Kinetic Stability of Si2C5H2 Isomer with a Planar Tetracoordinate Carbon Atom

 , , and
, , and 
Abstract
1. Introduction
2. Computational Details
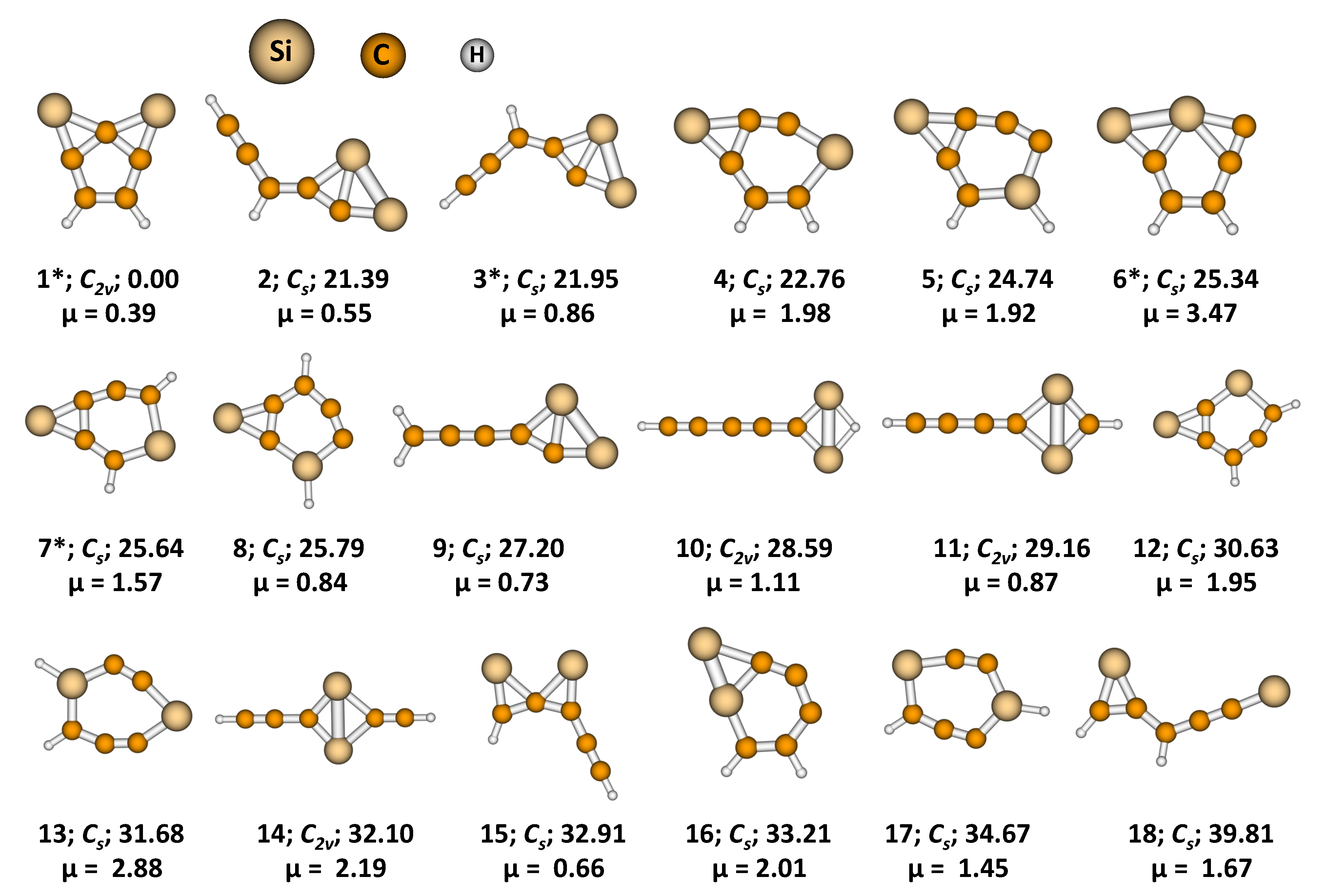
3. Results and Discussion
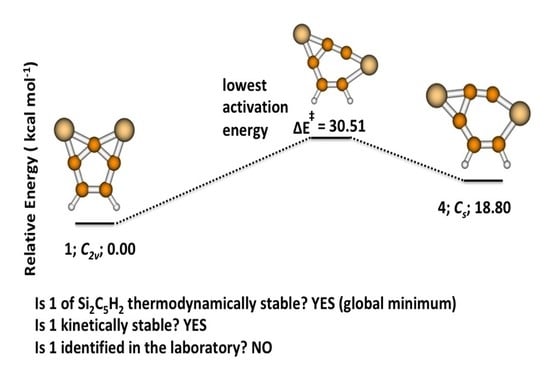
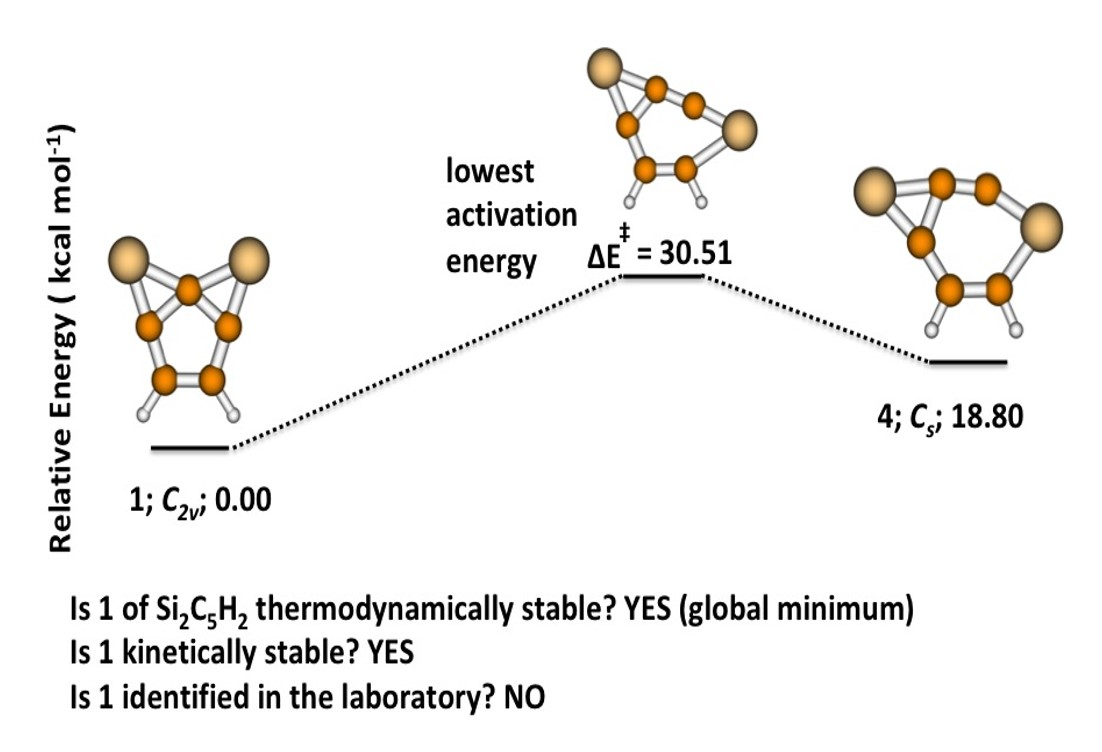
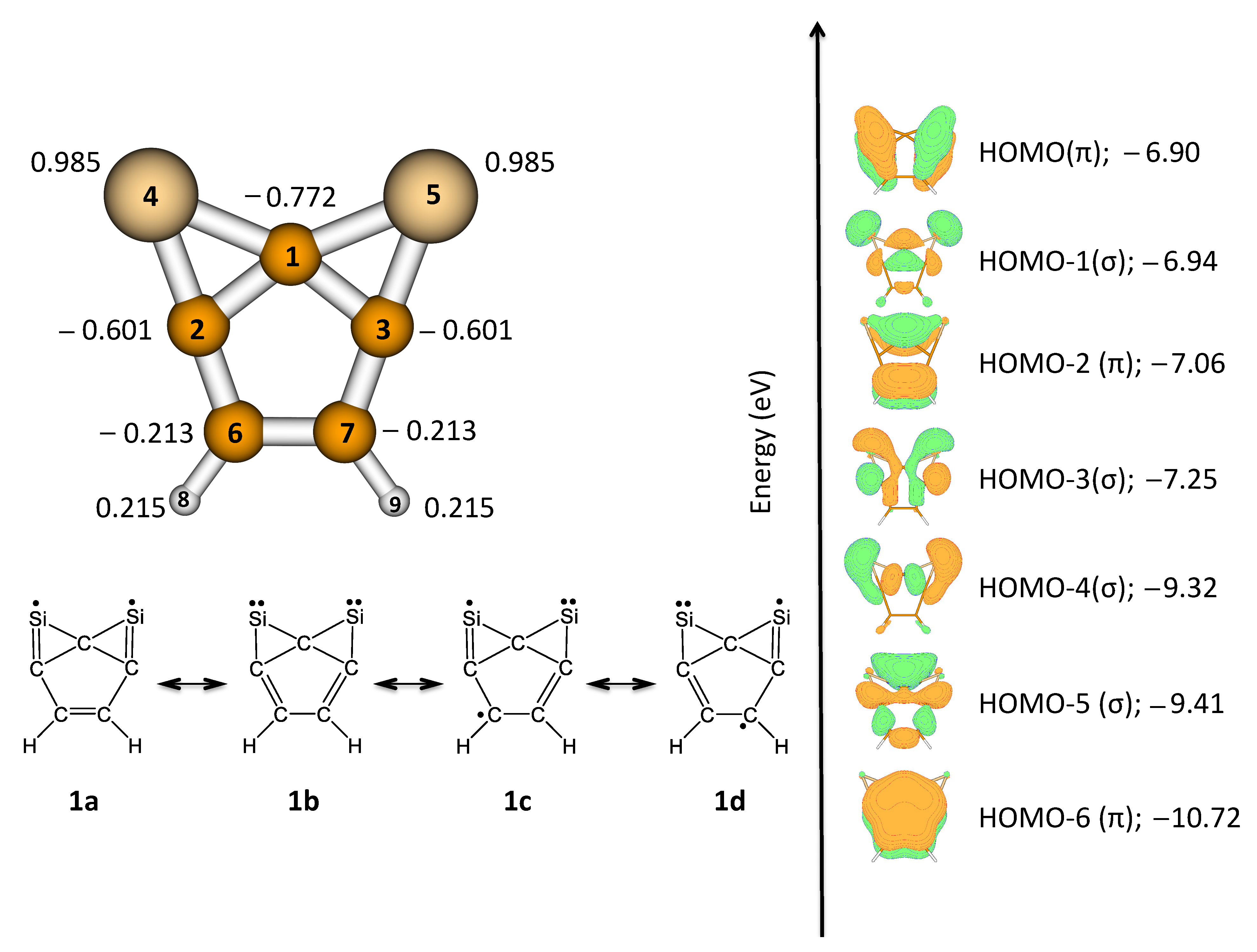
3.1. 2,7-Disilatricyclo[4.1.0.0]hept-2,4,6-trien-2,7-diyl (1)
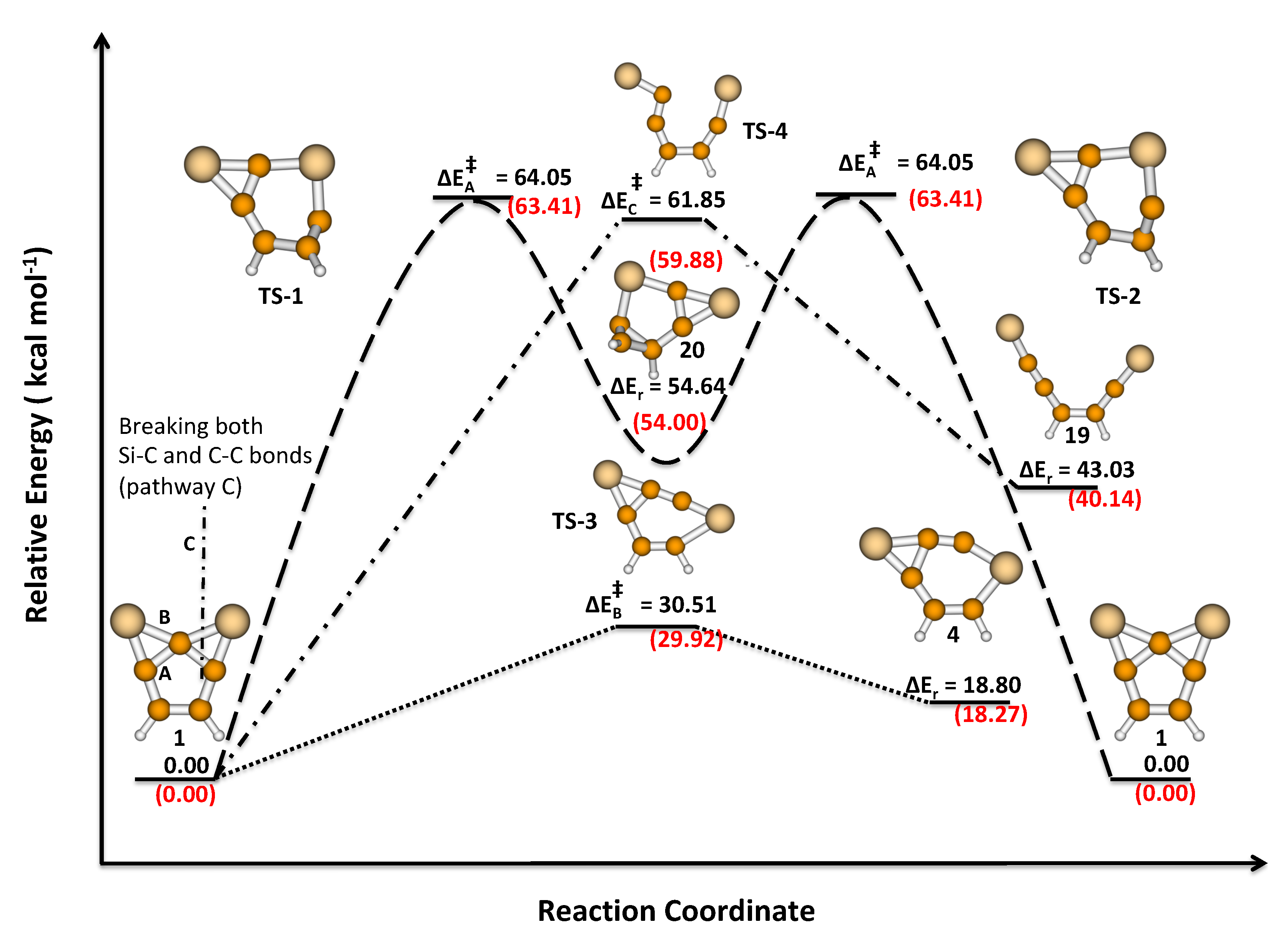
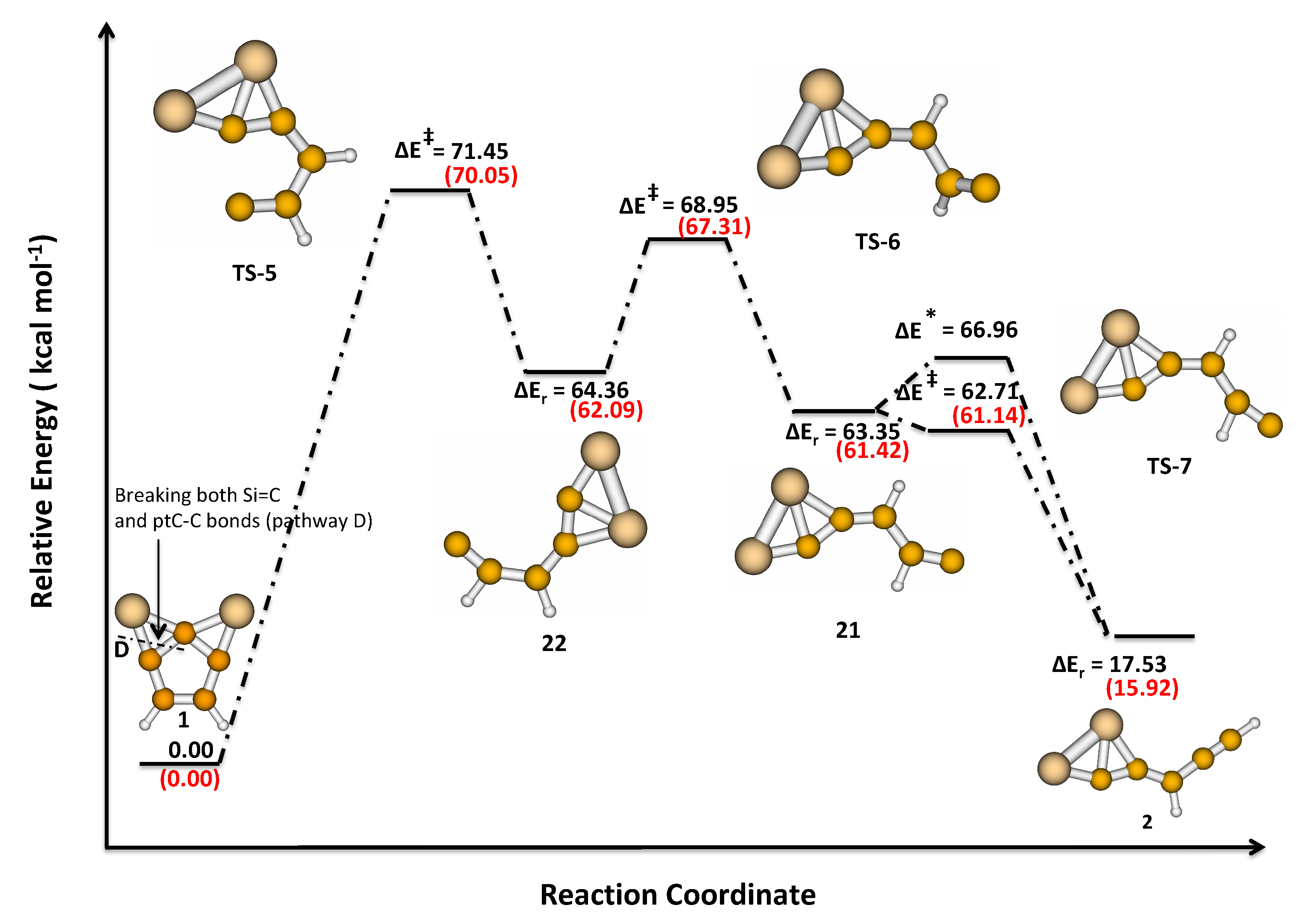
3.2. Activation and Reaction Energies
3.3. Isomerization of 1 to 2
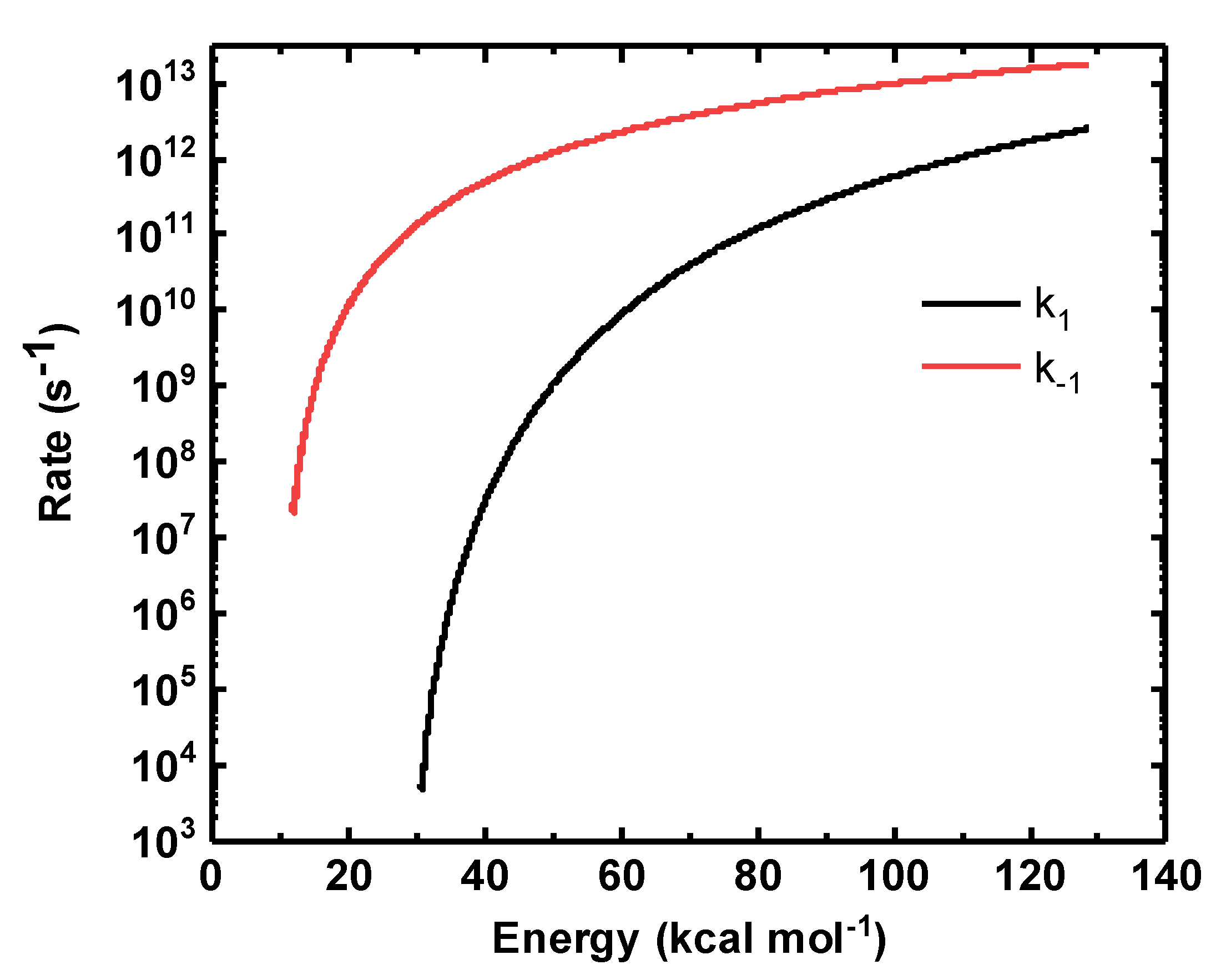
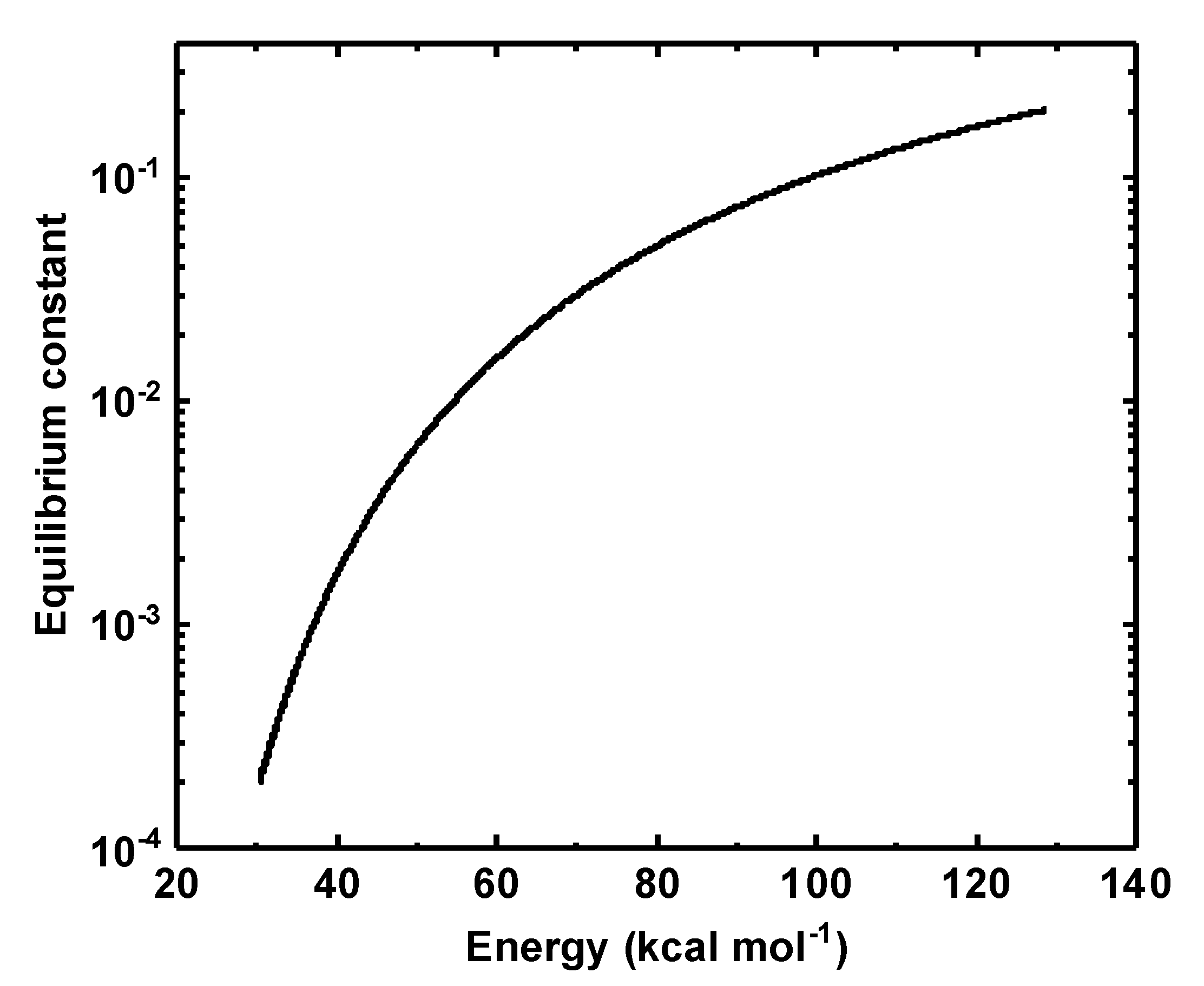
3.4. Rate Co-Efficient for the Isomerization Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ae | all-electron |
| fc | frozen-core |
| DFT | Density functional theory |
| CCSD | Coupled-cluster singles, doubles |
| CCSD(T) | Coupled-cluster singles, doubles including perturbative triples |
| ptC | planar tetracoordinate carbon |
| ptSi | planar tetracoordinate silicon |
References
- Hoffmann, R.; Alder, R.W.; Wilcox, C.F. Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1970, 92, 4992–4993. [Google Scholar] [CrossRef]
- Collins, J.B.; Dill, J.D.; Jemmis, E.D.; Apeloig, Y.; Schleyer, P.V.R.; Seeger, R.; Pople, J.A. Stabilization of Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1976, 98, 5419–5427. [Google Scholar] [CrossRef]
- Keese, R. Carbon Flatland: Planar Tetracoordinate Carbon and Fenestranes. Chem. Rev. 2006, 106, 4787–4808. [Google Scholar] [CrossRef] [PubMed]
- Radom, L.; Rasmussen, D.R. The Planar Carbon Story. Pure Appl. Chem. 1998, 70, 1977. [Google Scholar] [CrossRef]
- Merino, G.; Méndez-Rojas, M.A.; Beltrán, H.I.; Corminboeuf, C.; Heine, T.; Vela, A. Theoretical Analysis of the Smallest Carbon Cluster Containing a Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 2004, 126, 16160–16169. [Google Scholar] [CrossRef]
- Pancharatna, P.D.; Méndez-Rojas, M.A.; Merino, G.; Vela, A.; Hoffmann, R. Planar Tetracoordinate Carbon in Extended Systems. J. Am. Chem. Soc. 2004, 126, 15309–15315. [Google Scholar] [CrossRef]
- Roy, D.; Corminboeuf, C.; Wannere, C.S.; King, R.B.; Schleyer, P.V.R. Planar Tetracoordinate Carbon Atoms Centered in Bare Four-membered Rings of Late Transition Metals. Inorg. Chem. 2006, 45, 8902–8906. [Google Scholar] [CrossRef]
- Nandula, A.; Trinh, Q.T.; Saeys, M.; Alexandrova, A.N. Origin of Extraordinary Stability of Square-Planar Carbon Atoms in Surface Carbides of Cobalt and Nickel. Angew. Chem. Int. Ed. 2015, 54, 5312–5316. [Google Scholar] [CrossRef]
- Yang, L.M.; Ganz, E.; Chen, Z.; Wang, Z.X.; Schleyer, P.v.R. Four Decades of the Chemistry of Planar Hypercoordinate Compounds. Angew. Chem. Int. Ed. 2015, 54, 9468–9501. [Google Scholar] [CrossRef]
- Yañez, O.; Báez-Grez, R.; Garza, J.; Pan, S.; Barroso, J.; Vásquez-Espinal, A.; Merino, G.; Tiznado, W. Embedding a Planar Hypercoordinate Carbon Atom into a [4n+2] π-System. ChemPhysChem 2020, 21, 145–148. [Google Scholar] [CrossRef]
- Guo, J.C.; Feng, L.Y.; Dong, C.; Zhai, H.J. Planar Pentacoordinate versus Tetracoordinate Carbons in Ternary CBe4Li4 and CBe4 Clusters. J. Phys. Chem. A 2018, 122, 8370–8376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.F.; Han, S.J.; Wu, Y.B.; Lu, H.G.; Lu, G. Thermodynamic Stability versus Kinetic Stability: Is the Planar Hexacoordinate Carbon Species D3h CN3 Viable? J. Phys. Chem. A 2014, 118, 3319–3325. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.B.; Duan, Y.; Lu, H.G.; Li, S.D. CAl2 and Its Salt Complex LiCAl2: Anionic Global Minima with Planar Pentacoordinate Carbon. J. Phys. Chem. A 2012, 116, 3290–3294. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.C.; Feng, L.Y.; Zhang, X.Y.; Zhai, H.J. Star-Like CBe5 Cluster: Planar Pentacoordinate Carbon, Superalkali Cation, and Multifold (π and σ) Aromaticity. J. Phys. Chem. A 2018, 122, 1138–1145. [Google Scholar] [CrossRef]
- Wang, M.H.; Dong, X.; Ding, Y.H.; Cui, Z.H. Avoided Spin Coupling: An Unexpected σ-σ Diradical in Global Planar Pentacoordinate Carbon. Chem. Commun. 2020, 56, 7285–7288. [Google Scholar] [CrossRef]
- Guo, J.C.; Feng, L.Y.; Barroso, J.; Merino, G.; Zhai, H.J. Planar or Tetrahedral? A Ternary 17-Electron CBe5 Cluster with Planar Pentacoordinate Carbon. Chem. Commun. 2020, 56, 8305–8308. [Google Scholar] [CrossRef]
- Wang, Y.; Li, F.; Li, Y.; Chen, Z. Semi-Metallic Be5C2 Monolayer Global Minimum with Quasi-Planar Pentacoordinate Carbons and Negative Poisson’s Ratio. Nat. Commun. 2016, 7, 11488. [Google Scholar] [CrossRef]
- Li, Y.; Li, F.; Zhou, Z.; Chen, Z. SiC2 Silagraphene and Its One-Dimensional Derivatives: Where Planar Tetracoordinate Silicon Happens. J. Am. Chem. Soc. 2011, 133, 900–908. [Google Scholar] [CrossRef]
- Wang, M.H.; Dong, X.; Cui, Z.H.; Orozco-Ic, M.; Ding, Y.H.; Barroso, J.; Merino, G. Planar Pentacoordinate Silicon and Germanium Atoms. Chem. Commun. 2020, 56, 13772–13775. [Google Scholar] [CrossRef]
- Jimenez-Izal, E.; Saeys, M.; Alexandrova, A.N. Metallic and Magnetic 2D Materials Containing Planar Tetracoordinated C and N. J. Phys. Chem. C 2016, 120, 21685–21690. [Google Scholar] [CrossRef]
- Zhang, C.; Tian, Z.; Jia, W.; Mo, Y. Rational Design of Porous Organic Molecules (POMs) Based on B-Heterocyclic Carbenes. Mol. Syst. Des. Eng. 2021. [Google Scholar] [CrossRef]
- Röttger, D.; Erker, G. Compounds Containing Planar-Tetracoordinate Carbon. Angew. Chem. Int. Ed. Engl. 1997, 36, 812–827. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.S.; Boldyrev, A.I.; Simons, J. Tetracoordinated Planar Carbon in the Al4C− Anion. A Combined Photoelectron Spectroscopy and ab Initio Study. J. Am. Chem. Soc. 1999, 121, 6033–6038. [Google Scholar] [CrossRef]
- Wang, L.S.; Boldyrev, A.I.; Li, X.; Simons, J. Experimental Observation of Pentaatomic Tetracoordinate Planar Carbon-Containing Molecules. J. Am. Chem. Soc. 2000, 122, 7681–7687. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.F.; Wang, L.S.; Geske, G.; Boldyrev, A. Pentaatomic Tetracoordinate Planar Carbon, [CAl4]2−: A New Structural Unit and Its Salt Complexes. Angew. Chem. Int. Ed. 2000, 39, 3630–3632. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Yu, S.; Ding, Y.H.; Bowen, K.H. Identifying the Hydrogenated Planar Tetracoordinate Carbon: A Combined Experimental and Theoretical Study of CAl4H and CAl4H−. J. Phys. Chem. Lett. 2017, 8, 2263–2267. [Google Scholar] [CrossRef]
- Priyakumar, U.D.; Reddy, A.S.; Sastry, G.N. The Design of Molecules Containing Planar Tetracoordinate Carbon. Tetrahedron Lett. 2004, 45, 2495–2498. [Google Scholar] [CrossRef]
- Perez, N.; Heine, T.; Barthel, R.; Seifert, G.; Vela, A.; Mendez-Rojas, M.A.; Merino, G. Planar Tetracoordinate Carbons in Cyclic Hydrocarbons. Org. Lett. 2005, 7, 1509–1512. [Google Scholar] [CrossRef]
- Sateesh, B.; Srinivas Reddy, A.; Narahari Sastry, G. Towards Design of the Smallest Planar Tetracoordinate Carbon and Boron Systems. J. Comput. Chem. 2007, 28, 335–343. [Google Scholar] [CrossRef]
- Cui, Z.H.; Ding, Y.H.; Cabellos, J.L.; Osorio, E.; Islas, R.; Restrepo, A.; Merino, G. Planar Tetracoordinate Carbons with A Double Bond in CAl3E Clusters. Phys. Chem. Chem. Phys. 2015, 17, 8769–8775. [Google Scholar] [CrossRef]
- Suresh, C.H.; Frenking, G. Direct 1-3 Metal-Carbon Bonding and Planar Tetracoordinated Carbon in Group 6 Metallacyclobutadienes. Organometallics 2010, 29, 4766–4769. [Google Scholar] [CrossRef]
- Cui, Z.H.; Contreras, M.; Ding, Y.H.; Merino, G. Planar Tetracoordinate Carbon versus Planar Tetracoordinate Boron: The Case of CB4 and Its Cation. J. Am. Chem. Soc. 2011, 133, 13228–13231. [Google Scholar] [CrossRef] [PubMed]
- Thimmakondu, V.S.; Thirumoorthy, K. Si3C2H2 Isomers with A Planar Tetracoordinate Carbon or Silicon atom(s). Comput. Theor. Chem. 2019, 1157, 40–46. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Karton, A.; Thimmakondu, V.S. From High-Energy C7H2 Isomers with A Planar Tetracoordinate Carbon Atom to An Experimentally Known Carbene. J. Phys. Chem. A 2018, 122, 9054–9064. [Google Scholar] [CrossRef] [PubMed]
- Thirumoorthy, K.; Thimmakondu, V.S. Flat Crown Ethers with Planar Tetracoordinate Carbon Atoms. Int. J. Quantum Chem. 2020, e26479. [Google Scholar] [CrossRef]
- Raghunathan, S.; Yadav, K.; Rojisha, V.C.; Jaganade, T.; Prathyusha, V.; Bikkina, S.; Lourderaj, U.; Priyakumar, U.D. Transition Between [R]- and [S]-Stereoisomers without Bond Breaking. Phys. Chem. Chem. Phys. 2020, 22, 14983–14991. [Google Scholar] [CrossRef]
- Job, N.; Karton, A.; Thirumoorthy, K.; Cooksy, A.L.; Thimmakondu, V.S. Theoretical Studies of SiC4H2 Isomers Delineate Three Low-Lying Silylidenes Are Missing in the Laboratory. J. Phys. Chem. A 2020, 124, 987–1002. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, P.; Liang, J.; Jia, W.; Cao, Z. Theoretical Study on a Family of Organic Molecules with Planar Tetracoordinate Carbon. J. Mol. Struct. THEOCHEM 2010, 941, 41–46. [Google Scholar] [CrossRef]
- Yañez, O.; Vásquez-Espinal, A.; Báez-Grez, R.; Rabanal-León, W.A.; Osorio, E.; Ruiz, L.; Tiznado, W. Carbon Rings Decorated with Group 14 Elements: New Aromatic Clusters Containing Planar Tetracoordinate Carbon. New J. Chem. 2019, 43, 6781–6785. [Google Scholar]
- Van’t Hoff, J.H. A Suggestion Looking to the Extension Into Space of the Structural Formulas at Present Used in Chemistry. And A Note Upon the Relation Between the Optical Activity and the Chemical Constitution of Organic Compounds. Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445–454. [Google Scholar]
- Le-Bel, J.A. On the Relations Which Exist Between the Atomic Formulas of Organic Compounds and the Rotatory Power of Their Solutions. Bull. Soc. Chim. Fr. 1874, 22, 337–347. [Google Scholar]
- Würthwein, E.U.; von Ragué Schleyer, P. Planar Tetracoordinate Silicon. Angew. Chem. Int. Ed. Engl. 1979, 18, 553–554. [Google Scholar] [CrossRef]
- Guo, J.C.; Miao, C.Q.; Ren, G.M. Planar Tetracoordinate Si and Ge in π-Aromatic X3 (X=Si, Ge) Cations. Comput. Theor. Chem. 2014, 1032, 7–11. [Google Scholar] [CrossRef]
- Guo, J.C.; Wu, H.X.; Ren, G.M.; Miao, C.Q.; Li, Y.X. D3h X3 (X=C, Si and Ge): Superalkali Cations Containing Three Planar Tetracoordinate X Atoms. Comput. Theor. Chem. 2016, 1083, 1–6. [Google Scholar] [CrossRef]
- Li, S.D.; Miao, C.Q.; Guo, J.C.; Ren, G.M. Planar Tetra-, Penta-, Hexa-, Hepta-, and Octacoordinate Silicons: A Universal Structural Pattern. J. Am. Chem. Soc. 2004, 126, 16227–16231. [Google Scholar] [CrossRef]
- Sui, J.J.; Xu, J.; Ding, Y.H. A Template for A Planar Tetracoordinate Heavier Group 14 Atom: A Global Study of C2Si2Xq (X = C, Si, Ge, Sn, Pb; q = +1, 0, −1). Dalton Trans. 2016, 45, 56–60. [Google Scholar] [CrossRef]
- Xu, J.; Ding, Y.H. Pentaatomic Planar Tetracoordinate Silicon with 14 Valence Electrons: A Large-Scale Global Search of SiXn (n + m = 4; q = 0, ±1, −2; X, Y = Main Group Elements From H to Br). J. Comput. Chem. 2015, 36, 355–360. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Mo, Y.; Cao, Z. Planar Tetracoordinate Silicon in Organic Molecules As Carbenoid-Type Amphoteric Centers: A Computational Study. Chem. Eur. J. 2020, 26, 1–9. [Google Scholar] [CrossRef]
- Ebner, F.; Greb, L. Calix[4]pyrrole Hydridosilicate: The Elusive Planar Tetracoordinate Silicon Imparts Striking Stability to Its Anionic Silicon Hydride. J. Am. Chem. Soc. 2018, 140, 17409–17412. [Google Scholar] [CrossRef]
- Ghana, P.; Rump, J.; Schnakenburg, G.; Arz, M.I.; Filippou, A.C. Planar Tetracoordinated Silicon (ptSi): Room-Temperature Stable Compounds Containing Anti-van’t Hoff/Le Bel Silicon. J. Am. Chem. Soc. 2020. [Google Scholar] [CrossRef]
- Yañez, O.; Vásquez-Espinal, A.; Pino-Rios, R.; Ferraro, F.; Pan, S.; Osorio, E.; Merino, G.; Tiznado, W. Exploiting Electronic Strategies to Stabilize A Planar Tetracoordinate Carbon in Cyclic Aromatic Hydrocarbons. Chem. Commun. 2017, 53, 12112–12115. [Google Scholar] [CrossRef] [PubMed]
- Reusch, E.; Kaiser, D.; Schleier, D.; Buschmann, R.; Krueger, A.; Hermann, T.; Engels, B.; Fischer, I.; Hemberger, P. Pentadiynylidene and Its Methyl-Substituted Derivates: Threshold Photoelectron Spectroscopy of R1-C5-R2 Triplet Carbon Chains. J. Phys. Chem. A 2019, 123, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, J.; Bodi, A.; Hemberger, P. Nitrogen Matters: The Difference Between PANH and PAH Formation. Phys. Chem. Chem. Phys. 2018, 20, 29910–29917. [Google Scholar] [CrossRef] [PubMed]
- Henkel, S.; Huynh, Y.A.; Neuhaus, P.; Winkler, M.; Sander, W. Tunneling Rearrangement of 1-Azulenylcarbene. J. Am. Chem. Soc. 2012, 134, 13204–13207. [Google Scholar] [CrossRef]
- Fulara, J.; Nagy, A.; Filipkowski, K.; Thimmakondu, V.S.; Stanton, J.F.; Maier, J.P. Electronic Transitions of C6 Isomers: Neon Matrix and Theoretical Studies. J. Phys. Chem. A 2013, 117, 13605–13615. [Google Scholar] [CrossRef]
- Thimmakondu, V.S. MgC2H2 isomers—Simple Penta-Atomic Molecules Missing in the Laboratory. Chem. Phys. 2020, 538, 110899. [Google Scholar] [CrossRef]
- Pandey, A.P.; Padidela, U.K.; Thulasiraman, L.K.; Sethu, R.; Vairaprakash, P.; Thimmakondu, V.S. MgC6H2 Isomers: Potential Candidates for Laboratory and Radioastronomical Studies. J. Phys. Chem. A 2020, 124, 7518–7525. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Cooksy, A.; Thimmakondu, V.S. Si2C5H2 Isomers—Search Algorithms Versus Chemical Intuition. Phys. Chem. Chem. Phys. 2020, 22, 5865–5872. [Google Scholar] [CrossRef]
- Saunders, M. Stochastic Search for Isomers on a Quantum Mechanical Surface. J. Comput. Chem. 2004, 25, 621–626. [Google Scholar] [CrossRef]
- Bera, P.P.; Sattelmeyer, K.W.; Saunders, M.; Schaefer, H.F.; Schleyer, P.V.R. Mindless Chemistry. J. Phys. Chem. A 2006, 110, 4287–4290. [Google Scholar] [CrossRef]
- Zhao, J.; Xie, R.H. Genetic Algorithms for the Geometry Optimization of Atomic and Molecular Clusters. J. Comput. Theor. Nanosci. 2004, 1, 117–131. [Google Scholar] [CrossRef]
- Grande-Aztatzi, R.; Martínez-Alanis, P.R.; Cabellos, J.L.; Osorio, E.; Martínez, A.; Merino, G. Structural Evolution of Small Gold Clusters Doped by One and Two Boron Atoms. J. Comput. Chem. 2014, 35, 2288–2296. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Manzanares, A.; Peña, J.; Azpiroz, J.M.; Merino, G. A Hierarchical Algorithm for Molecular Similarity (H-FORMS). J. Comput. Chem. 2015, 36, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Yañez, O.; Báez-Grez, R.; Inostroza, D.; Rabanal-León, W.A.; Pino-Rios, R.; Garza, J.; Tiznado, W. AUTOMATON: A Program That Combines a Probabilistic Cellular Automata and a Genetic Algorithm for Global Minimum Search of Clusters and Molecules. J. Chem. Theory Comput. 2019, 15, 1463–1475. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Fukui, K. The Path of Chemical Reactions—The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Chapter 10—Finding Minima, Transition States, and Following Reaction Pathways On Ab Initio Potential Energy Surfaces. In Theory and Applications of Computational Chemistry; Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 195–249. [Google Scholar]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Fallah-Bagher-Shaidaei, H.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.V.R. Which NICS Aromaticity Index for Planar π Rings Is Best? Org. Lett. 2006, 8, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Seal, P.; Chakrabarti, S. Is Nucleus-Independent Chemical Shift Scan a Reliable Aromaticity Index for Planar Heteroatomic Ring Systems? J. Phys. Chem. A 2007, 111, 9988–9994. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Stability Analysis for Solutions of the Closed Shell Kohn–Sham Equation. J. Chem. Phys. 1996, 104, 9047–9052. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Purvis, G.D.; Bartlett, R.J. A Full Coupled-Cluster Singles and Doubles Model: The Inclusion of Disconnected Triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A Fifth-Order Perturbation Comparison of Electron Correlation Theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Watts, J.; Kucharski, S.; Noga, J. Non-Iterative Fifth-Order Triple and Quadruple Excitation Energy Corrections in Correlated Methods. Chem. Phys. Lett. 1990, 165, 513–522. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use In Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Peterson, K.A.; Dunning, T.H. Accurate Correlation Consistent Basis Sets for Molecular Core–Valence Correlation Effects: The Second Row Atoms Al-Ar, and the First Row Atoms B-Ne Revisited. J. Chem. Phys. 2002, 117, 10548–10560. [Google Scholar] [CrossRef]
- Stanton, J.F.; Gauss, J.; Cheng, L.; Harding, M.E.; Matthews, D.A.; Szalay, P.G. CFOUR, Coupled-Cluster Techniques for Computational Chemistry, a Quantum-Chemical Program Package. With Contributions from A.A. Auer, R.J. Bartlett, U. Benedikt, C. Berger, D.E. Bernholdt, Y.J. Bomble, O. Christiansen, F. Engel, R. Faber, M. Heckert, O. Heun, M. Hilgenberg, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, T. Kirsch, K. Klein, W.J. Lauderdale, F. Lipparini, T. Metzroth, L.A. Mück, D.P. O’Neill, D.R. Price, E. Prochnow, C. Puzzarini, K. Ruud, F. Schiffmann, W. Schwalbach, C. Simmons, S. Stopkowicz, A. Tajti, J. Vázquez, F. Wang, J.D. Watts and the integral packages MOLECULE (J. Almlöf and P.R. Taylor), PROPS (P.R. Taylor), ABACUS (T. Helgaker, H.J. Aa. Jensen, P. Jørgensen, and J. Olsen), and ECP routines by A. V. Mitin and C. van Wüllen. For the Current Version. Available online: http://www.cfour.de (accessed on 11 December 2020).
- Gauss, J.; Stanton, J.F. Analytic CCSD(T) Second Derivatives. Chem. Phys. Lett. 1997, 276, 70–77. [Google Scholar] [CrossRef]
- Spackman, P.R.; Jayatilaka, D.; Karton, A. Basis Set Convergence of CCSD(T) Equilibrium Geometries Using a Large and Diverse Set of Molecular Structures. J. Chem. Phys. 2016, 145, 104101. [Google Scholar] [CrossRef]
- Feller, D.; Peterson, K.A. Probing the Limits of Accuracy in Electronic Structure Calculations: Is Theory Capable of Results Uniformly Better Than “Chemical Accuracy”? J. Chem. Phys. 2007, 126, 114105. [Google Scholar] [CrossRef]
- Wang, S.; Schaefer, H.F. The Small Planarization Barriers for the Amino Group in the Nucleic Acid Bases. J. Chem. Phys. 2006, 124, 044303. [Google Scholar] [CrossRef] [PubMed]
- Bak, K.L.; Gauss, J.; Jørgensen, P.; Olsen, J.; Helgaker, T.; Stanton, J.F. The Accurate Determination of Molecular Equilibrium Structures. J. Chem. Phys. 2001, 114, 6548. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Taylor, P.R. Structure and Vibrations of Small Carbon Clusters from Coupled-Cluster Calculations. J. Phys. Chem. 1996, 100, 6047. [Google Scholar] [CrossRef]
- Xie, Y.; Scuseria, G.E.; Yates, B.F.; Yamaguchi, Y.; Schaefer, H.F. Methylnitrene: Theoretical Predictions of its Molecular Structure and Comparison with the Conventional C-N Single Bond in Methylamine. J. Am. Chem. Soc. 1989, 111, 5181. [Google Scholar] [CrossRef]
- Thimmakondu, V.S. The Equilibrium Geometries of Heptatriynylidene, Cyclohepta-1,2,3,4-tetraen-6-yne, and Heptahexaenylidene. Comput. Theor. Chem. 2016, 1079, 1–10. [Google Scholar] [CrossRef]
- Thimmakondu, V.S.; Karton, A. Energetic and Spectroscopic Properties of the Low-Lying C7H2 Isomers: A High-Level Ab Initio Perspective. Phys. Chem. Chem. Phys. 2017, 19, 17685–17697. [Google Scholar] [CrossRef]
- Thimmakondu, V.S.; Karton, A. The Quest for the Carbene Bent-Pentadiynylidene Isomer of C5H2. Chem. Phys. 2018, 515, 411–417. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Viji, M.; Pandey, A.P.; Netke, T.G.; Sekar, B.; Yadav, G.; Deshpande, S.; Thimmakondu, V.S. Many Unknowns Below or Close to the Experimentally Known Cumulene Carbene—A Case Study of C9H2 Isomers. Chem. Phys. 2019, 527, 110496. [Google Scholar] [CrossRef]
- Thimmakondu, V.S.; Ulusoy, I.; Wilson, A.K.; Karton, A. Theoretical Studies of Two Key Low-Lying Carbenes of C5H2 Missing in the Laboratory. J. Phys. Chem. A 2019, 123, 6618–6627. [Google Scholar] [CrossRef] [PubMed]
- Thimmakondu, V.S. Molecules in Laboratory and in Interstellar Space? In Proceedings of the 71st International Symposium on Molecular Spectroscopy, Urbana, IL, USA, 20–24 June 2016; p. RH05. [Google Scholar]
- Tsipis, A.C.; Depastas, I.G.; Tsipis, C.A. Diagnosis of the σ-, π-and (σ+ π)-Aromaticity by the Shape of the NICSzz-Scan Curves and Symmetry-Based Selection Rules. Symmetry 2010, 2, 284–319. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, M.; Kanatomi, Y.; Minoura, M.; Hatanaka, M.; Morokuma, K.; Ishimura, K.; Saito, M. Double Aromaticity Arising From σ- and π-Rings. Commun. Chem. 2018, 1, 60. [Google Scholar] [CrossRef]
- Kalita, A.J.; Rohman, S.S.; Kashyap, C.; Ullah, S.S.; Guha, A.K. Double Aromaticity in a BBe6 Cluster with A Planar Hexacoordinate Boron Structure. Chem. Commun. 2020, 56, 12597–12599. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.J.; Taylor, P.R. A Diagnostic for Determining the Quality of Single-Reference Electron Correlation Methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef]
- Petrie, S.; Herbst, E. Some Interstellar Reactions Involving Electrons and Neutral Species: Attachment and Isomerization. Astrophys. J. 1997, 491, 210–215. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, X.; Goddard, W.A. Doubly Hybrid Density Functional for Accurate Descriptions of Nonbond Interactions, Thermochemistry, and Thermochemical Kinetics. Proc. Natl. Acad. Sci. USA 2009, 106, 4963–4968. [Google Scholar] [CrossRef]
- Zhang, I.Y.; Wu, J.; Xu, X. Extending the Reliability and Applicability of B3LYP. Chem. Commun. 2010, 46, 3057–3070. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical Hybrid Density Functional with Perturbative Second-Order Correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef]
- Grimme, S.; Neese, F. Double-Hybrid Density Functional Theory for Excited Electronic States of Molecules. J. Chem. Phys. 2007, 127, 154116. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.; Swinehart, D.F. Algorithm 448: Number of Multiply-Restricted Partitions. Commun. ACM 1973, 16, 379. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | B3LYP | CCSD | CCSD(T) | CCSD | CCSD(T) | CCSD(T) |
|---|---|---|---|---|---|---|
| 6-311++G(2d,2p) | cc-pVDZ | cc-pVTZ | cc-pwCVTZ | |||
| R(CSi;CSi) | 1.9290 | 1.9549 | 1.9612 | 1.9177 | 1.9238 | 1.9081 |
| R(CC;CC) | 1.4679 | 1.4777 | 1.4875 | 1.4677 | 1.4781 | 1.4727 |
| R(CSi;CSi) | 1.7506 | 1.7679 | 1.7803 | 1.7488 | 1.7612 | 1.7475 |
| R(CC;CC) | 1.4179 | 1.4379 | 1.4393 | 1.4228 | 1.4244 | 1.4197 |
| R(CC) | 1.3813 | 1.3914 | 1.3993 | 1.3792 | 1.3876 | 1.3832 |
| R(CH;CH) | 1.0800 | 1.0946 | 1.0968 | 1.0796 | 1.0820 | 1.0806 |
| (CCC) | 104.00 | 104.49 | 104.21 | 104.44 | 104.11 | 104.10 |
| (CCSi;CCSi) | 60.24 | 60.10 | 60.32 | 60.49 | 60.69 | 60.64 |
| (CCC;CCC) | 108.81 | 108.57 | 108.66 | 108.48 | 108.60 | 108.63 |
| (CCH;CCH) | 124.87 | 124.82 | 124.78 | 124.71 | 124.67 | 124.69 |
| Mode | Isomer 1 | Isotopic Shifts (cm) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Symmetry | Frequency | Intensity | Si–Si | C–C | H–D | ||||||
| cm | km mol | Si(4) | SiCH | C(1) | C(2) | C(6) | SiCH | H(8) | SiCD | ||
| 1 | 195.2 | 0.0 | 0.4 | 0.7 | 0.0 | 1.9 | 1.0 | 5.4 | 4.3 | 7.7 | |
| 2 | 219.4 | 14.7 | 0.3 | 0.6 | 3.9 | 1.0 | 0.3 | 6.6 | 3.5 | 8.0 | |
| 3 | 232.8 | 2.0 | 1.8 | 3.6 | 0.6 | 0.1 | 0.1 | 1.1 | 0.1 | 0.3 | |
| 4 | 427.7 | 106.7 | 0.7 | 1.4 | 3.3 | 4.0 | 1.0 | 12.7 | 7.9 | 14.9 | |
| 5 | 513.2 | 17.8 | 2.9 | 5.7 | 0.6 | 0.3 | 2.7 | 6.6 | 5.8 | 9.8 | |
| 6 | 576.7 | 0.0 | 0.0 | 0.0 | 0.0 | 5.3 | 5.0 | 19.0 | 37.6 | 56.1 | |
| 7 | 606.8 | 0.0 | 0.0 | 0.0 | 10.6 | 4.7 | 0.3 | 22.9 | 4.4 | 26.7 | |
| 8 | 616.0 | 43.6 | 2.6 | 5.4 | 5.3 | 2.0 | 1.4 | 11.5 | 4.8 | 11.0 | |
| 9 | 787.9 | 0.9 | 0.6 | 1.1 | 5.9 | 5.2 | 6.9 | 27.4 | 85.0 | 118.1 | |
| 10 | 829.8 | 20.6 | 0.0 | 0.0 | 0.0 | 0.5 | 3.2 | 7.3 | 50.6 | 76.7 | |
| 11 | 887.0 | 0.1 | 0.1 | 0.1 | 2.9 | 7.1 | 8.4 | 33.0 | 43.5 | 125.1 | |
| 12 | 915.7 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 4.6 | 9.4 | 33.7 | 96.6 | |
| 13 | 991.3 | 30.4 | 0.0 | 0.1 | 16.5 | 9.2 | 0.1 | 36.6 | 104.1 | 142.1 | |
| 14 | 1004.5 | 17.3 | 0.3 | 0.7 | 21.1 | 0.8 | 3.5 | 32.4 | 9.4 | 90.5 | |
| 15 | 1103.8 | 2.3 | 0.0 | 0.0 | 0.1 | 1.7 | 3.8 | 11.2 | 80.2 | 99.1 | |
| 16 | 1272.8 | 13.3 | 0.4 | 0.7 | 2.2 | 9.3 | 1.6 | 19.3 | 54.5 | 171.6 | |
| 17 | 1340.0 | 48.5 | 0.6 | 1.2 | 0.0 | 17.9 | 5.6 | 48.6 | 9.9 | 24.1 | |
| 18 | 1461.5 | 4.2 | 0.0 | 0.0 | 0.0 | 1.6 | 24.5 | 47.2 | 23.2 | 45.3 | |
| 19 | 1484.7 | 8.6 | 0.3 | 0.6 | 0.8 | 11.1 | 8.1 | 49.1 | 13.5 | 28.7 | |
| 20 | 3208.9 | 2.0 | 0.0 | 0.0 | 0.0 | 0.0 | 6.4 | 9.9 | 825.3 | 841.2 | |
| 21 | 3227.9 | 21.1 | 0.0 | 0.0 | 0.0 | 0.0 | 4.3 | 11.4 | 9.4 | 828.5 | |
| Isomer | Dissociation | |||
|---|---|---|---|---|
| Pathway | kcal mol | kcal mol | kcal mol | |
| 1 | A | 64.05 (TS-1;TS-2) | 54.64 (20) | 0.00 |
| B | 30.51 (TS-3) | 18.80 (4) | 22.76 | |
| C | 61.85 (TS-4) | 43.03 (19) | – | |
| D | 71.45 (TS-5) | 64.36 (22) | – | |
| 68.94 (TS-6) | 63.35 (21) | – | ||
| 62.71 (TS-7) | 17.53 (2) | 21.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thirumoorthy, K.; Chandrasekaran, V.; Cooksy, A.L.; Thimmakondu, V.S. Kinetic Stability of Si2C5H2 Isomer with a Planar Tetracoordinate Carbon Atom. Chemistry 2021, 3, 13-27. https://doi.org/10.3390/chemistry3010002
Thirumoorthy K, Chandrasekaran V, Cooksy AL, Thimmakondu VS. Kinetic Stability of Si2C5H2 Isomer with a Planar Tetracoordinate Carbon Atom. Chemistry. 2021; 3(1):13-27. https://doi.org/10.3390/chemistry3010002
Chicago/Turabian StyleThirumoorthy, Krishnan, Vijayanand Chandrasekaran, Andrew L. Cooksy, and Venkatesan S. Thimmakondu. 2021. "Kinetic Stability of Si2C5H2 Isomer with a Planar Tetracoordinate Carbon Atom" Chemistry 3, no. 1: 13-27. https://doi.org/10.3390/chemistry3010002
APA StyleThirumoorthy, K., Chandrasekaran, V., Cooksy, A. L., & Thimmakondu, V. S. (2021). Kinetic Stability of Si2C5H2 Isomer with a Planar Tetracoordinate Carbon Atom. Chemistry, 3(1), 13-27. https://doi.org/10.3390/chemistry3010002