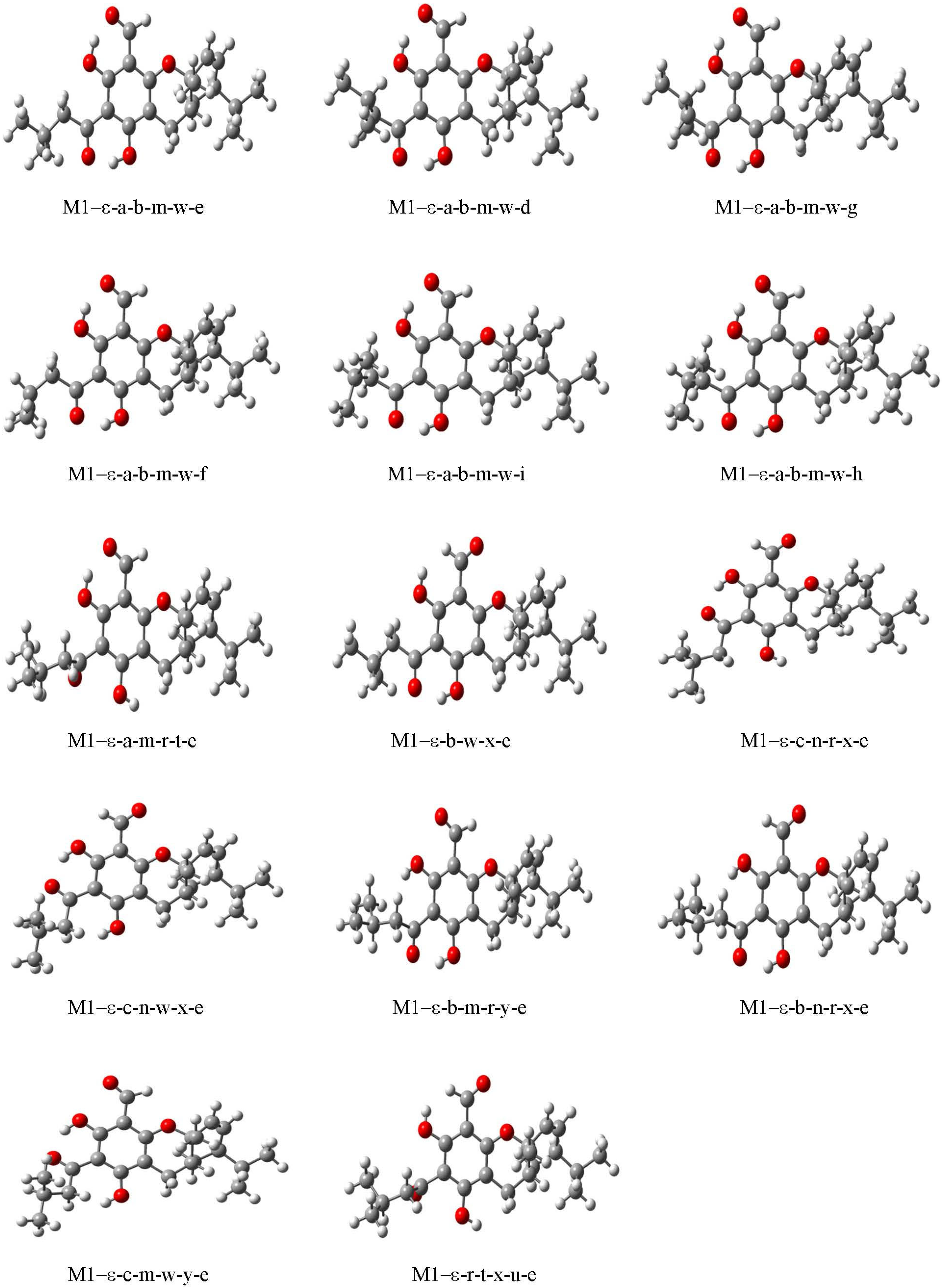
3.2. Conformational Preferences and Energetics
3.2.1. Conformers’ Geometrical Characteristics
3.2.2. Conformers’ Relative Energies and Factors Influencing Them
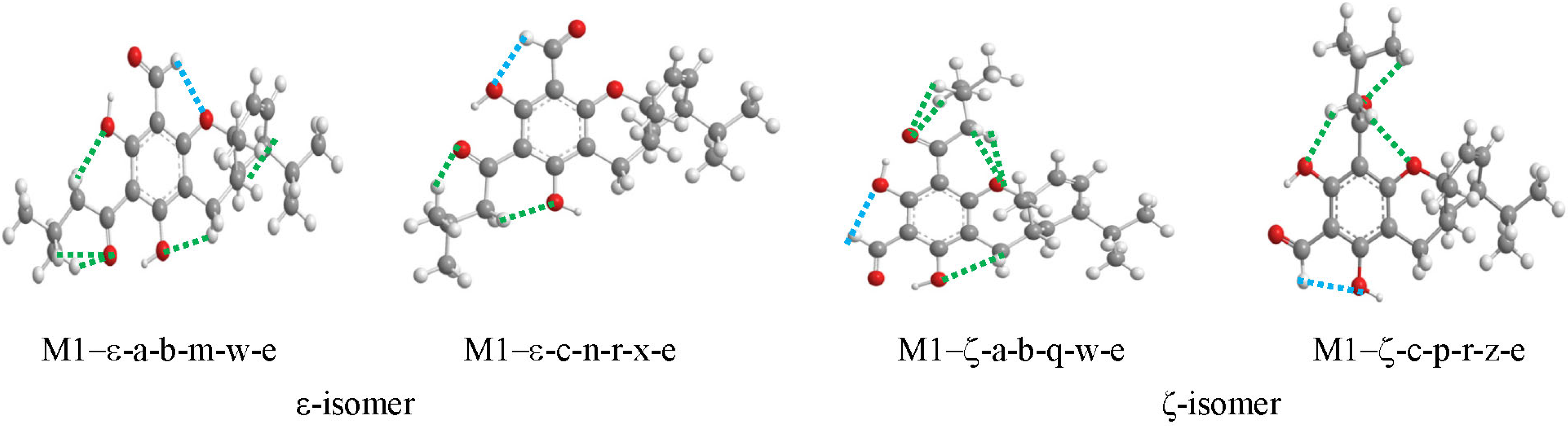
3.3. Characteristics of the IHBs
3.3.1. Characteristics of the O–H···O IHBs
Parameters of the O–H···O IHBs
Energy Increase Accompanying the Removal of O–H···O IHBs
Vibrational Frequency Decreases Associated with O–H···O IHBs
3.3.2. Characteristics of the C–H···O IHBs
Parameters of the C–H···O IHBs
Vibrational Frequency Increases Associated with C–H···O IHBs
3.4. Other Molecular Properties
3.4.1. Energies of the Frontier Molecular Orbitals and Selected Derived Quantities
3.4.2. Dipole Moments of the Conformers
3.4.3. The Mulliken Charges on Relevant Atoms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 30 February 2022).
- World Health Organization. World Cancer Report 2019. Available online: https://www.who.int/healthtopics/cancer#tab=tab_1 (accessed on 15 December 2021).
- Chen, S.; Cao, Z.; Prettner, K.; Kuhn, M.; Yang, J.; Jiao, L.; Wang, Z.; Li, W.; Geldsetzer, P.; Bärnighausen, T.; et al. Estimates and projections of the global economic cost of 29 cancers in 204 countries and territories from 2020 to 2050. JAMA Oncol. 2023, 9, 465. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Rahman, M.; Hasan, M.R. Cancer metabolism and drug resistance. Metabolites 2015, 5, 571–600. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Niero, E.L.; Rocha-Sales, B.; Lauand, C.; Cortez, B.A.; Souza, M.M.; Rezende-Teixeira, P.; Machado-Santelli, G.M. The multiple facets of drug resistance: One history, different approaches. J. Exp. Clin. Cancer Res. 2014, 33, 37. [Google Scholar] [CrossRef] [PubMed]
- Mann, J. Natural products in cancer chemotherapy: Past, present and future. Nat. Rev. Cancer 2002, 2, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S. New aspects of natural products in drug discovery. Trends Microbiol. 2007, 15, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, M.; Kapulnik, Y.; Koltai, H. Plant derived substances with anti-cancer activity: From folklore to practice. Front. Plant Sci. 2015, 6, 799. [Google Scholar] [CrossRef] [PubMed]
- Gurnani, N.; Mehta, D.; Gupta, M.; Mehta, B.K. Natural products: Source of potential drugs. Aust. J. Basic Appl. Sci. 2014, 6, 171–186. [Google Scholar]
- Cragg, G.M.; Newman, D.J. Natural product drug discovery in the next millennium. Pharm. Biol. 2001, 39 (Suppl. S1), 8–17. [Google Scholar] [PubMed]
- Singh, I.P.; Bharate, S.B. Phloroglucinol Compounds of Natural Origin. Nat. Prod. Rep. 2006, 23, 558–591. [Google Scholar] [CrossRef] [PubMed]
- Mammino, L.; Kabanda, M.M. Model structures for the study of acylated phloroglucinols and computational study of the caespitate molecule. J. Mol. Struct. Theochem 2007, 805, 39–52. [Google Scholar] [CrossRef]
- Mammino, L.; Kabanda, M.M. A study of the intramolecular hydrogen bond in acylphloroglucinols. J. Molec. Struct. Theochem 2009, 901, 210–219. [Google Scholar] [CrossRef]
- Kabanda, M.M.; Mammino, L. The conformational preferences of acylphloroglucinols—A promising class of biologically active compounds. Int. J. Quantum Chem. 2012, 112, 3691–3702. [Google Scholar] [CrossRef]
- Yunta, M.J.R. Is it important to compute intramolecular hydrogen bonding in drug design? Am. J. Model. Optim. 2017, 5, 24–57. [Google Scholar]
- Fersht, A.R. The hydrogen bond in molecular recognition. Trend. Biochem. Sci. 1987, 12, 301–304. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Seto, N.O.L.; Cai, Y.; Leinala, E.K.; Borisova, S.N.; Palcic, M.M.; Evans, S.V. The influence of an intramolecular hydrogen bond in differential recognition of inhibitory acceptor analogs by human ABO(H) blood group A and B glycosyltransferases. J. Biol. Chem. 2003, 278, 49191–49195. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, M.; Konoshima, T.; Kozuka, M.; Tokuda, H. Anti-tumor-promoting activities of euglobals from Eucalyptus plants. Biol. Pharm. Bull. 1995, 18, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, M.; Konoshima, T.; Fujitani, K.; Yoshida, S.; Nishimura, H.; Tokuda, H.; Kozuka, M. Inhibitors of Skin-Tumor Promotion. VIII.: Inhibitory Effects of Euglobals and Their Related Compounds on Epstein-Barr Virus Activation. Chem. Pharm. Bull. 1990, 38, 2737–2739. [Google Scholar] [CrossRef]
- Takasaki, M.; Konoshima, T.; Etoh, H.; Singh, I.P.; Tokuda, H.; Nishino, H. Cancer chemopreventive activity of euglobal-G1 from leaves of Eucalyptus grandis. Cancer Lett. 2000, 155, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F.; Salvetti, M. Epstein-Barr virus and multiple sclerosis: Supporting causality. Lancet Neurol. 2022, 21, 300–301. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Soldan, S.S.; Lieberman, P.M. Epstein-Barr virus and multiple sclerosis. Nat. Rev. Microbiol. 2022, 21, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Thomas, O.G.; Bronge, M.; Tengvall, K.; Akpinar, B.; Nilsson, O.B.; Holmgren, E.; Hessa, T.; Gafvelin, G.; Khademi, M.; Alfredsson, L.; et al. Cross-reactive EBNA1 immunity targets alpha-crystallin B and is associated with multiple sclerosis. Sci. Adv. 2023, 9, eadg3032. [Google Scholar] [CrossRef]
- Bharate, S.B.; Bhutani, K.K.; Khan, S.I.; Tekwani, B.L.; Jacob, M.R.; Khan, I.A.; Singh, I.P. Biomimetic synthesis, antimicrobial, antileishmanial and antimalarial activities of euglobals and their analogues. Bioorg. Med. Chem. 2006, 14, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Snyder, J.K.; Nakanishi, K. Strongadials A and B from Eucalyptus Stronga. J. Am. Chem. Soc. 1984, 106, 734–736. [Google Scholar] [CrossRef]
- Zhang, Y.; He, X.Z.; Yang, H.; Liu, H.Y.; An, L.-K. Strongadial A and B from Eucalyptus Globulus Labill. and their Anticancer Activity as Selective Tyrosyl-Dna Phosphodiesterase 2 Inhibitors. Phytother Res. 2021, 35, 5282–5289. [Google Scholar] [CrossRef] [PubMed]
- Al-Snafi, A.E. The pharmacological and therapeutic importance of Eucalyptus species grown in Iraq. IOSR J. Pharm. 2017, 7, 72–91. [Google Scholar] [CrossRef]
- Sawada, T.; Kozuka, M.; Komiya, T.; Amano, T.; Goto, M. Euglobal-III, a novel granulation inhibiting agent from Eucalyptus globulus Labill. Chem. Pharm. Bull. 1980, 28, 2546–2548. [Google Scholar] [CrossRef]
- Kozuka, M.; Sawada, T.; Kasahara, F.; Mizuta, E.; Amano, T.; Komiya, T.; Goto, M. The granulation-inhibiting principles from Eucalyptus globulus Labill. II. The structures of euglobal-Ia1,-Ia2,-Ib,-Ic,-IIa,-IIb and-IIc. Chem. Pharm. Bull. 1982, 30, 1952–1963. [Google Scholar] [CrossRef]
- Singh, I.P.; Sidana, J.; Bharatea, S.B.; Foley, W.J. Phloroglucinol Compounds of Natural Origin: Synthetic Aspects. Nat. Prod. Rep. 2010, 27, 393–416. [Google Scholar] [CrossRef]
- John, J.W. (Ed.) Eucalyptus: The Genus Eucalyptus. Coppen Euglobal III Glycyrrhetic Acid. Available online: https://books.google.co.za/books?id=0dRlDMvlhQ0C&pg=PA286&lpg=PA286&dq=Eucalyptus:+The+Genus+Eucalyptus,+Edited+By+John+J.W.+Coppen+Euglobal+Iii+Glycyrrhetic+Acid&source=bl&ots=rHKwQl8bvI&sig=ACfU3U2W0OANK688YiPU7PA70V7X2zzhEQ&hl=en&sa=X&ved=2ahUKEwih-LX_y8j3AhVlQvEDHZaeBv8Q6AF6BAgCEAM#v=onepage&q=Eucalyptus%3A%20The%20Genus%20Eucalyptus%2C%20Edited%20By%20John%20J.W.%20Coppen%20Euglobal%20Iii%20Glycyrrhetic%20Acid&f=false (accessed on 19 November 2021).
- Begleiter, A.; Lin, D.; Larson, K.K.; Lang, J.; Wu, X.; Cabral, T.; Taylor, H.; Guziec, L.J.; Kerr, P.D.; Hasinoff, B.B.; et al. Structure-activity studies with cytotoxic anthrapyrazoles. Oncol. Rep. 2006, 15, 1575–1580. [Google Scholar] [CrossRef] [PubMed]
- Perlovich, G.L.; Volkova, T.V.; Manin, A.N.; Bauer-Brandl, A. Influence of position and size of substituents on the mechanism of partitioning: A thermodynamic study on acetaminophens, hydroxybenzoic acids, and parabens. AAPS PharmSciTech 2008, 9, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.L.; Marusich, J.A.; Huffman, J.W. Moving around the molecule: Relationship between chemical structure and in vivo activity of synthetic cannabinoids. Life Sci. 2014, 97, 55–63. [Google Scholar] [CrossRef]
- Dean, J.A.; Liu, D. Mechanisms of Molecular Activity; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Durrant, M.C.; Oprea, T.I. Quantitative structure-activity relationship analysis in drug discovery. J. Med. Chem. 2017, 60, 1283–1300. [Google Scholar]
- Patle, S.; Saini, S. Hydrophobic effect on drug stability and oral bioavailability. J. Pharm. Sci. 2016, 105, 4037–4049. [Google Scholar]
- Cremer, D. Møller–Plesset perturbation theory: From small molecule methods to methods for thousands of atoms. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 509–530. [Google Scholar] [CrossRef]
- Mammino, L.; Kabanda, M.M. A computational study of the effects of different solvents on the characteristics of the intramolecular hydrogen bond in acylphloroglucinols. J. Phys. Chem. A 2009, 113, 15064–15077. [Google Scholar] [CrossRef]
- Mammino, L.; Kabanda, M.M. Computational study of the patterns of weaker intramolecular hydrogen bonds stabilizing acylphloroglucinols. Int. J. Quantum Chem. 2012, 112, 2650–2658. [Google Scholar] [CrossRef]
- Mammino, L.; Kabanda, M.M. The role of additional O–H∙∙∙O intramolecular hydrogen bonds for acylphloroglucinols’ conformational preferences in vacuo and in solution. Mol. Simul. 2013, 39, 1–13. [Google Scholar] [CrossRef]
- Polo, V.; Kraka, E.; Cremer, D. Some thoughts about the stability and reliability of commonly used exchange–correlation functionals–coverage of dynamic and nondynamic correlation effects. Theor. Chem. Acc. 2002, 107, 291–303. [Google Scholar] [CrossRef]
- Chiodo, S.G.; Leopoldini, M.; Russo, N.; Toscano, M. The inactivation of lipid peroxide radical by quercetin, A theoretical insight. Phys. Chem. Chem. Phys. 2010, 12, 7662–7670. [Google Scholar] [CrossRef]
- Baker, J.; Autschbach, J.; Zheng, J. Performance of DFT methods for molecular properties of polarizable molecules. J. Chem. Theory Comput. 2010, 7, 26–35. [Google Scholar]
- Merrick, J.P.; Moran, D.; Radom, L. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef]
- Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Scalmani, G.; Frisch, M.J.; Barone, V.; et al. Gaussian 09, Revision E. 01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Dennington, R.D.I.I.; Keith, T.; Millam, J. GaussView, Version 4.1. 2; Semichem Inc.: Shawnee Mission, KS, USA, 2007. [Google Scholar]
- ChemDraw Ultra. 6.0 And Chem3D Ultra; Cambridge Soft Corporation: Cambridge, MA, USA, 2001. [Google Scholar]
- Mammino, L. A Computational Study of Euglobal G1–An Acylphloroglucinol with Anticancer Activity. Curr. Bioact. Compd. 2014, 10, 163–180. [Google Scholar] [CrossRef]
- Mammino, L. A Computational Study of Euglobal G4. In Proceedings of the 10th Theoretical Chemistry Conference in Africa and Eastern and Southern Africa Environmental Chemistry Conference; Mammino, L., van Ree, T., Eds.; Kalahari Productions and Booksellers: Thohoyandou, South Africa, 2016; pp. 29–55. [Google Scholar]
- Spoliti, M.; Bencivenni, L.; Quirante, J.J.; Ramondo, F. Molecular conformations and harmonic force field of 1,3,5-benzenetriol molecule from ab initio and density functional theory investigations. J. Mol. Struct. THEOCHEM 1997, 390, 139–148. [Google Scholar] [CrossRef]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the beta-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 2. Intercorrelation between crystal structure and spectroscopic parameters in eight intramolecularly hydrogen bonded 1, 3-diaryl-1, 3-propanedione enols. J. Am. Chem. Soc. 1991, 113, 4917–4925. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Gallegos, M.; Valentin-Rodriguez, M.A.; Costales, A.; Rocha-Rinza, T.; Pendás, A.M. On the relationships between hydrogen bond strength and the formation energy in resonance-assisted hydrogen bonds. Molecules 2021, 26, 4196. [Google Scholar] [CrossRef] [PubMed]
- Grosch, A.A.; van der Lubbe, S.C.C.; Guerra, C.F. Nature of intramolecular resonance assisted hydrogen bonding in malonalehyde and its saturated analogue. J. Phys. Chem. A 2018, 122, 1813–1820. [Google Scholar] [CrossRef]
- Barnes, A.J. Blue-shifting hydrogen bonds—Are they improper or proper? J. Molec. Struct. 2004, 704, 3–9. [Google Scholar] [CrossRef]
- Das, M.; Ghosh, S.K. A computational investigation of the red and blue shifts in hydrogen bonded systems. J. Chem. Sci. 2017, 129, 975–981. [Google Scholar] [CrossRef]
- Joseph, J.; Jemmis, E.D. Red-, Blue-, or No-Shift in Hydrogen Bonds: A Unified Explanation. J. Am. Chem. Soc. 2007, 129, 4620–4632. [Google Scholar] [CrossRef]
- Struble, M.D.; Kelly, C.; Siegler, M.A.; Lectka, T. Search for a Strong, Virtually “No-Shift” Hydrogen Bond: A Cage Molecule with an Exceptional OH⋅⋅⋅F Interaction. Angew. Chem. Int. Ed. 2014, 53, 8924–8928. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. The C-H⋅⋅⋅O hydrogen bond: Structural implications and supramolecular design. Acc. Chem. Res. 1996, 29, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.L.; Kar, T.; Scheiner, S. Fundamental properties of the CH⋅⋅⋅O interaction: Is it a true hydrogen bond? J. Am. Chem. Soc. 1999, 121, 9411–9422. [Google Scholar] [CrossRef]
- An, N.T.; Duong, N.T.; Tri, N.N.; Trung, N.T. Role of O–H/O/S conventional hydrogen bonds in considerable Csp2–H blue-shift in the binary systems of acetaldehyde and thioacetaldehyde with substituted carboxylic and thiocarboxylic acids. RSC Adv. 2022, 12, 35309–35319. [Google Scholar] [CrossRef]
- Cuc, N.T.T.; Phan, C.D.; Nhung, N.T.A.; Nguyen, M.T.; Trung, N.T.; Ngan, V.T. Theoretical Aspects of Nonconventional Hydrogen Bonds in the Complexes of Aldehydes and Hydrogen Chalcogenides. J. Phys. Chem. A 2021, 125, 10291–10302. [Google Scholar] [CrossRef]
- Tabayashi, K.; Chohda, M.; Yamanaka, T.; Tsutsumi, Y.; Takahashi, O.; Yoshida, H.; Taniguchi, M. Hydrogen bonding interaction of small acetaldehyde clusters studied with core-electron excitation spectroscopy in the oxygen K-edge region. J. Phys. Conf. Ser. 2010, 235, 012017. [Google Scholar] [CrossRef]
- Trung, N.T.; Hung, N.P.; Hue, T.T.; Nguyen, M.T. Existence of both blue-shifting hydrogen bond and Lewis acid–base interaction in the complexes of carbonyls and thiocarbonyls with carbon dioxide. Phys. Chem. Chem. Phys. 2011, 13, 14033–14042. [Google Scholar] [CrossRef]
- Tang, Q.; Huang, T.; Huang, R.; Cao, H.; Wang, L.; Zheng, X. Theoretical Researches On Binding Modes and Stability of Hydrogen Bonds Between Uracil and Formic Acid. Res. Square 2022. [Google Scholar] [CrossRef]
- Scheiner, S.; Kar, T.; Gu, Y. Strength of the CαH···O Hydrogen Bond of Amino Acid Residues. J. Biol. Chem. 2001, 276, 9832–9837. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. The Hydrogen Bond. A Hundred Years and Counting. 2019. Available online: https://digitalcommons.usu.edu/cgi/viewcontent (accessed on 16 March 2023).
- Chen, J.C.; Li, J.; Qian, L.; Zheng, K.C. Electronic structures and SARs of the isomeric complexes α-, β-, γ- [Ru(mazpy)2Cl2] with different antitumor activities. J. Molec. Struct. THEOCHEM 2005, 728, 93–101. [Google Scholar] [CrossRef]
- Hossan, A.; Alrefaei, A.F.; Katouah, H.A.; Bayazeed, A.; Asghar, B.H.; Shaaban, F.; El-Metwaly, N.M. Synthesis, anticancer activity, and molecular docking of new pyrazolo[1,5-a]pyrimidine derivatives. J. Saudi Chem. Soc. 2023, 27, 101599. [Google Scholar] [CrossRef]
- Karrouchi, K.; Brandan, S.A.; Sert, Y.; El-marzouqi, H.; Radi, S.; Ferbinteanu, M.; Faouzi, M.E.A.; Garcia, Y.; Ansar, M. Synthesis, X-ray structure, vibrational spectroscopy, DFT, biological evaluation and molecular docking studies of (E)-N’-(4-(dimethylamino)benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2020, 1219, 128541. [Google Scholar] [CrossRef]
- Rahmouni, N.T.; el Houda Bensiradj, N.; Megatli, S.A.; Djebbar, S.; Baitich, O.B. New mixed amino acids complexes of iron(III) and zinc(II) with isonitrosoacetophenone: Synthesis, spectral characterization, DFT study and anticancer activity. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 213, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Kumer, A.M.; Sarker, N.; Paul, S. The theoretical investigation of HOMO, LUMO, thermophysical properties and QSAR study of some aromatic carboxylic acids using HyperChem programming. Int. J. Chem. Technol. 2019, 3, 26–37. [Google Scholar] [CrossRef]
- Javed, F.; Sirajuddin, M.; Ali, S.; Khalid, N.; Tahir, M.N.; Shah, N.A.; Rasheed, Z.; Khan, M.R. Organotin(IV) Derivatives of o-isobutyl carbonodithioate: Synthesis, spectroscopic characterization, X-ray structure, HOMO/LUMO and in vitro biological activities. Polyhedron 2016, 104, 80–90. [Google Scholar] [CrossRef]
- Salihović, M.; Huseinović, Š.; Špirtović-Halilović, S.; Osmanović, A.; Dedić, A.; Ašimović, Z.; Završnik, D. DFT Study and Biological Activity of Some Methylxanthines. Bull. Chem. Technol. Bosnia Herzeg. 2014, 42, 31–36. [Google Scholar]
- Kirishnamaline, G.; Daisy Magdaline, J.; Chithambarathanu, T.; Aruldhas, D.; Ronaldo Anuf, A. Theoretical investigation of structure, anticancer activity and molecular docking of thiourea derivatives. J. Molec. Struct. 2021, 1225, 129118. [Google Scholar] [CrossRef]
- Lima, F.C.; So, Y.A.O.; Gargano, R.; Fujimori, M.; França, E.L.; Honorio-França, A.C.; Gatto, C.C. Synthesis, theoretical calculation and anticancer activity of 4,6-diacetylresorcinol-dithiocarbazates and their Copper(II) complexes. J. Molec. Struct. 2020, 1212, 128083. [Google Scholar] [CrossRef]
- Manoj, K.P.; Elangovan, N.; Chandrasekar, S. Synthesis, XRD, hirshfeld surface analysis, ESP, HOMO-LUMO, quantum chemical modeling and anticancer activity of di(p-methyl benzyl)(dibromo) (1,10-phenanthroline) tin(IV) complex, Inorg. Chem. Commun. 2022, 139, 109324. [Google Scholar] [CrossRef]
- Kostal, J.; Voutchkova-Kostal, A.; Anastas, P.T.; Zimmerman, J.B. Identifying and designing chemicals with minimal acute aquatic toxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 6289–6294. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraj, R.; Subramanian, V.; Chattaraj, P.K. Comparison of global reactivity descriptors calculated using various density functionals: A QSAR perspective. J. Chem. Theory Comput. 2009, 5, 2744–2753. [Google Scholar] [CrossRef] [PubMed]
- Mebi, C.A. DFT study on structure, electronic properties, and reactivity of cis-isomers of [(NC5H4-S)2Fe(CO)2]. J. Chem. Sci. 2011, 123, 727–731. [Google Scholar] [CrossRef]
- Domingo, L.R.; Mar Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Tanış, E.; Cankaya, N.; Yalçın, S. Synthesis, characterization, computation of global reactivity descriptors and antiproliferative activity of N-(4-nitrophenyl) acrylamide. Russian J. Phys. Chem. B 2019, 13, 49–61. [Google Scholar] [CrossRef]
- Miar, M.; Shiroudi, A.; Pourshamsian, K.; Oliaey, A.R.; Hatamjafari, F. Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo[d]thiazole-2(3H)-imine and its para-substituted derivatives: Solvent and substituent effects. J. Chem. Res. 2021, 45, 147–158. [Google Scholar]
- Kaya, S.; Kaya, C. A new method for calculation of molecular hardness: A theoretical study. Comput. Theor. Chem. 2015, 1060, 66–70. [Google Scholar] [CrossRef]
- Oláh, J.; Van Alsenoy, C.; Sannigrahi, A.B. Condensed Fukui Functions Derived from Stockholder Charges: Assessment of Their Performance as Local Reactivity Descriptors. J. Phys. Chem. A 2002, 106, 3885–3890. [Google Scholar] [CrossRef]
- Vektariene, A.; Vektaris, G.; Svoboda, J. A theoretical approach to the nucleophilic behavior of benzofused thieno[3,2-b]furans using DFT and HF based reactivity descriptors. Arkivoc 2009, 2009, 311–329. [Google Scholar] [CrossRef]
- Mahmoud, S.; Said, M.S.; Najim, Z.A. A Theoretical Approach to Relate the Reactivity Descriptors and Mulliken Charges with Carcinogenity of Some Methylated Benzo[a]Anthracene. Pak. J. Anal. Environ. Chem. 2012, 13, 40–47. [Google Scholar]
- Babu, N.S.; Jayaprakash, D. Studies for reactivity indexes of cyanuric acid tautomers in different solvents by ab initio Hartree–Fock (HF) theory. J. Adv. Chem. 2015, 11, 3828–3837. [Google Scholar]
- Mendoza Huizar, L.H.; Rios-Reyes, C.H.; Olvera-Maturano, N.J.; Robles, J.; Rodriguez, J.A. Chemical reactivity of quinclorac employing the HSAB local principle—Fukui function. Open Chem. 2015, 13, 52–60. [Google Scholar] [CrossRef]
- Prasad, S.; Ojha, D.P. Geometric structures, vibrational spectroscopic and global reactivity descriptors of nematogens containing strong polar group- A comparative analysis using DFT, HF and MP2 methods. Molec. Cryst. Liq. Cryst. 2019, 666, 12–28. [Google Scholar] [CrossRef]
- Das, A.; Das, A.; Banik, B.K. Influence of dipole moments on the medicinal activities of diverse organic compounds. J. Indian Chem. Soc. 2021, 98, 100005. [Google Scholar] [CrossRef]
- Bushelyev, S.N.; Stepanov, N.F. Elektronnaya Struktura y Biologhicheskaya Aktivnost Molecul; Khimiya, Snanye: Moscow, Russia, 1989. [Google Scholar]
- Dashnau, J.L.; Sharp, K.A.; Vanderkooi, J.M. Carbohydrate intramolecular hydrogen bonding cooperativity and its effect on water structure. J. Phys. Chem. B 2005, 109, 24152–24159. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Weinhold, F. Natural Localized Molecular Orbitals. J. Chem. Phys. 1985, 83, 1736–1741. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 0.6: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terpene Moiety | ε-Type Molecule | ζ-Type Molecule | |||
|---|---|---|---|---|---|
| Molecule | Acronym | Molecule | Acronym | ||
| monoterpene | α-phellandrene | Euglobal IIC | M1-ε | Euglobal T1 | M1-ζ |
| α-pinane | Euglobal G2 | M2-ε | Euglobal G1 | M2-ζ | |
| β-pinane | Euglobal G3 | M3-ε | Euglobal G4 | M3-ζ | |
| pinane | Euglobal G5 | M4-ε | [model] | M4*-ζ | |
| γ- terpinene | Euglobal G6 | M5-ε | Euglobal G7 | M5-ζ | |
| γ- terpinene | Euglobal G8 | M6-ε | [model] | M6*-ζ | |
| α-terpinene | Euglobal G9 | M7-ε | Euglobal G10 | M7-ζ | |
| α-terpinene | Euglobal G11 | M8-ε | [model] | M8*-ζ | |
| terpinolene | [model] | M9*-ε | Euglobal G12 | M9-ζ | |
| sesquiterpene | bicyclogermacrane | Euglobal VII | M10-ε | [model] | M10*-ζ |
| Feature Category | Symbol | Meaning of Symbol |
|---|---|---|
| Molecule type | ε | Isomer with the acyl group with R = H attached at C1 (ε-type) |
| ζ | Isomer with the acyl group with R = isobutyl attached at C1 (ζ-type) | |
| * | Molecule not found in the literature, but used as model to complete a pair | |
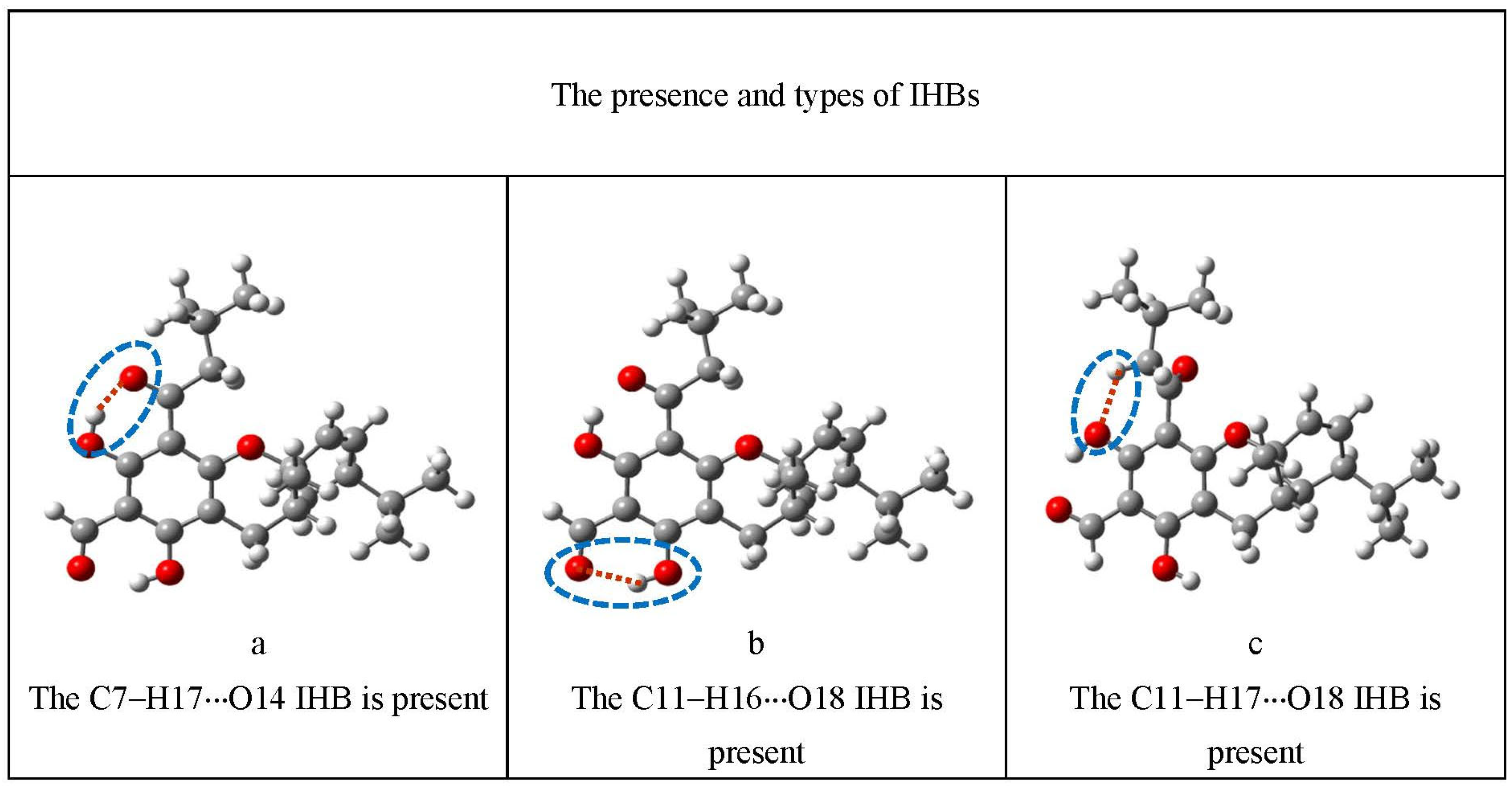
| IHBs present | a | The H17···O14 IHB is present |
| b | The H16···O18 IHB is present | |
| c | The H17···O18 IHB is present | |
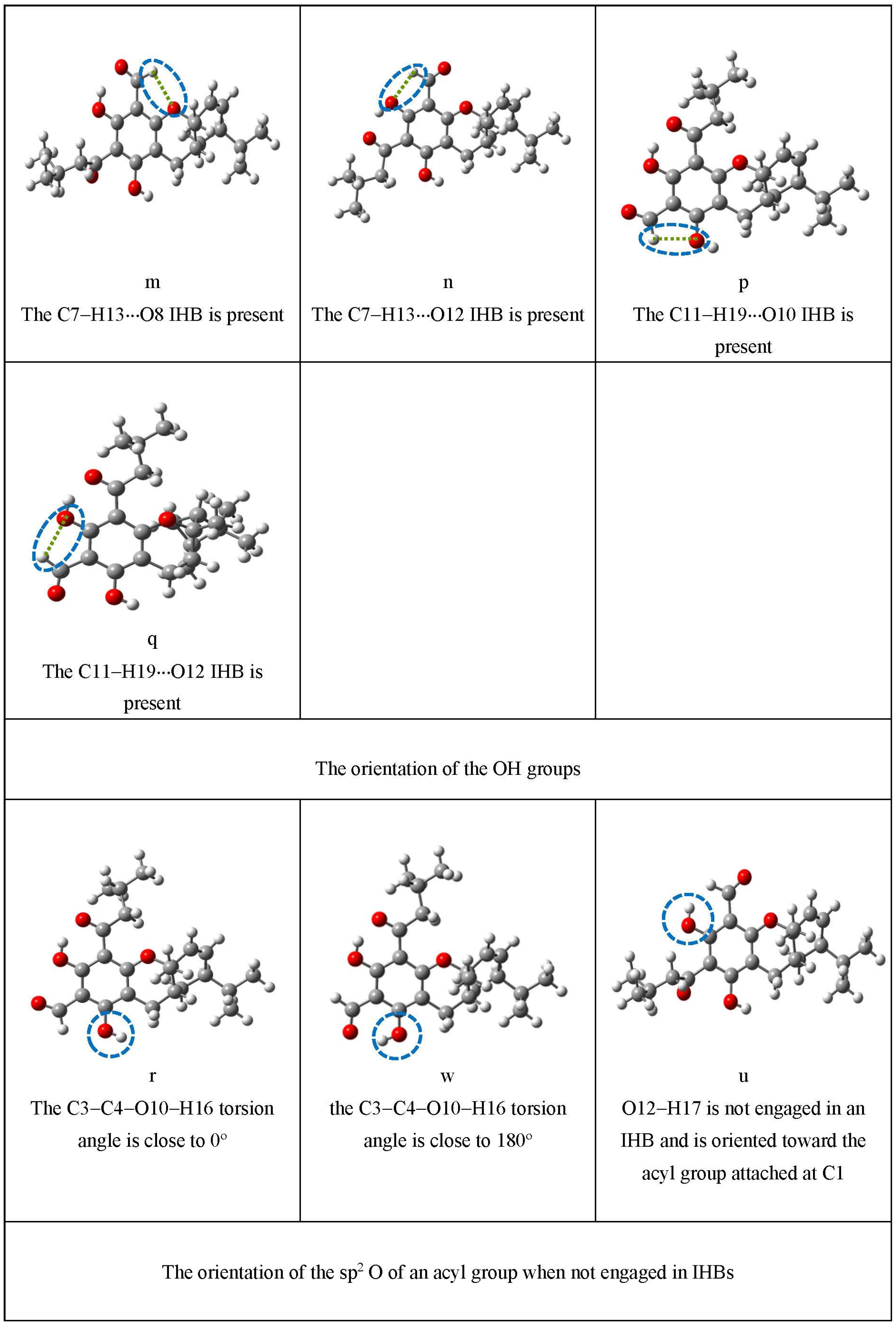
| m | The C7–H13···O8 IHB is present | |
| n | The C7–H13···O12 IHB is present | |
| p | The C11–H19···O10 IHB is present | |
| q | The C11–H19···O12 IHB is present | |
| Orientation of OHs | r | the C3–C4–O10–H16 torsion angle is close to 0° |
| w | the C3–C4–O10–H16 torsion angle is close to 180° | |
| u | O12–H17 is not engaged in an IHB and is oriented toward the acyl group attached at C1 | |
| Orientation of sp2 O not engaged in an IHB (determined by the orientation of the acyl group) | x | O14 is not engaged in an IHB and is oriented towards O8 |
| y | O14 is not engaged in an IHB and is oriented towards O12 | |
| v | O14 is not engaged in an IHB and is off-plane, ‘towards us’ | |
| z | O14 is not engaged in an IHB and is off-plane, ‘away from us’ | |
| j | O18 is not engaged in an IHB and is oriented towards O10 | |
| k | O18 is not engaged in an IHB and is oriented towards O12 | |
| s | O18 is not engaged in an IHB and is off-plane, ‘towards us’ | |
| t | O18 is not engaged in an IHB and is off-plane, ‘away from us’ | |
| Orientation of the isobutylchain | e | the two methyls of the isobutyl group are oriented on the other side with respect to the sp2 O of that group |
| f | the two methyls of the isobutyl group are oriented towards the sp2 O of that group | |
| d | the isobutyl group is oriented ‘towards us’, with the H atom attached to C20 facing towards O12 (ε-isomer) or towards O8 (ζ-isomer) | |
| i | the isobutyl group is oriented ‘towards us’, with the H atom attached to C20 facing away from O12 (ε-isomer) or from O8 (ζ-isomer) | |
| g | the isobutyl group is oriented ‘away from us’, with the H atom attached to C20 facing towards O12 (ε-isomer) or towards O8 (ζ-isomer) | |
| h | the isobutyl group is oriented away from us, with the H atom attached to C20 facing away from O12 (ε-isomer) or from O8 (ζ-isomer) |
| ε-Type Molecule | ζ-Type Molecule | ||||
|---|---|---|---|---|---|
| Conformer Type | O–H···O IHBs Present | C–H···O IHBs Present | Conformer Type | O–H···O IHBs Present | C–H···O IHBs Present |
| a-b-m-w-e | H17···O14 H16···O18 | C7–H13···O8 C19–H19a···O12 C19–H19b···O12 C22–H22a···O18 C20–H20···O18 C9–H9···O10 | a-b-q-w-e | H17···O14 H16···O18 | C11–H19···O12 C13–H13a···O8 C13–H13b···O8 C22–H22a···O14 C20–H20···O14 C9–H9···O10 |
| a-b-m-w-d | H17···O14 H16···O18 | C7–H13···O8 C19–H19a···O12 C19–H19b···O10 C20–H20···O12 C9–H9···O10 | a-b-q-w-g | H17···O14 H16···O18 | C11–H19···O12 C13–H13a···O12 C13–H13b···O8 C20–H20···O8 C9–H9···O10 |
| a-b-m-w-g | H17···O14 H16···O18 | C7–H13···O8 C19–H19a···O10 C19–H19b···O12 C20–H20···O12 C9–H9···O10 | a-b-q-w-d | H17···O14 H16···O18 | C11–H19···O12 C13–H13a···O12 C13–H13b···O8 C20–H20···O8 C9–H9···O10 |
| a-b-m-w-f | H17···O14 H16···O18 | C7–H13···O8 C19–H19a···O12 C19–H19b···O12 C9–H9···O10 | a-b-q-w-f | H17···O14 H16···O18 | C11–H19···O12 C13–H13a···O8 C13–H13b···O8 C9–H9···O10 |
| a-b-m-w-i | H17···O14 H16···O18 | C7–H13···O8 C19–H19a···O12 C19–H19b···O10 C21–H21a···O12 C22–H22a···O10 C9–H9···O10 | a-b-q-w-h | H17···O14 H16···O18 | C11–H19···O12 C13–H13a···O12 C13–H13b···O8 C22–H22a···O12 C9–H9···O10 |
| a-b-m-w-h | H17···O14 H16···O18 | C7–H13···O8 C19–H19a···O10 C19–H19b···O12 C21–H21a···O10 C22–H22a···O12 C9–H9···O10 | a-b-q-w-i | H17···O14 H16···O18 | C11–H19···O12 C13–H13a···O12 C13–H13b···O8 C21–H21a···O12 C9–H9···O10 |
| a-m-r-t-e | H17···O14 | C7–H13···O8 C19–H19a···O10 C19–H19b···O12 C20–H20···O18 C22–H22···O18 | c-p-r-z-e | H17···O18 | C11–H19···O10 C13–H13a···O12 C20–H20···O14 C21–H21a···O14 |
| a-m-r-s-e | H17···O14 | C7–H13···O8 C19–H19a···O10 C19–H19b···O12 C20–H20···O18 C22–H22···O18 | c-p-r-v-e | H17···O18 | C11–H19···O10 C13–H13a···O12 C22–H22a···O14 C20–H20···O14 |
| b-w-x-e | H16···O18 | C19–H19a···O12 C19–H19b···O12 C22–H22a···O18 C20–H20···O18 C9–H9···O10 | a-q-r-j-e | H17···O14 | C11–H19···O12 C13–H13a···O12 C13–H13b···O12 C20–H20···O14 C21–H21a···O14 |
| c-n-r-e | H17···O18 | C7–H13···O12 C19–H19a···O10 C19–H19b···O10 C20–H20···O18 C22–H22···O18 | q-r-e | H17···O14 | C11–H19···O12 C13–H13a···O8 C13–H13b···O8 C22–H22a···O14 C20–H20···O14 |
| c-n-r-x-e | H17···O18 | C7–H13···O12 C19–H19a···O10 C19–H19b···O10 C22–H22a···O18 C20–H20···O18 | a-w-k-e | H17···O14 | C11–H19···O12 C13–H13a···O8 C13–H13b···O8 C22–H22a···O14 C20–H20···O14 C9–H9···O10 |
| c-n-w-x-e | H17···O18 | C7–H13···O12 C22–H22a···O18 C20–H20···O18 C9–H9···O10 | c-r-x-e | H17···O18 | C13–H13a···O8 C13–H13b···O12 C22–H22a···O14 C9–H9···O10 |
| b-m-w-y-e | H16···O18 | C7–H13···O8 C19–H19a···O12 C19–H19b···O10 C22–H22a···O18 C20–H20···O18 C9–H9···O10 | c-w-z-e | H17···O18 | C13–H13a···O8 C22–H22a···O14 C20–H20···O14 C9–H9···O10 |
| b-n-w-x-e | H16···O18 | C7–H13···O12 C19–H19a···O12 C19–H19b···O10 C22–H22a···O18 C20–H20···O18 C9–H9···O10 | a-p-r-e | H17···O14 | C11–H19···O10 C13–H13a···O8 C13–H13b···O8 C22–H22a···O14 C20–H20···O14 |
| c-m-w-y-e | H17···O18 | C7–H13···O8 C22–H22a···O18 C20–H20···O18 C9–H9···O10 | a-p-r-k-e | H17···O14 | C13–H13a···O8 C13–H13b···O8 C22–H22a···O14 C20–H20···O14 C9–H9···O10 |
| c-m-r-y-e | H17···O18 | C7–H13···O8 C19–H19a···O10 C19–H19b···O10 C22–H22a···O18 C20–H20···O18 | p-r-k-z-e | none | C11–H19···O10 C13–H13a···O14 C20–H20···O14 |
| r-t-x-u-e | none | C19–H19a···O12 C19–H19b···O10 C22–H22a···O18 C20–H20···O18 | r-j-z-u-e | none | C13–H13a···O8 C22–H22a···O14 C20–H20···O14 |
| r-s-x-u-e | none | C19–H19a···O12 C20–H20···O18 C9–H9···O10 | r-j-v-u-e | none | C13–H13a···O12 C22–H22a···O14 C20–H20···O14 |
| Conformers | DFT | HF | MP2 | ||||
|---|---|---|---|---|---|---|---|
| ΔE (kcal/mol) | ΔEcorr (kcal/mol) | ΔG (kcal/mol) | ΔE (kcal/mol) | ΔEcorr (kcal/mol) | ΔG (kcal/mol) | ΔE (kcal/mol) | |
| M1–ε-a-b-m-w-e | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.632 |
| M1–ε-a-b-m-w-d | 0.365 | 0.707 | 0.547 | 0.781 | 1.057 | 0.855 | 0.128 |
| M1–ε-a-b-m-w-g | 0.385 | 0.632 | 0.493 | 0.785 | 1.063 | 0.857 | 0.000 |
| M1–ε-a-b-m-w-f | 1.884 | 2.015 | 2.198 | 1.757 | 1.844 | 1.832 | 1.972 |
| M1–ε-a-b-m-w-i | 2.884 | 3.256 | 3.197 | 3.591 | 3.943 | 3.535 | 2.033 |
| M1–ε-a-b-m-w-h | 2.896 | 3.197 | 3.181 | 3.593 | 3.944 | 3.519 | 1.905 |
| M1–ε-a-m-r-t-e | 16.906 | 16.448 | 15.566 | 13.792 | 12.858 | 12.048 | 14.636 |
| M1–ε-b-w-x-e | 17.384 | 16.551 | 15.579 | 17.619 | 16.650 | 15.857 | 17.024 |
| M1–ε-c-n-r-x-e | 17.770 | 17.155 | 15.980 | 16.153 | 15.334 | 14.548 | 16.783 |
| M1–ε-c-n-w-x-e | 19.167 | 18.735 | 17.897 | 18.601 | 17.856 | 17.121 | 17.901 |
| M1–ε-b-m-r-y-e | 19.879 | 19.707 | 19.159 | 19.097 | 18.539 | 18.049 | 17.930 |
| M1–ε-b-n-r-x-e | 20.025 | 19.698 | 18.621 | 19.376 | 18.721 | 18.087 | 17.899 |
| M1–ε-c-m-w-y-e | 20.432 | 20.068 | 19.164 | 19.757 | 19.057 | 18.345 | 18.810 |
| M1–ε-r-t-x-u-e | 32.141 | 30.599 | 28.737 | 29.239 | 27.110 | 25.270 | 29.017 |
| O−H⋅⋅⋅O IHBs Present | Isomer or Molecule | Relative Energy Ranges (kcal mol−1) | ||
|---|---|---|---|---|
| DFT | HF | MP2 | ||
| H17⋅⋅⋅O14 H16⋅⋅⋅O18 | ε | 0.000−2.931 | 0.000−3.680 | 0.000−2.094 |
| ζ | 0.000−3.925 | 0.000−4.375 | 0.000−3.785 | |
| G | 0.000–2.975 | 0.000–3.711 | 0.000–1.958 | |
| H17⋅⋅⋅O14 | ε | 16.014–19.741 | 13.071–19.277 | 13.709–16.934 |
| ζ | 16.330–20.519 | 14.650–19.446 | 14.992–19.457 | |
| G | 17.615–19.309 | 16.652–18.413 | 16.961–18.285 | |
| H16⋅⋅⋅O18 | ε | 16.434–21.074 | 16.156–20.399 | 13.467–19.078 |
| ζ | 14.400–15.458 | 11.408–13.848 | 11.058–13.124 | |
| G | - | - | - | |
| H17⋅⋅⋅O18 | ε | 16.025–22.475 | 14.430–21.986 | 14.445–20.489 |
| ζ | 12.197–17.849 | 9.549–17.353 | 9.606–17.279 | |
| G | 16.665–16.825 | 14.151–14.497 | 14.596–14.969 | |
| none | ε | 30.701–34.728 | 27.554–31.126 | 26.762–29.017 |
| ζ | 28.802–31.789 | 27.164–29.544 | 26.766–29.236 | |
| G | 31.660 | 29.404 | 29.045 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tshilande, N.; Mammino, L. Comparison of the Molecular Properties of Euglobals Differing by the Mutual Positions of the Two R–C=O Groups (R = H and CH2CH(CH3)2): A Computational Study. Chemistry 2023, 5, 2120-2154. https://doi.org/10.3390/chemistry5040144
Tshilande N, Mammino L. Comparison of the Molecular Properties of Euglobals Differing by the Mutual Positions of the Two R–C=O Groups (R = H and CH2CH(CH3)2): A Computational Study. Chemistry. 2023; 5(4):2120-2154. https://doi.org/10.3390/chemistry5040144
Chicago/Turabian StyleTshilande, Neani, and Liliana Mammino. 2023. "Comparison of the Molecular Properties of Euglobals Differing by the Mutual Positions of the Two R–C=O Groups (R = H and CH2CH(CH3)2): A Computational Study" Chemistry 5, no. 4: 2120-2154. https://doi.org/10.3390/chemistry5040144
APA StyleTshilande, N., & Mammino, L. (2023). Comparison of the Molecular Properties of Euglobals Differing by the Mutual Positions of the Two R–C=O Groups (R = H and CH2CH(CH3)2): A Computational Study. Chemistry, 5(4), 2120-2154. https://doi.org/10.3390/chemistry5040144