

Efficient Metal-Free Oxidative C–H Amination for Accessing Dibenzoxazepinones via μ-Oxo Hypervalent Iodine Catalysis

 , and
, and 
Abstract
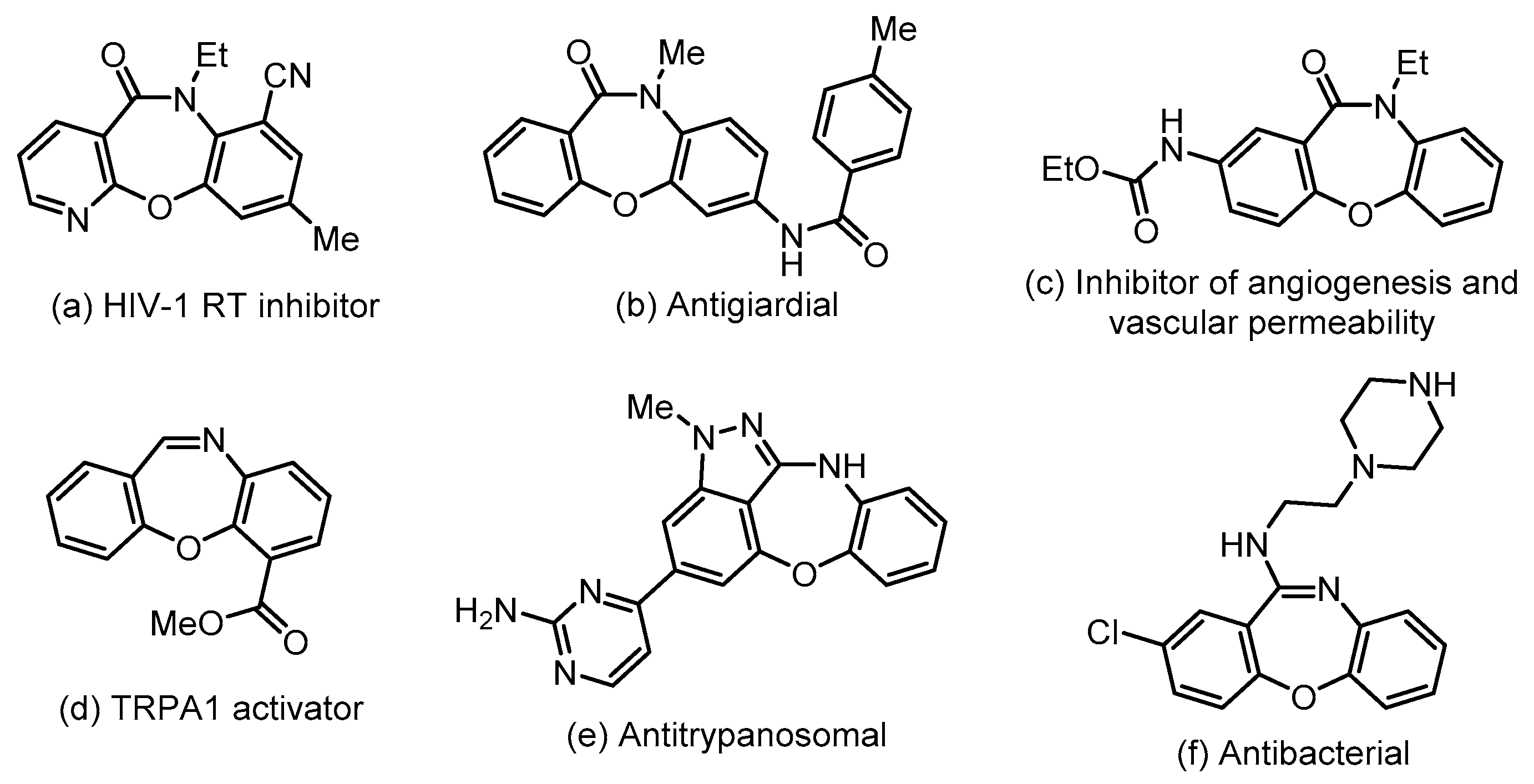
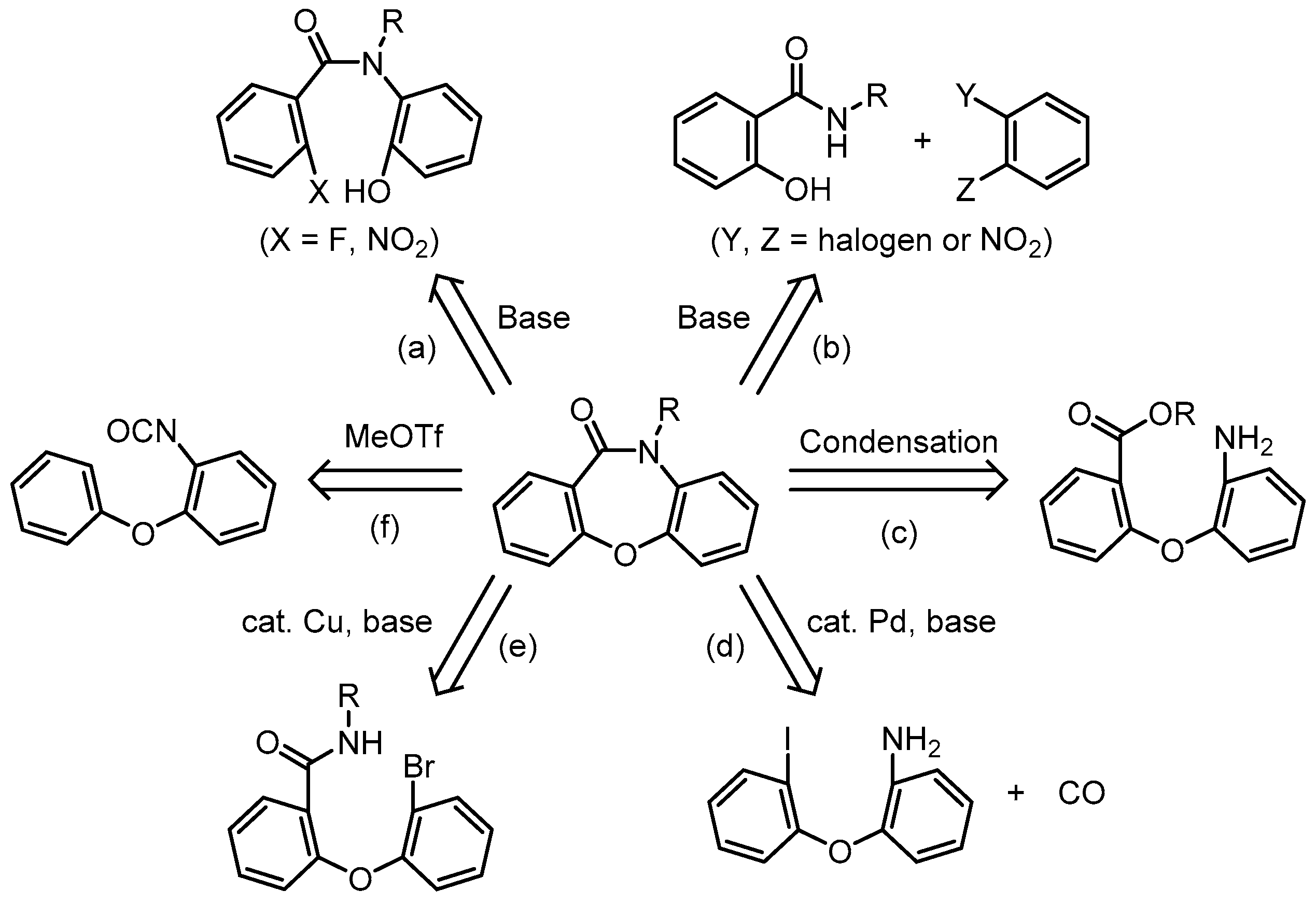
1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Synthesis of Amides 1
2.2.1. General Procedures for the Synthesis of 2-Phenoxybenzoic Acids
2.2.2. General Procedures for the Synthesis of Amides 1
2.3. Hypervalent Iodine-Catalyzed Oxidative C–H Amination Reaction
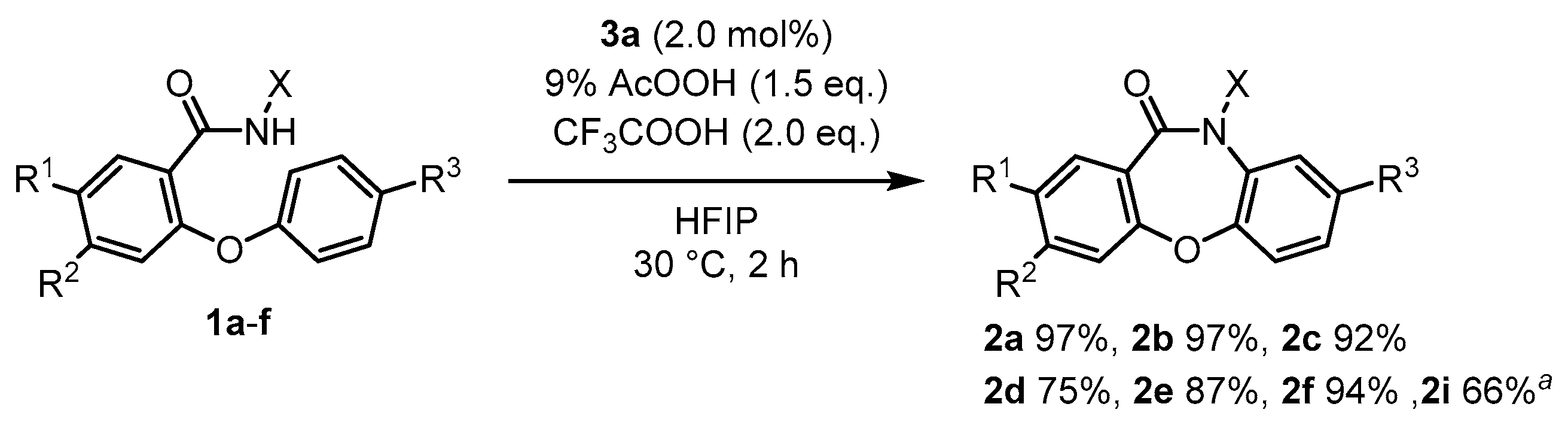
2.3.1. General Procedure A for the Synthesis of Dibenzoxazepinones 2
2.3.2. General Procedure B for the Synthesis of Dibenzoxazepinones 2
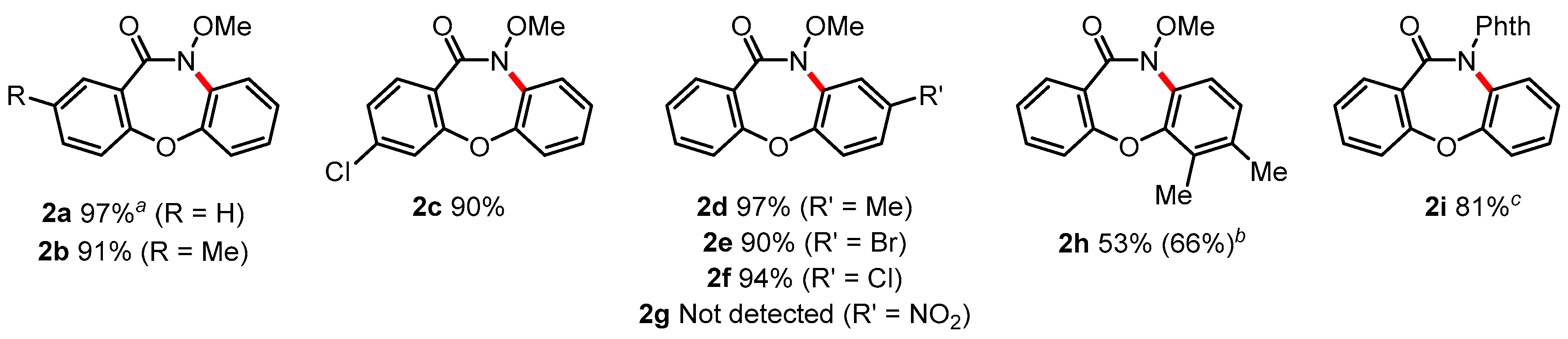
2.3.3. 10-Methoxy-dibenz[b,f][1,4]oxazepin-11(10H)-one (2a)
2.3.4. 10-Methoxy-2-methyl-dibenz[b,f][1,4]oxazepin-11(10H)-one (2b)
2.3.5. 3-Chloro-10-methoxy-dibenz[b,f][1,4]oxazepin-11(10H)-one (2c)
2.3.6. 10-Methoxy-8-methyl-dibenz[b,f][1,4]oxazepin-11(10H)-one (2d)
2.3.7. 8-Bromo-10-methoxy-dibenz[b,f][1,4]oxazepin-11(10H)-one (2e)
2.3.8. 8-Chloro-10-methoxy-dibenz[b,f][1,4]oxazepin-11(10H)-one (2f)
2.3.9. 10-Methoxy-6,7-dimethyl-dibenz[b,f][1,4]oxazepin-11(10H)-one (2h)
2.3.10. 2-(11-Oxodibenzo[b,f][1,4]oxazepin-10(11H)-yl)isoindoline-1,3-dione (2i)
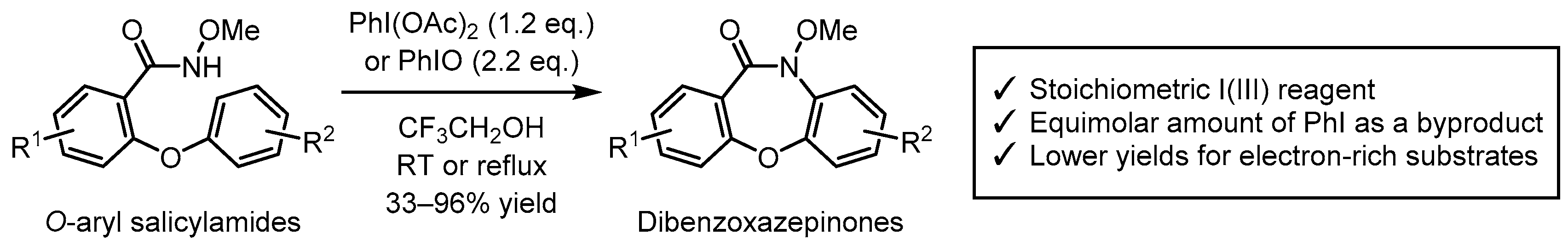
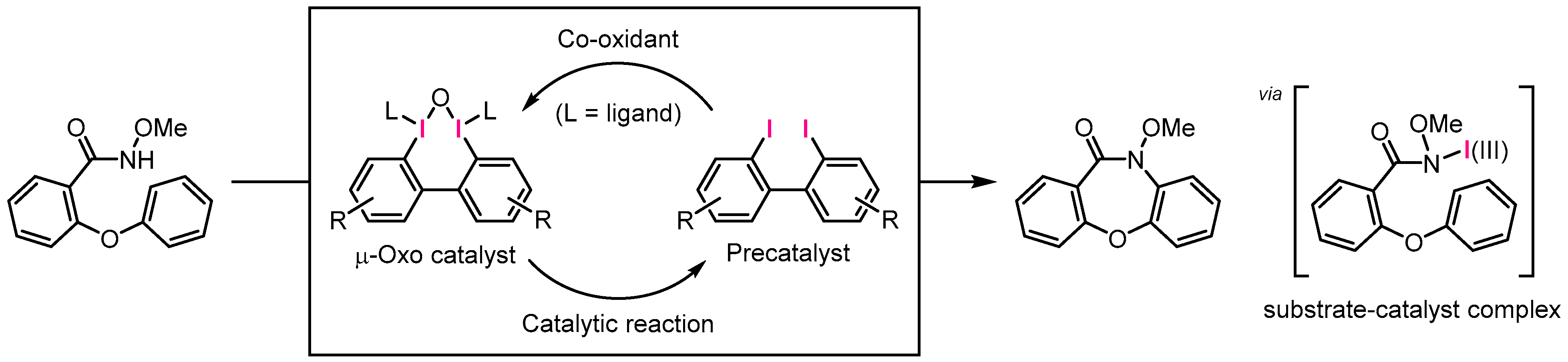
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klunder, J.M.; Hargrave, K.D.; West, M.; Cullen, E.; Pal, K.; Behnke, M.L.; Kapadia, S.R.; McNeil, D.W.; Wu, J.C.; Chow, G.C.; et al. Novel Non-Nucleoside Inhibitors of HIV-1 Reverse Transcriptase. 2. Tricyclic Pyridobenzoxazepinones and Dibenzoxazepinones. J. Med. Chem. 1992, 35, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Alhendi, A.M.N.; Yeh, M.-C.; Elahy, M.; Santiago, F.S.; Deshpande, N.P.; Wu, B.; Chan, E.; Inam, S.; Prado-Lourenco, L.; et al. Thermostable small-molecule inhibitor of angiogenesis and vascular permeability that suppresses a pERK-FosB/ΔFosB–VCAM-1 axis. Sci. Adv. 2020, 6, eaaz7815. [Google Scholar] [CrossRef] [PubMed]
- Riches, A.G.; Hart, C.J.S.; Schmit, M.; Debele, E.A.; Tiash, S.; Clapper, E.; Skinner-Adams, T.S.; Ryan, J.H. Structural reassignment of a dibenz[b,f][1,4]oxazepin-11(10H)-one with potent antigiardial activity. Aust. J. Chem. 2022, 75, 839–845. [Google Scholar] [CrossRef]
- Gijsen, H.J.M.; Berthelot, D.; Zaja, M.; Brône, B.; Geuens, I.; Mercken, M. Analogues of Morphanthridine and the Tear Gas Dibenz[b,f][1,4]oxazepine (CR) as Extremely Potent Activators of the Human Transient Receptor Potential Ankyrin 1 (TRPA1) Channel. J. Med. Chem. 2010, 53, 7011–7020. [Google Scholar] [CrossRef] [PubMed]
- Klug, D.M.; Tschiegg, L.; Diaz, R.; Rojas-Barros, D.; Perez-Moreno, G.; Ceballos, G.; García-Hernández, R.; Martinez-Martinez, M.S.; Manzano, P.; Ruiz, L.M.; et al. Hit-to-Lead Optimization of Benzoxazepinoindazoles As Human African Trypanosomiasis Therapeutics. J. Med. Chem. 2020, 63, 2527–2546. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-C.; Wu, Y.-L.; Hsu, C.-Y.; Lin, M.-Y.; Chen, L.-H.; Shiau, C.-W.; Chiu, H.-C. Discovery of antipsychotic loxapine derivatives against intracellular multidrug-resistant bacteria. RSC Med. Chem. 2022, 13, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Tamayo, N.; Kiselyov, A.S. Solid Support Synthesis of 2-Substituted Dibenz[b,f]oxazepin-11(10H)-ones via SNAr Methodology on AMEBA Resin. Tetrahedron 1999, 55, 2827–2834. [Google Scholar] [CrossRef]
- Hone, N.D.; Salter, J.I.; Reader, J.C. Solid-phase synthesis of dibenzoxazepinones. Tetrahedron Lett. 2003, 44, 8169–8172. [Google Scholar] [CrossRef]
- Samet, A.V.; Kislyi, K.A.; Marshalkin, V.N.; Semenov, V.V. Synthesis of dibenzo[b,f][1,4]oxazepin-11(10H)-ones from 2-nitrobenzoic acids. Russ. Chem. Bull. Int. Ed. 2006, 55, 549–553. [Google Scholar] [CrossRef]
- Liu, Y.; Chu, C.; Huang, A.; Zhan, C.; Ma, Y.; Ma, C. Regioselective Synthesis of Fused Oxazepinone Scaffolds through One-Pot Smiles Rearrangement Tandem Reaction. ACS Comb. Sci. 2011, 13, 547–553. [Google Scholar] [CrossRef]
- Kitching, M.O.; Hurst, T.E.; Snieckus, V. Copper-Catalyzed Cross-Coupling Interrupted by an Opportunistic Smiles Rearrangement: An Efficient Domino Approach to Dibenzoxazepinones. Angew. Chem. Int. Ed. 2012, 51, 2925–2929. [Google Scholar] [CrossRef] [PubMed]
- Sapegin, A.; Kalinin, S.; Angeli, A.; Supuran, C.T.; Krasavin, M. Unprotected primary sulfonamide group facilitates ring-forming cascade en route to polycyclic [1,4]oxazepine-based carbonic anhydrase inhibitors. Bioorg. Chem. 2018, 76, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Grintsevich, S.; Sapegin, A.; Reutskaya, E.; Peintner, S.; Erdélyi, M.; Krasavin, M. An Alternative Approach to the Hydrated Imidazoline Ring Expansion (HIRE) of Diarene-Fused [1.4]Oxazepines. Eur. J. Org. Chem. 2020, 2020, 5664–5676. [Google Scholar] [CrossRef]
- Brewster, K.; Clarke, R.J.; Harrison, J.M.; Inch, T.D.; Utley, D. Preparation of the Eight Monohydroxydibenz[b,f][1,4]oxazepin-l1(10H)-ones. J. Chem. Soc. Perkin Trans. 1 1976, 12, 1286–1290. [Google Scholar] [CrossRef]
- Bunce, R.A.; Schammerhorn, J.E. Dibenzo-fused Seven-membered Nitrogen Heterocycles by a Tandem Reduction-Lactamization Reaction. J. Heterocycl. Chem. 2006, 43, 1031–1035. [Google Scholar] [CrossRef]
- Tsvelikhovsky, D.; Buchwald, S.L. Concise Palladium-Catalyzed Synthesis of Dibenzodiazepines and Structural Analogues. J. Am. Chem. Soc. 2011, 133, 14228–14231. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kishore, D.; Singh, B. An Expedient Method for the Synthesis of 1,2,4-Triazolo-fused 1,5-Benzodiazepine, 1,5-Benzoxazepine, and 1,5-Benzothiazepine Scaffolds: A Novel Seven-membered Ring System of Biological Interest. J. Heterocycl. Chem. 2018, 55, 586–592. [Google Scholar] [CrossRef]
- Lu, S.-M.; Alper, H. Intramolecular Carbonylation Reactions with Recyclable Palladium-Complexed Dendrimers on Silica: Synthesis of Oxygen, Nitrogen, or Sulfur-Containing Medium Ring Fused Heterocycles. J. Am. Chem. Soc. 2005, 127, 14776–14784. [Google Scholar] [CrossRef]
- Yang, Q.; Cao, H.; Robertson, A.; Alper, H. Synthesis of Dibenzo[b,f][1,4]oxazepin-11(10H)-ones via Intramolecular Cyclocarbonylation Reactions Using PdI2/Cytop 292 as the Catalytic System. J. Org. Chem. 2010, 75, 6297–6299. [Google Scholar] [CrossRef]
- Hermange, P.; Lindhardt, A.T.; Taaning, R.H.; Bjerglund, K.; Lupp, D.; Skrydstrup, T. Ex Situ Generation of Stoichiometric and Substoichiometric 12CO and 13CO and Its Efficient Incorporation in Palladium Catalyzed Aminocarbonylations. J. Am. Chem. Soc. 2011, 133, 6061–6071. [Google Scholar] [CrossRef]
- Anchan, K.; Baburajan, P.; Puttappa, N.H.; Sarkar, S.K. One-pot synthesis of substituted dibenzoxazepinones and pyridobenzoxazepinones using octacarbonyldicobalt as an effective CO source. Synth. Commun. 2020, 50, 348–360. [Google Scholar] [CrossRef]
- Feng, Z.; Ma, J.-A.; Cheung, C.W. Ni-Catalysed intramolecular reductive aminocarbonylation of 2-haloaryl-tethered nitroarenes for the synthesis of dibenzazepine-based heterocycles. Org. Chem. Front. 2022, 9, 3869–3875. [Google Scholar] [CrossRef]
- Shi, J.; Wu, J.; Cui, C.; Dai, W.-M. Microwave-Assisted Intramolecular Ullmann Diaryl Etherification as the Post-Ugi Annulation for Generation of Dibenz[b,f][1,4]oxazepane Scaffold. J. Org. Chem. 2016, 81, 10392–10403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Dai, Z.; Ma, X.; Liu, Y.; Ma, X.; Li, W.; Ma, C. Cu-catalyzed one-pot synthesis of fused oxazepinone derivatives via sp2 C–H and O–H cross-dehydrogenative coupling. Org. Chem. Front. 2016, 3, 799–803. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, Q.; Zhang, R.; Hu, J.; Zhou, Y.; Xu, L.; Pan, X. Ligand-Controlled Chemoselective One-Pot Synthesis of Dibenzothiazepinones and Dibenzoxazepinones via Twice Copper-Catalyzed Cross-Coupling. Synlett 2017, 28, 1201–1208. [Google Scholar] [CrossRef][Green Version]
- Wang, S.; Shao, P.; Du, G.; Xi, C. MeOTf- and TBD-Mediated Carbonylation of ortho-Arylanilines with CO2 Leading to Phenanthridinones. J. Org. Chem. 2016, 81, 6672–6676. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Zhang, Z.; Chen, C.; Xi, C. MeOTf-Catalyzed Intramolecular Acyl-Cyclization of Aryl Isocyanates: Efficient Access to Phenanthridin-6(5H)-one and 3,4-Dihydroisoquinolin-1(2H)-one Derivatives. Asian J. Org. Chem. 2021, 10, 355–359. [Google Scholar] [CrossRef]
- Jiao, J.; Murakami, K.; Itami, K. Catalytic Methods for Aromatic C−H Amination: An Ideal Strategy for Nitrogen-Based Functional Molecules. ACS Catal. 2016, 6, 610–633. [Google Scholar] [CrossRef]
- Kim, H.; Chang, S. Transition-Metal-Mediated Direct C−H Amination of Hydrocarbons with Amine Reactants: The Most Desirable but Challenging C−N Bond-Formation Approach. ACS Catal. 2016, 6, 2341–2351. [Google Scholar] [CrossRef]
- Park, Y.; Kim, Y.; Chang, S. Transition Metal-Catalyzed C−H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. [Google Scholar] [CrossRef]
- Henry, M.C.; Mostafa, M.A.B.; Sutherland, A. Recent Advances in Transition-Metal-Catalyzed, Directed Aryl C–H/N–H Cross-Coupling Reactions. Synthesis 2017, 49, 4586–4598. [Google Scholar] [CrossRef]
- Timsina, Y.N.; Gupton, B.F.; Ellis, K.C. Palladium-Catalyzed C−H Amination of C(sp2) and C(sp3)−H Bonds: Mechanism and Scope for N-Based Molecule Synthesis. ACS Catal. 2018, 8, 5732–5776. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, D.; Vessally, E. Direct Amination of Aromatic C–H Bonds with Free Amines. Top. Curr. Chem. 2020, 378, 37. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Sathish, E.; Gupta, A.K.; Goyal, S. 3d-transition metal catalyzed C–H to C–N bond formation: An update. Tetrahedron 2021, 100, 132474. [Google Scholar] [CrossRef]
- Guo, X.; Zhang-Negreriea, D.; Du, Y. Iodine(III)-mediated construction of the dibenzoxazepinone skeleton from 2-(aryloxy) benzamides through oxidative C–N formation. RSC Adv. 2015, 5, 94732–94736. [Google Scholar] [CrossRef]
- Liang, D.; Yu, W.; Nguyen, N.; Deschamps, J.R.; Imler, G.H.; Li, Y.; MacKerell, A.D., Jr.; Jiang, C.; Xue, F. Iodobenzene-Catalyzed Synthesis of Phenanthridinones via Oxidative C−H Amidation. J. Org. Chem. 2017, 82, 3589–3596. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Sasa, H.; Dochi, M.; Yasui, C.; Kita, Y. Oxidative Coupling of N-Methoxyamides and Related Compounds toward Aromatic Hydrocarbons by Designer μ-Oxo Hypervalent Iodine Catalyst. Synthesis 2019, 51, 1185–1195. [Google Scholar] [CrossRef]
- Sasa, H.; Mori, K.; Kikushima, K.; Kita, Y.; Dohi, T. μ-Oxo-Hypervalent-Iodine-Catalyzed Oxidative C–H Amination for Synthesis of Benzolactam Derivatives. Chem. Pharm. Bull. 2022, 70, 106–110. [Google Scholar] [CrossRef]
- Dohi, T.; Takenaga, N.; Fukushima, K.; Uchiyama, T.; Kato, D.; Shiro, M.; Fujioka, H.; Kita, Y. Designer μ-oxo-bridged hypervalent iodine(III) organocatalysts for greener oxidations. Chem. Commun. 2010, 46, 7697–7699. [Google Scholar] [CrossRef]
- Dohi, T.; Mochizuki, E.; Yamashita, D.; Miyazaki, K.; Kita, Y. Efficient Oxidative Spirolactamization by μ-Oxo Bridged Heterocyclic Hypervalent Iodine Compound. Heterocycles 2014, 88, 245. [Google Scholar]
- Lee, C.-K.; Mak, T.C.W.; Li, W.-K.; Kirner, J.F. Iodobenzene diacetate. Acta. Cryst. 1977, B33, 1620–1622. [Google Scholar] [CrossRef]
- Kikushima, K.; Miyamoto, N.; Watanabe, K.; Koseki, D.; Kita, Y.; Dohi, T. Ligand- and Counterion-Assisted Phenol O-Arylation with TMP-Iodonium(III) Acetates. Org. Lett. 2022, 24, 1924–1928. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Precatalyst | Solvent | Yield of 2a b | Recovery of 1a b |
| 1 | 3a (1.0 mol%) | CH2Cl2 | 25% | 72% |
| 2 | 3a (1.0 mol%) | EtOH | N.D. | 77% |
| 3 | 3a (1.0 mol%) | CF3CH2OH | 35% | 63% |
| 4 | 3a (1.0 mol%) | HFIP | >99% (98%) c | N.D. |
| 5 d | 3a (1.0 mol%) | HFIP | 5% | 94% |
| 6 | 3a (0.5 mol%) | HFIP | 64% | 33% |
| 7 e | 3a (0.5 mol%) | HFIP | 97% | N.D. |
| 8 f | 3a (0.25 mol%) | HFIP | 82% | 15% |
| 9 g | 3a (0.1 mol%) | HFIP | 87% | 6% |
| 10 | 3b (0.5 mol%) | HFIP | 10% | 86% |
| 11 | 3c (0.5 mol%) | HFIP | 11% | 88% |
 | ||
|---|---|---|
| Precatalyst | Yield of 2a a | Recovery of 1a a |
| 3d | 60% | 32% |
| 3e | 25% | 72% |
| 3f | 5% | 85% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sasa, H.; Hamatani, S.; Hirashima, M.; Takenaga, N.; Hanasaki, T.; Dohi, T. Efficient Metal-Free Oxidative C–H Amination for Accessing Dibenzoxazepinones via μ-Oxo Hypervalent Iodine Catalysis. Chemistry 2023, 5, 2155-2165. https://doi.org/10.3390/chemistry5040145
Sasa H, Hamatani S, Hirashima M, Takenaga N, Hanasaki T, Dohi T. Efficient Metal-Free Oxidative C–H Amination for Accessing Dibenzoxazepinones via μ-Oxo Hypervalent Iodine Catalysis. Chemistry. 2023; 5(4):2155-2165. https://doi.org/10.3390/chemistry5040145
Chicago/Turabian StyleSasa, Hirotaka, Syotaro Hamatani, Mayu Hirashima, Naoko Takenaga, Tomonori Hanasaki, and Toshifumi Dohi. 2023. "Efficient Metal-Free Oxidative C–H Amination for Accessing Dibenzoxazepinones via μ-Oxo Hypervalent Iodine Catalysis" Chemistry 5, no. 4: 2155-2165. https://doi.org/10.3390/chemistry5040145
APA StyleSasa, H., Hamatani, S., Hirashima, M., Takenaga, N., Hanasaki, T., & Dohi, T. (2023). Efficient Metal-Free Oxidative C–H Amination for Accessing Dibenzoxazepinones via μ-Oxo Hypervalent Iodine Catalysis. Chemistry, 5(4), 2155-2165. https://doi.org/10.3390/chemistry5040145